
Combined Modeling Study of the Binding Characteristics of Natural Compounds, Derived from Psoralea Fruits, to β-Amyloid Peptide Monomer


Abstract
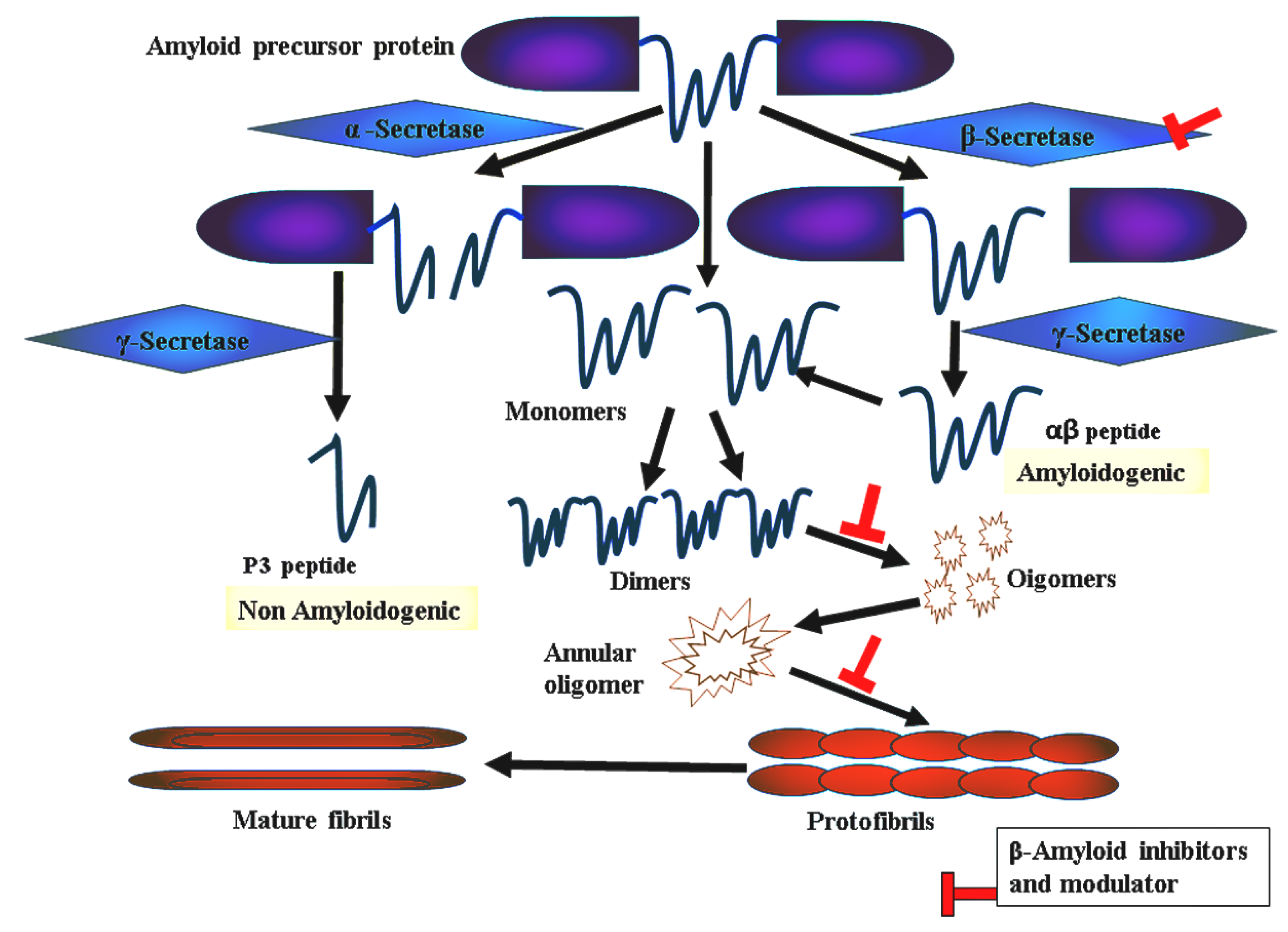
:1. Introduction
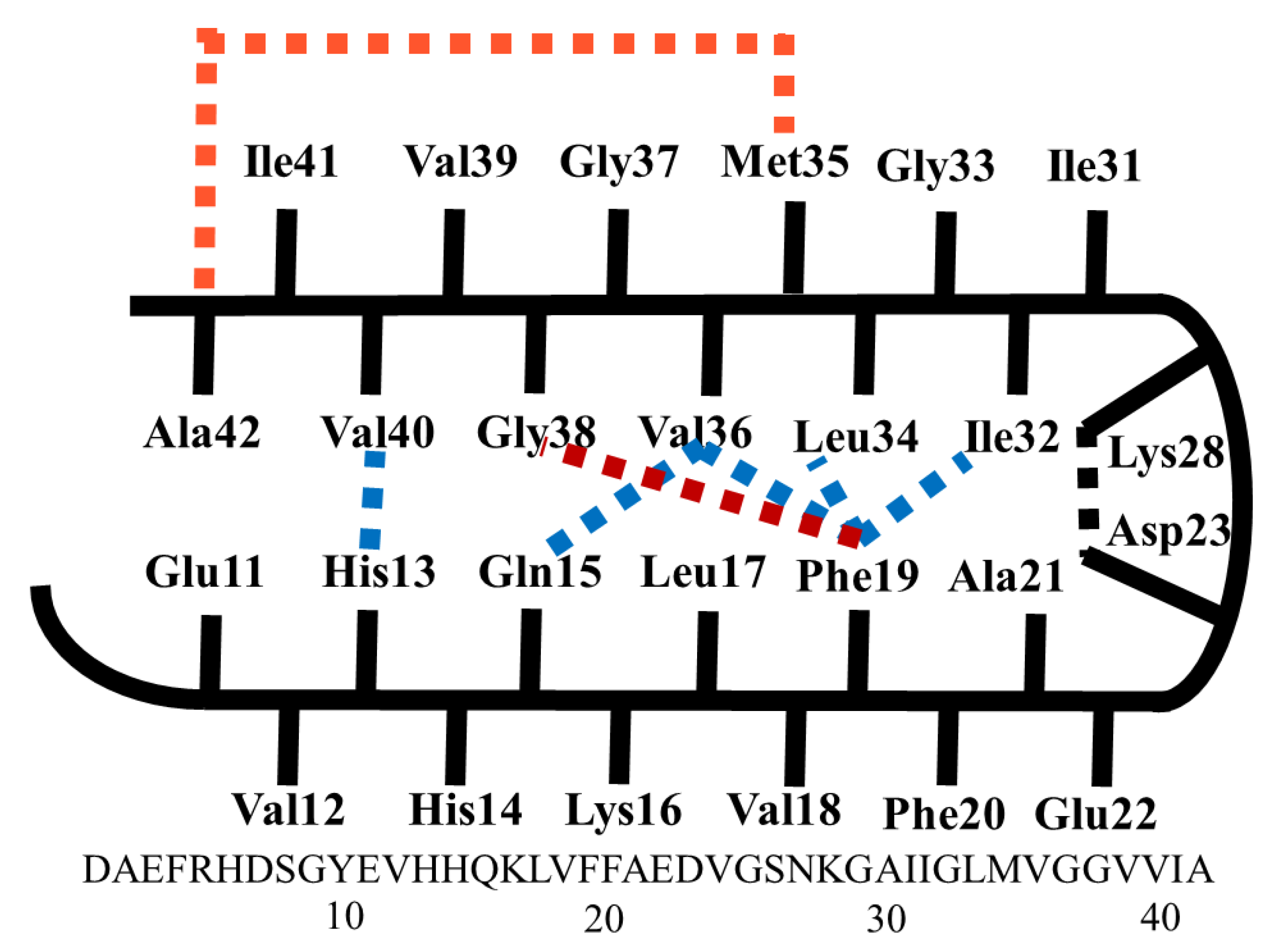
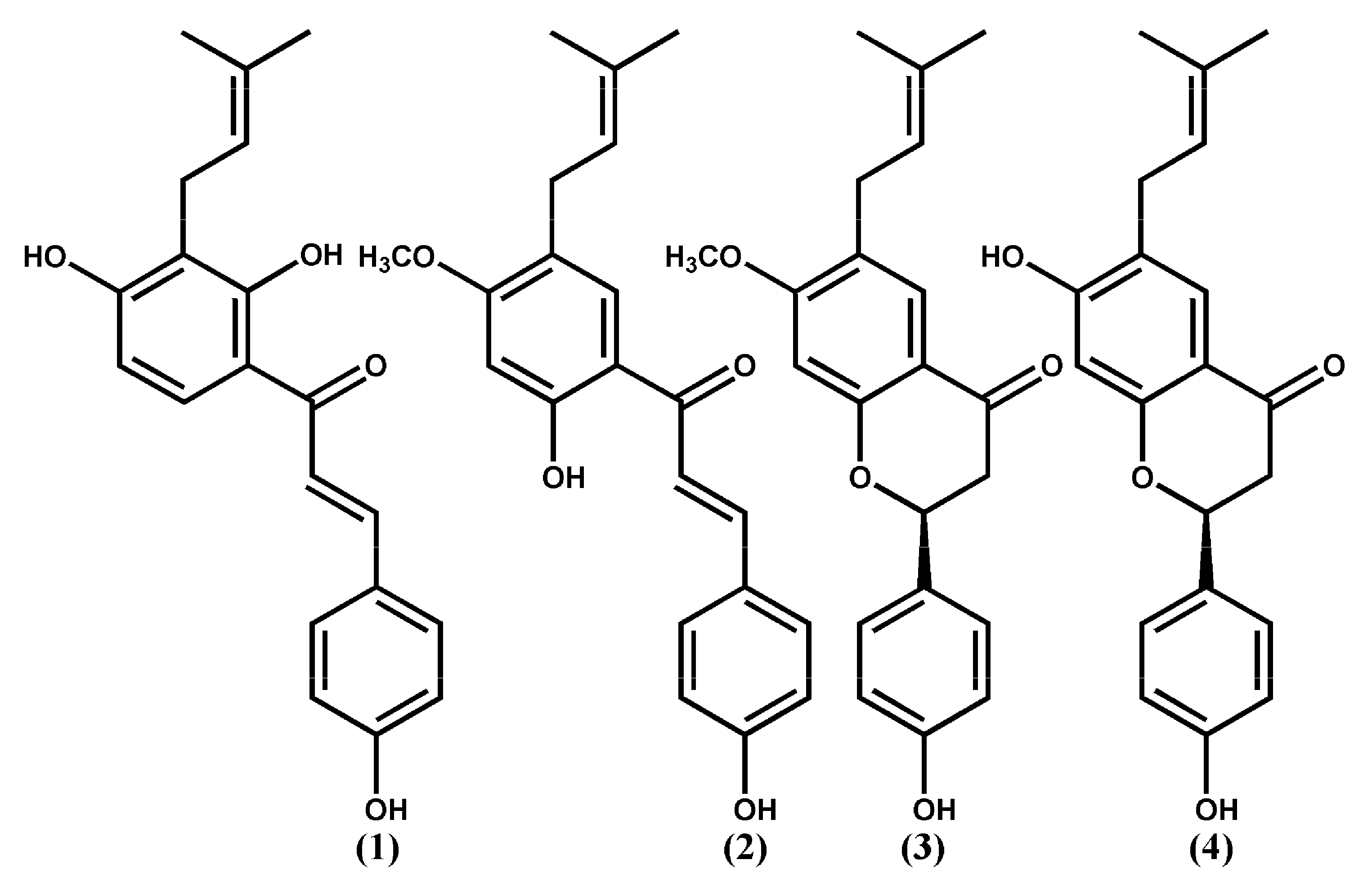
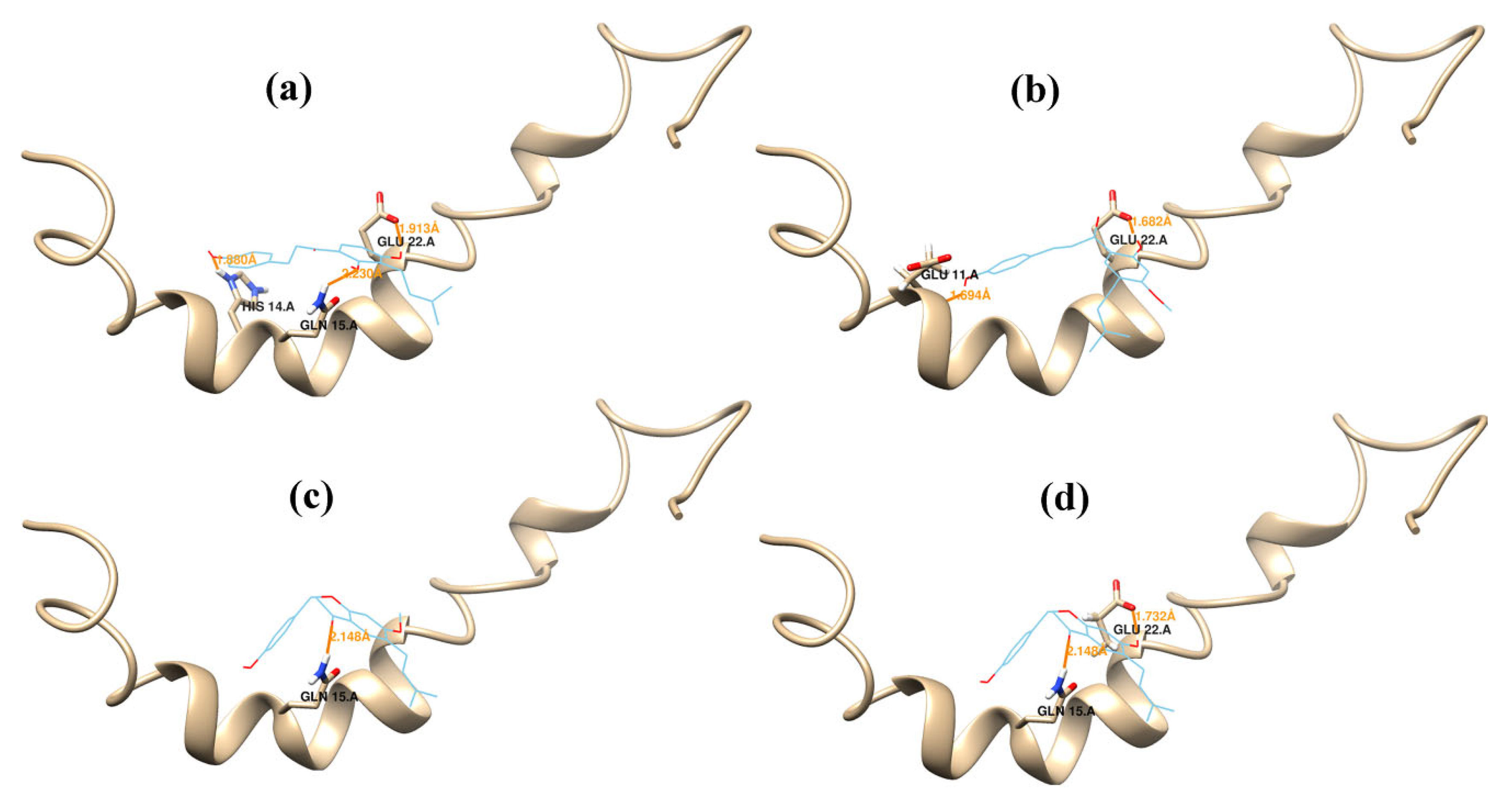
2. Results and Discussions
2.1. Docking Study
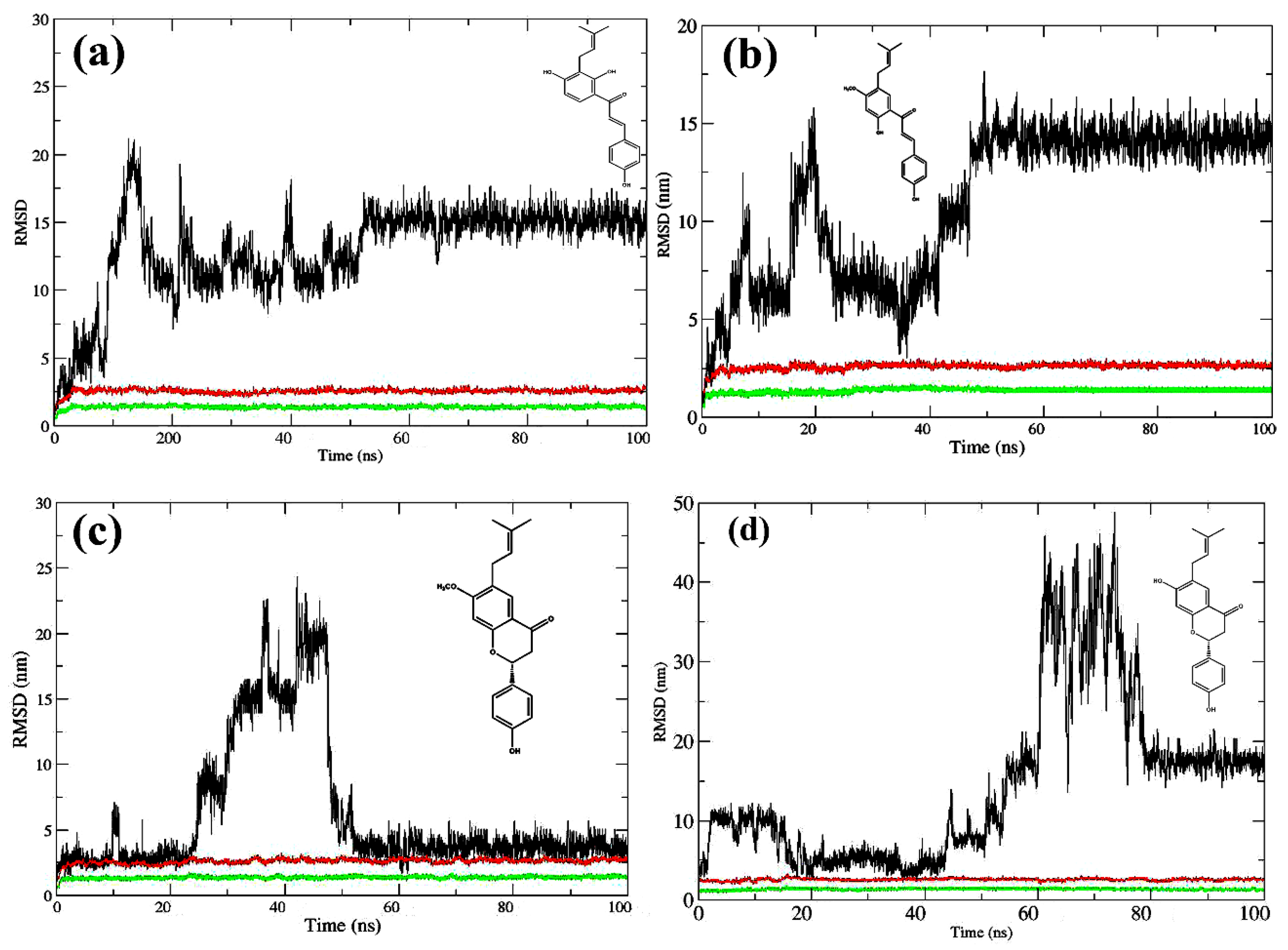
2.2. Molecular Dynamics Simulations
3. Methods
3.1. Docking Study
3.2. MD Simulations
3.3. Binding Free Energy Calculation
3.4. Clustering Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association Report. Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2010, 6, 158–194. [Google Scholar] [CrossRef]
- Alzheimer’s Association Report. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2015, 11, 332–384. [Google Scholar] [CrossRef]
- Santana, I.; Farinha, F.; Freitas, S.; Rodrigues, V.; Carvalho, Å. The epidemiology of dementia and Alzheimer’s disease in Portugal: Estimations of prevalence and treatment costs. Acta Méd. Port. 2015, 28, 182–188. [Google Scholar] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Arlington, VA, USA, 2013; Available online: http://www.dsm5.org/about/pages/dsmvoverview.aspx (accessed on 1 July 2013).
- Grasso, G.; Danani, A. Molecular simulations of amyloid beta assemblies. Adv. Phys. X 2020, 5, 1–27. [Google Scholar]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar]
- Tycko, R. Molecular structure of amyloid fibrils: Insights from solid-state NMR. Q. Rev. Biophys. 2006, 39, 1–55. [Google Scholar]
- Paravastua, A.K.; Leapman, R.D.; Yau, W.M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-β(1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar]
- Masuda, Y.; Uemura, S.; Nakanishi, A.; Ohashi, R.; Takegoshi, K.; Shimizu, T.; Shirasawa, T.; Irie, K. Verification of the C-terminal intramolecular β-sheet in Aβ42 aggregates using solid-state NMR: Implications for potent neurotoxicity through the formation of radicals. Bioorg. Med. Chem. Lett. 2008, 18, 3206–3210. [Google Scholar]
- Barten, D.M.; Albright, C.F. Therapeutic strategies for Alzheimer’s disease. Mol. Neurobiol. 2008, 37, 171–186. [Google Scholar]
- De Oliveira, O.V.; Gonçalves, A.S.; de Almeida, N.E.C. Insights into β-amyloid transition prevention by cucurbit[7]uril from molecular modeling. J. Biomol. Struct. Dyn. 2021, 2021, 1–11. [Google Scholar]
- Shuaib, S.; Goyal, B. Scrutiny of the mechanism of small molecule inhibitor preventing conformational transition of amyloid–b42 monomer: Insights from molecular dynamics simulations. J. Biomol. Struct. Dyn. 2018, 36, 663–678. [Google Scholar] [PubMed]
- Saini, R.K.; Shuaib, S.; Goyal, D.; Goyal, B. Insights into the inhibitory mechanism of a resveratrol and clioquinol hybrid against Ab42 aggregation and protofibril destabilization: A molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2019, 37, 3183–3197. [Google Scholar]
- Liu, F.; Ma, Z.; Sang, J.; Lu, F. Edaravone inhibits the conformational transition of amyloid–b42: Insights from molecular dynamics simulations. J. Biomol. Struct. Dyn. 2020, 38, 2377–2388. [Google Scholar] [PubMed]
- Zhao, L.; Wu, M.; Xiang, B. Analysis of Psoralea corylifolia L fruits in different regions. Chem. Pharm. Bull. 2005, 53, 1054–1057. [Google Scholar]
- Olivares, D.; Deshpande, V.K.; Shi, Y.; Lahiri, D.K.; Greig, N.H.; Rogers, J.T.; Huang, X. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr. Alzheimer Res. 2012, 9, 746–758. [Google Scholar] [PubMed]
- Nagata, T.; Shinagawa, S.; Nakajima, S.; Noda, Y.; Mimura, M. Pharmacotherapeutic combinations for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2022, 1, 1–11. [Google Scholar]
- Zhang, X.N.; Zhao, W.W.; Wang, Y.; Lu, J.J.; Chen, X.P. The chemical constituents and bioactivities of Psoralea corylifolia Linn.: A review. Am. J. Chin. Med. 2016, 44, 35–60. [Google Scholar]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008, 372, 216–223. [Google Scholar]
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, R.P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: Randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012, 11, 597–604. [Google Scholar] [PubMed]
- Ferreira, J.P.S.; Albuquerque, H.M.T.; Cardoso, S.M.; Silva, A.M.S.; Silva, V.L.M. Dual-target compounds for Alzheimer’s disease: Natural and synthetic AChE and BACE-1 dual-inhibitors and their structure-activity relationship (SAR). Eur. J. Med. Chem. 2021, 221, 113492. [Google Scholar] [PubMed]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar]
- Cui, Y.M.; Taniguchi, S.; Kuroda, T.; Hatano, T. Constituents of Psoralea corylifolia fruits and their effects on methicillin-resistant Staphylococcus aureus. Molecules 2015, 20, 12500–12511. [Google Scholar]
- Chen, Z.J.; Yang, Y.F.; Zhang, Y.T.; Yang, D.H. Dietary total prenylflavonoids from the fruits of Psoralea corylifolia L prevents age-related cognitive deficits and down-regulates Alzheimer’s markers in SAMP8 mice. Molecules 2018, 23, 196. [Google Scholar]
- Epperly, T.; Dunay, M.A.; Boice, J.L. Alzheimer Disease: Pharmacologic and Nonpharmacologic Therapies for Cognitive and Functional Symptoms. Am. Fam. Physician 2017, 95, 771–778. [Google Scholar] [PubMed]
- Chen, X.M.; Yang, Y.F.; Zhang, Y.T. Isobavachalcone and bavachinin from Psoraleae Fructus modulate Aβ42 aggregation process through different mechanisms in vitro. FEBS Lett. 2013, 587, 2930–2935. [Google Scholar]
- Xu, Q.X.; Hu, Y.; Li, G.Y.; Xu, W.; Zhang, Y.T.; Yang, X.W. Multi-target anti-Alzheimer activities of four prenylated compounds from Psoralea Fructus. Molecules 2018, 23, 614. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar]
- Hernández-Rodríguez, M.; Correa-Basurto, J.; Nicolás-Vázquez, M.I.; Miranda-Ruvalcaba, R.; Benítez-Cardoza, C.G.; Reséndiz-Albor, A.A.; Méndez-Méndez, J.V.; Rosales-Hernández, M.C. Virtual and in vitro screens reveal a potential pharmacophore that avoids the fibrillization of Aβ1–42. PLoS ONE 2015, 10, e0130263. [Google Scholar]
- Radwan, A.; Mahrous, G.M. Docking studies and molecular dynamics simulations of the binding characteristics of waldiomycin and its methyl ester analog to Staphylococcus aureus histidine kinase. PLoS ONE 2020, 15, e0234215. [Google Scholar]
- Bai, B.; Zou, R.; Chan, H.C.S.; Li, H.; Yuan, S. MolADI: A Web Server for Automatic Analysis of Protein–Small Molecule Dynamic Interactions. Molecules 2021, 26, 4625. [Google Scholar] [PubMed]
- De Boer, M.; Gouridis, G.; Vietrov, R.; Begg, S.L.; Schuurman-Wolters, G.K.; Husada, F.; Eleftheriadis, N.; Poolman, B.; McDevitt, C.A.; Cordes, T. Conformational and dynamic plasticity in substrate-binding proteins underlies selective transport in ABC importers. eLife 2019, 8, e44652. [Google Scholar] [PubMed]
- Salmaso, V.; Moro, S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An Overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [PubMed] [Green Version]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [PubMed]
- Itoh, S.G.; Okumura, H. Promotion and inhibition of amyloid-β peptide aggregation: Molecular Dynamics Studies. Int. J. Mol. Sci. 2021, 22, 1859. [Google Scholar]
- Salamanova, E.; Atanasova, M.; Dimitrov, I.; Doytchinova, I. Effects of curcumin and ferulic acid on the folding of amyloid-β peptide. Molecules 2021, 26, 2815. [Google Scholar]
- Shruthila, N.; Narayan, P.; Kumar, D.J.; Nagendra, H.G. Docking studies of plant polyphenols with Aβ fragments suggests determinants that enable design of inhibitors towards preventing aggregation events during Alzheimer’s. Int. J. Pharm. Sci. Drug Res. 2013, 5, 170–174. [Google Scholar]
- Pagano, K.; Tomaselli, S.; Molinari, H.; Ragona, L. Natural Compounds as inhibitors of Aβ peptide aggregation: Chemical requirements and molecular mechanisms. Front. Neurosci. 2020, 14, 619667. [Google Scholar]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D. Amber 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar]
- Yue, Y.; Sergio, E.W.; Felice, C.L. Understanding a substrate’s product regioselectivity in a family of enzymes: A case study of acetaminophen binding in cytochrome P450s. PLoS ONE 2014, 9, e87058. [Google Scholar]
- Turner, M.; Mutter, S.T.; Kennedy-Britten, O.D.; Platts, J.A. Molecular dynamics simulation of aluminium binding to amyloid-β and its effect on peptide structure. PLoS ONE 2019, 14, e0217992. [Google Scholar]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [PubMed]
- Lee, V.S.; Tue-ngeun, P.; Nangola, S.; Kitidee, K.; Jitonnom, J.; Nimmanpipug, P.; Jiranusornkul, S.; Tayapiwatana, C. Pairwise decomposition of residue interaction energies of single chain Fv with HIV-1 p17 epitope variants. Mol. Immunol. 2010, 47, 982–990. [Google Scholar] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | ∆G (Kcal Mol−1) | Estimated Ki (µM) | Inhibitory Rate % on Aβ142 Aggregations | H.b a.a | VdW a.a |
|---|---|---|---|---|---|
| 1 | −5.23 | 147.06 | 98 | His14, Gln15, Glu22 | Glu11, Val18, Phe19 |
| 2 | −4.78 | 315.52 | 90 | Glu11, Glu22 | His14, Gln15, Val18, Phe19 |
| 3 | −4.27 | 747.57 | 68 | Gln15 | Glu11, His14, Val18, Phe19, Phe29, Glu22, Asp23, Asn27 |
| 4 | −4.00 | 147.06 | 19 | Gln15, Glu22 | Glu11, His14, Val18, Phe19, Asp23 |
| Compounds | ΔGvdw | ΔGelec | ΔGpolarb | ΔGSurfc | ΔGMMGBSA |
|---|---|---|---|---|---|
| 1 | −23.8391 | −36.6043 | 40.9619 | −3.9710 | −23.4525 |
| 2 | −10.5171 | −45.8234 | 40.6926 | −3.4022 | −19.0388 |
| 3 | −19.8804 | −16.0076 | 23.9728 | −3.4274 | −15.3399 |
| 4 | −19.0178 | −1.8406 | 8.9947 | −2.2198 | −14.0815 |
| Aβ42 Residues | Glu11 | His14 | Gln15 | Leu17 | Val18 | Phe19 | Glu22 | Asn27 |
|---|---|---|---|---|---|---|---|---|
| 1 | −0.099 | −2.661 | −0.896 | −0.137 | −3.954 | −1.879 | −5.157 | −3.072 |
| 2 | −0.103 | −3.669 | −2.055 | −2.568 | −3.754 | −0.099 | −8.321 | −0.403 |
| 3 | −3.233 | v1.306 | −2.790 | −0.166 | −4.619 | −3.206 | −2.569 | −3.619 |
| 4 | −5.109 | −0.740 | −7.221 | −0.174 | −2.243 | −3.324 | −3.446 | −1.369 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radwan, A.; Alanazi, F. Combined Modeling Study of the Binding Characteristics of Natural Compounds, Derived from Psoralea Fruits, to β-Amyloid Peptide Monomer. Int. J. Mol. Sci. 2022, 23, 3546. https://doi.org/10.3390/ijms23073546
Radwan A, Alanazi F. Combined Modeling Study of the Binding Characteristics of Natural Compounds, Derived from Psoralea Fruits, to β-Amyloid Peptide Monomer. International Journal of Molecular Sciences. 2022; 23(7):3546. https://doi.org/10.3390/ijms23073546
Chicago/Turabian StyleRadwan, Awwad, and Fars Alanazi. 2022. "Combined Modeling Study of the Binding Characteristics of Natural Compounds, Derived from Psoralea Fruits, to β-Amyloid Peptide Monomer" International Journal of Molecular Sciences 23, no. 7: 3546. https://doi.org/10.3390/ijms23073546