
Molecular Lego of Human Cytochrome P450: The Key Role of Heme Domain Flexibility for the Activity of the Chimeric Proteins

 , , ,
, , , 
Abstract
: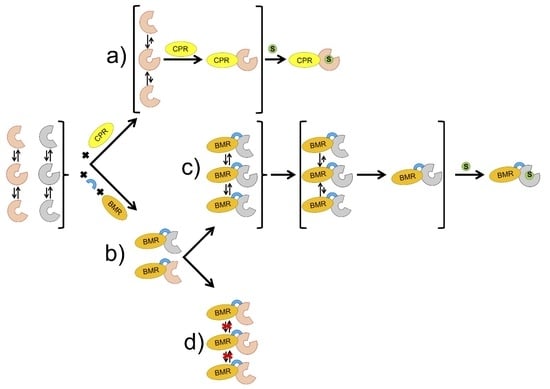
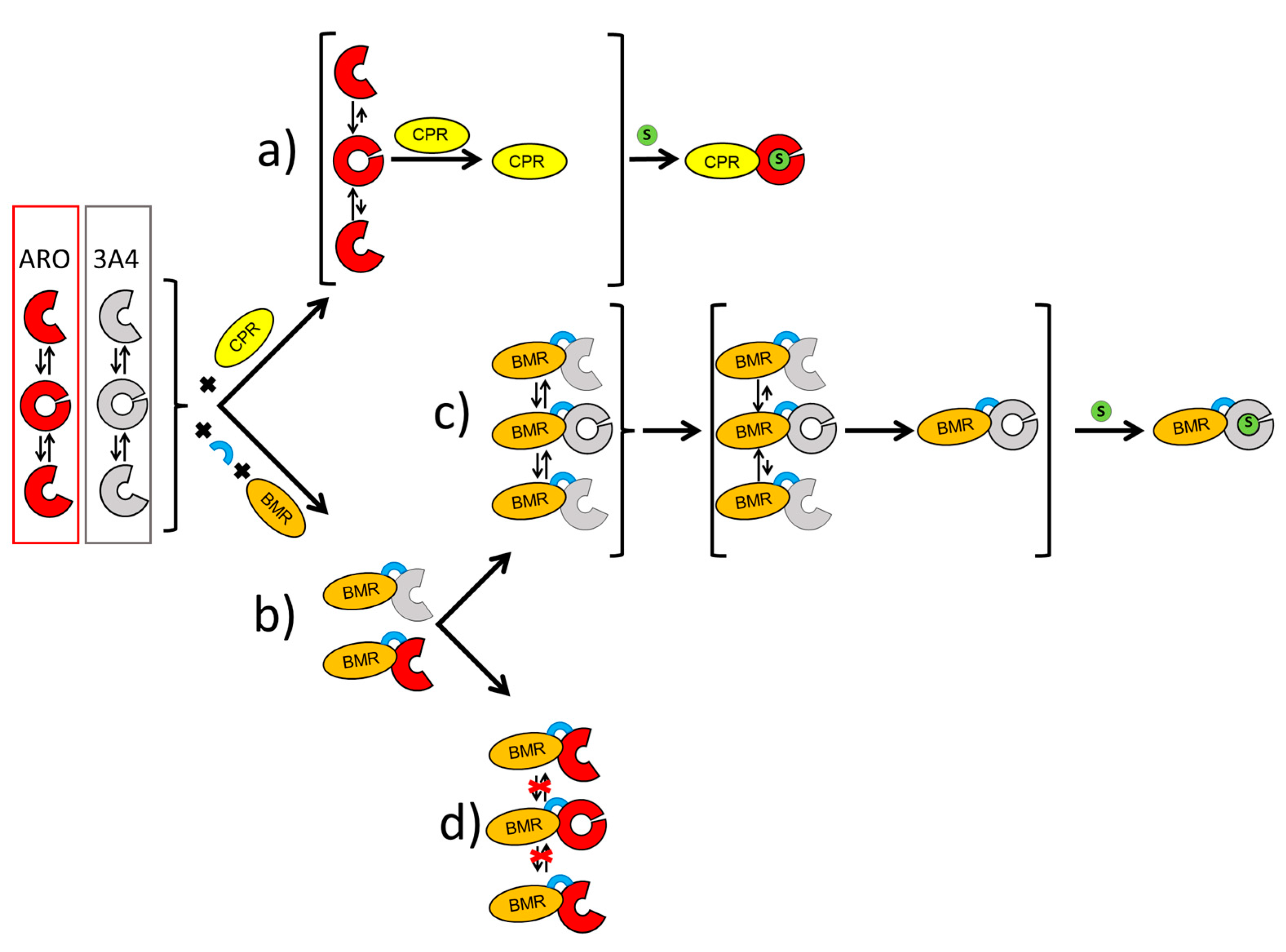
1. Introduction
2. Results
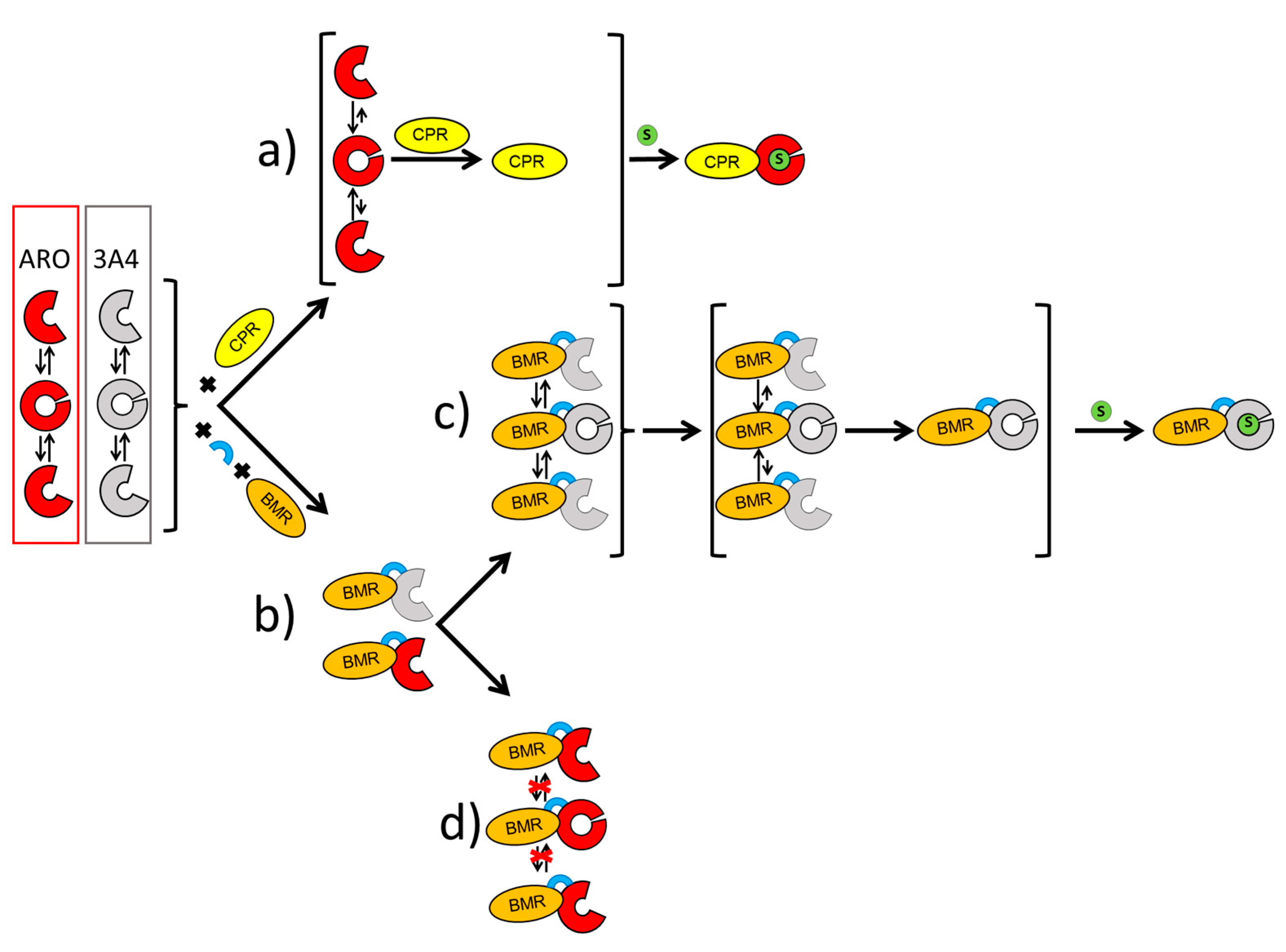
2.1. Characterization and Activity of the ARO-BMR Chimeras
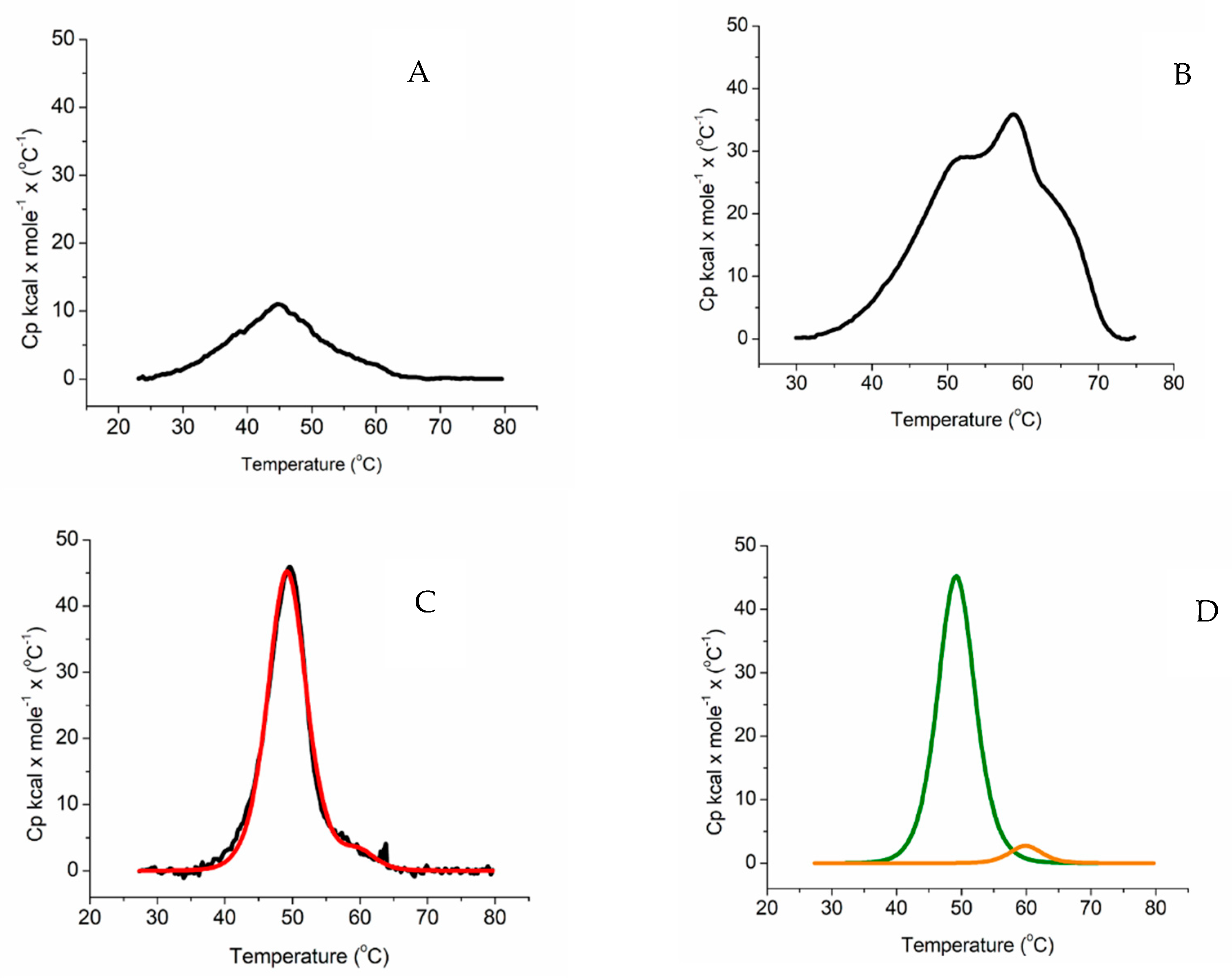
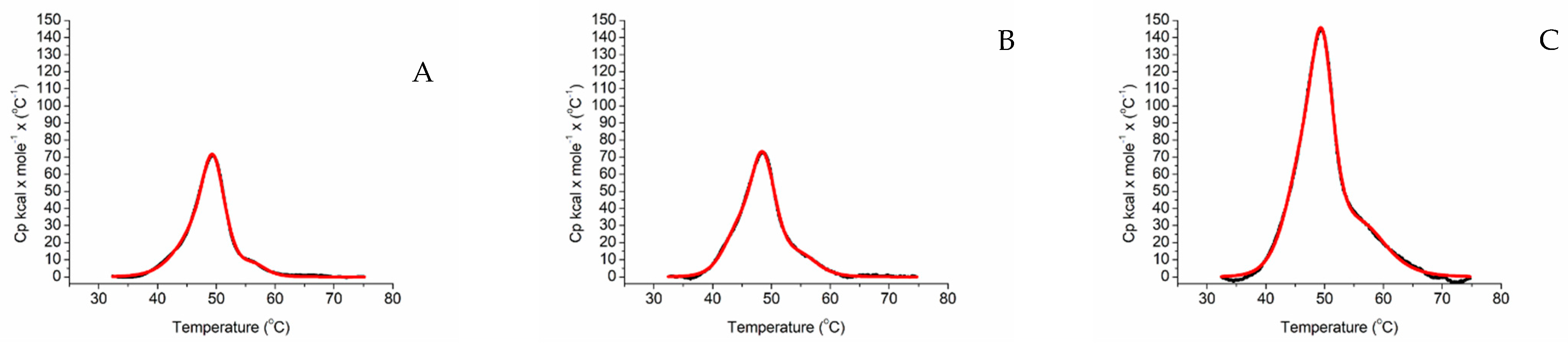
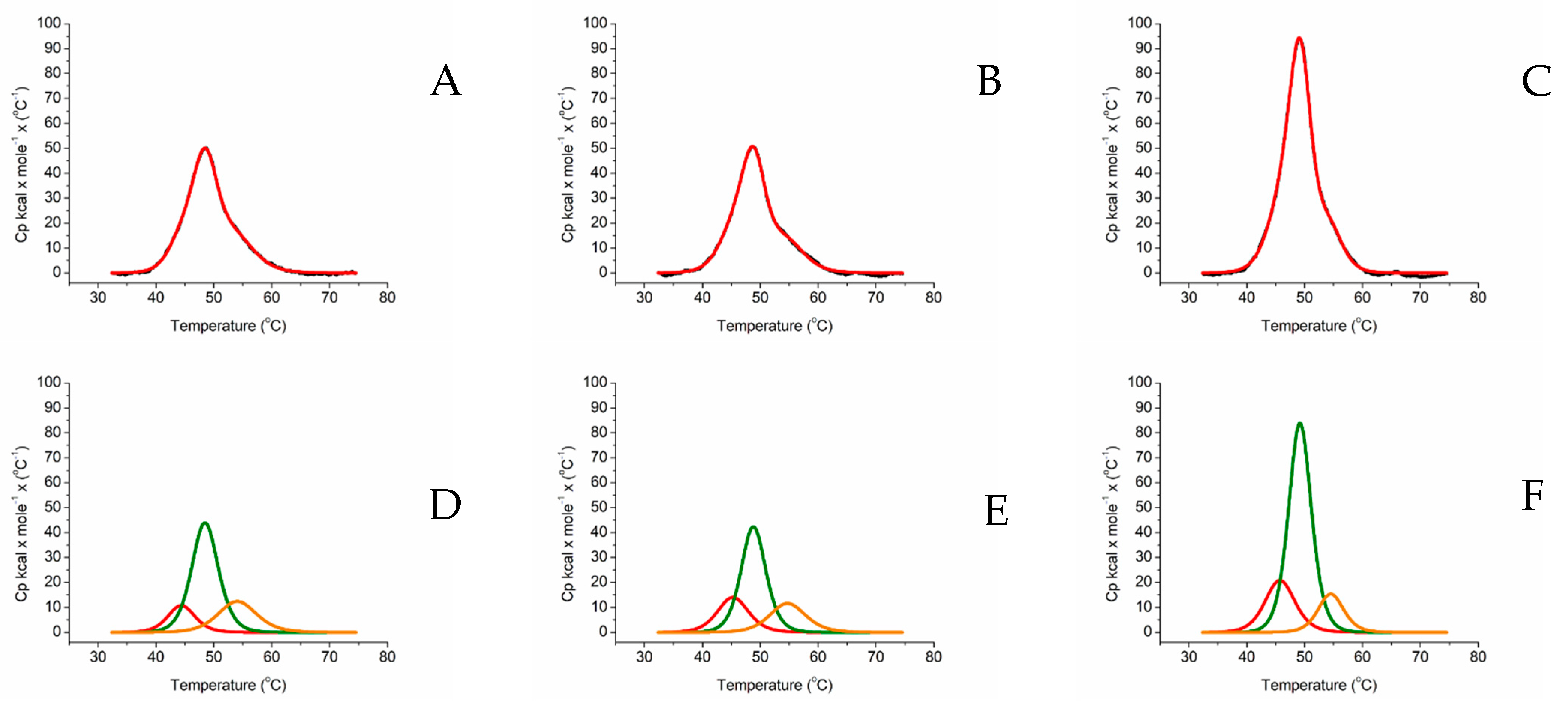
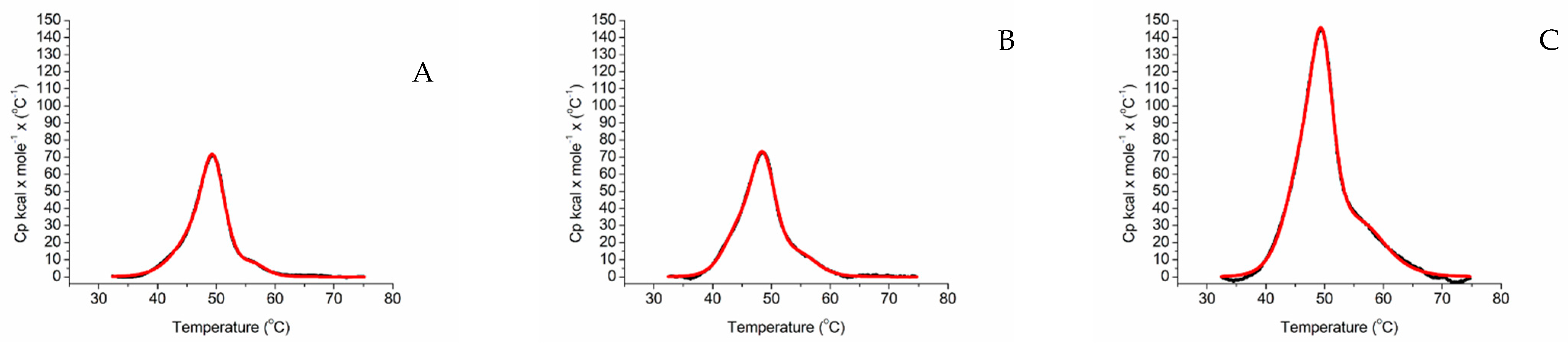
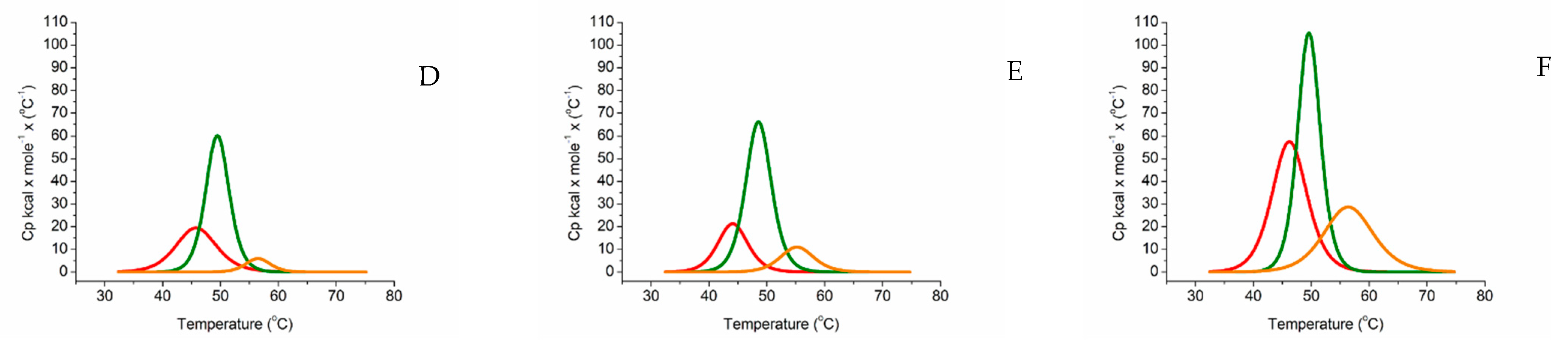
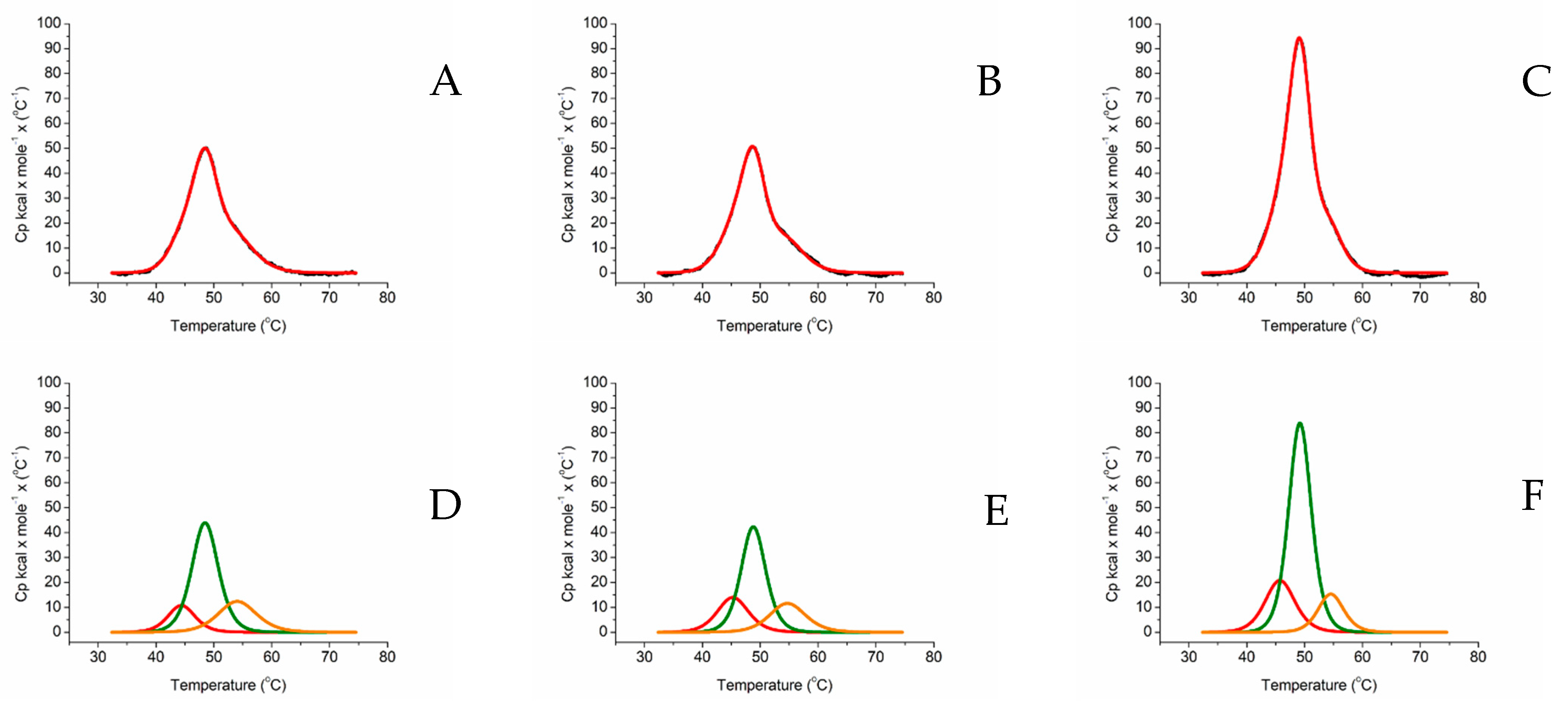
2.2. Differential Scanning Calorimetry
3. Discussion
- The transfer of electrons from NADPH to FMN via FAD reduction within the BMR domain.
- The transfer of electrons from FMN to the heme domain involving the formation of a functional P450-BMR electron-transfer complex.
- The binding of the substrate to the heme domain to form a catalytically competent complex.
4. Materials and Methods
4.1. Materials
4.2. Cloning of the BMR Domain of CYP450 BM3
4.3. Expression and Purification of BMR
4.4. Engineering of ARO-BMR Chimeras
4.5. Expression and Purification of Aromatase, CYP3A4, ARO-BMR, and 3A4-BMR Chimeras
4.6. Rapid Kinetics
4.7. Substrate Binding and Aromatase Activity Assay
4.8. Differential Scanning Calorimetry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Rendic, S.; Carlo, F.J.D. Human Cytochrome P450 Enzymes: A Status Report Summarizing Their Reactions, Substrates, Inducers, and Inhibitors. Drug Metab. Rev. 1997, 29, 413–580. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Common and Uncommon Cytochrome P450 Reactions Related to Metabolism and Chemical Toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Isin, E.M.; Guengerich, F.P. Complex Reactions Catalyzed by Cytochrome P450 Enzymes. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2007, 1770, 314–329. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Munro, A.W. Unusual Cytochrome P450 Enzymes and Reactions. J. Biol. Chem. 2013, 288, 17065–17073. [Google Scholar] [CrossRef] [Green Version]
- Di Nardo, G.; Gilardi, G. Natural Compounds as Pharmaceuticals: The Key Role of Cytochromes P450 Reactivity. Trends Biochem. Sci. 2020, 45, 511–525. [Google Scholar] [CrossRef]
- Das, A.; Sligar, S.G. Modulation of the Cytochrome P450 Reductase Redox Potential by the Phospholipid Bilayer. Biochemistry 2009, 48, 12104–12112. [Google Scholar] [CrossRef] [Green Version]
- Ortiz de Montellano, P.R. (Ed.) Cytochrome P450; Springer International Publishing: Cham, Switzerland, 2015; ISBN 978-3-319-12107-9. [Google Scholar]
- Sevrioukova, I.F.; Peterson, J.A. NADPH-P-450 Reductase: Structural and Functional Comparisons of the Eukaryotic and Prokaryotic Isoforms. Biochimie 1995, 77, 562–572. [Google Scholar] [CrossRef]
- Sevrioukova, I.; Shaffer, C.; Ballou, D.P.; Peterson, J.A. Equilibrium and Transient State Spectrophotometric Studies of the Mechanism of Reduction of the Flavoprotein Domain of P450BM-3. Biochemistry 1996, 35, 7058–7068. [Google Scholar] [CrossRef]
- Gorsky, L.D.; Koop, D.R.; Coon, M.J. On the Stoichiometry of the Oxidase and Monooxygenase Reactions Catalyzed by Liver Microsomal Cytochrome P-450. Products of Oxygen Reduction. J. Biol. Chem. 1984, 259, 6812–6817. [Google Scholar] [CrossRef]
- Hiroya, K.; Ishigooka, M.; Shimizu, T.; Hatano, M. Role of Glu318 and Thr319 in the Catalytic Function of Cytochrome P450 d (P4501A2): Effects of Mutations on the Methanol Hydroxylation. FASEB J. 1992, 6, 749–751. [Google Scholar] [CrossRef] [PubMed]
- Grinkova, Y.V.; Denisov, I.G.; McLean, M.A.; Sligar, S.G. Oxidase Uncoupling in Heme Monooxygenases: Human Cytochrome P450 CYP3A4 in Nanodiscs. Biochem. Biophys. Res. Commun. 2013, 430, 1223–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsunaga, I.; Yamada, M.; Kusunose, E.; Miki, T.; Ichihara, K. Further Characterization of Hydrogen Peroxide-Dependent Fatty Acid -Hydroxylase from Sphingomonas Paucimobilis. J. Biochem. 1998, 124, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, I.; Ueda, A.; Fujiwara, N.; Sumimoto, T.; Ichihara, K. Characterization of the YbdT Gene Product of Bacillus Subtilis: Novel Fatty Acid β-Hydroxylating Cytochrome P450. Lipids 1999, 34, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Girhard, M.; Schuster, S.; Dietrich, M.; Dürre, P.; Urlacher, V.B. Cytochrome P450 Monooxygenase from Clostridium Acetobutylicum: A New α-Fatty Acid Hydroxylase. Biochem. Biophys. Res. Commun. 2007, 362, 114–119. [Google Scholar] [CrossRef]
- Munro, A.W.; McLean, K.J.; Grant, J.L.; Makris, T.M. Structure and Function of the Cytochrome P450 Peroxygenase Enzymes. Biochem. Soc. Trans. 2018, 46, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Ciaramella, A.; Catucci, G.; Gilardi, G.; Di Nardo, G. Crystal Structure of Bacterial CYP116B5 Heme Domain: New Insights on Class VII P450s Structural Flexibility and Peroxygenase Activity. Int. J. Biol. Macromol. 2019, 140, 577–587. [Google Scholar] [CrossRef]
- Ciaramella, A.; Catucci, G.; Di Nardo, G.; Sadeghi, S.J.; Gilardi, G. Peroxide-Driven Catalysis of the Heme Domain of A. Radioresistens Cytochrome P450 116B5 for Sustainable Aromatic Rings Oxidation and Drug Metabolites Production. New Biotechnol. 2020, 54, 71–79. [Google Scholar] [CrossRef]
- Hannemann, F.; Bichet, A.; Ewen, K.M.; Bernhardt, R. Cytochrome P450 Systems—Biological Variations of Electron Transport Chains. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2007, 1770, 330–344. [Google Scholar] [CrossRef]
- Minerdi, D.; Sadeghi, S.J.; Di Nardo, G.; Rua, F.; Castrignanò, S.; Allegra, P.; Gilardi, G. CYP116B5: A New Class VII Catalytically Self-Sufficient Cytochrome P450 from A Cinetobacter Radioresistens That Enables Growth on Alkanes: Oxidation of n -Alkanes by a Novel Self-Suffcient Bacterial Cytochrome P450. Mol. Microbiol. 2015, 95, 539–554. [Google Scholar] [CrossRef]
- Correddu, D.; Di Nardo, G.; Gilardi, G. Self-Sufficient Class VII Cytochromes P450: From Full-Length Structure to Synthetic Biology Applications. Trends Biotechnol. 2021, 39, 1184–1207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xie, Z.; Liu, Z.; Zhou, S.; Ma, L.; Liu, W.; Huang, J.-W.; Ko, T.-P.; Li, X.; Hu, Y.; et al. Structural Insight into the Electron Transfer Pathway of a Self-Sufficient P450 Monooxygenase. Nat. Commun. 2020, 11, 2676. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Li, H.; Zhang, H.; Peterson, J.A.; Poulos, T.L. Structure of a Cytochrome P450-Redox Partner Electron-Transfer Complex. Proc. Natl. Acad. Sci. USA 1999, 96, 1863–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, P.A.; Shen, A.L.; Paschke, R.; Kasper, C.B.; Kim, J.-J.P. NADPH-Cytochrome P450 Oxidoreductase: STRUCTURAL BASIS FOR HYDRIDE AND ELECTRON TRANSFER. J. Biol. Chem. 2001, 276, 29163–29170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shumyantseva, V.V.; Ivanov, Y.D.; Bistolas, N.; Scheller, F.W.; Archakov, A.I.; Wollenberger, U. Direct Electron Transfer of Cytochrome P450 2B4 at Electrodes Modified with Nonionic Detergent and Colloidal Clay Nanoparticles. Anal. Chem. 2004, 76, 6046–6052. [Google Scholar] [CrossRef] [PubMed]
- Laursen, T.; Jensen, K.; Møller, B.L. Conformational Changes of the NADPH-Dependent Cytochrome P450 Reductase in the Course of Electron Transfer to Cytochromes P450. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2011, 1814, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Hanukoglu, I. Electron Transfer Proteins of Cytochrome P450 Systems. In Advances in Molecular and Cell Biology; Elsevier: Amsterdam, The Netherlands, 1996; Volume 14, pp. 29–56. ISBN 978-0-7623-0113-3. [Google Scholar]
- Porter, T.D. The Roles of Cytochromeb5 in Cytochrome P450 Reactions. J. Biochem. Mol. Toxicol. 2002, 16, 311–316. [Google Scholar] [CrossRef]
- Valetti, F.; Sadeghi, S.J.; Meharenna, Y.T.; Leliveld, S.R.; Gilardi, G. Engineering Multi-Domain Redox Proteins Containing Flavodoxin as Bio-Transformer: Preparatory Studies by Rational Design. Biosens. Bioelectron. 1998, 13, 675–685. [Google Scholar] [CrossRef]
- Fairhead, M.; Giannini, S.; Gillam, E.M.J.; Gilardi, G. Functional Characterisation of an Engineered Multidomain Human P450 2E1 by Molecular Lego. J. Biol. Inorg. Chem. 2005, 10, 842–853. [Google Scholar] [CrossRef]
- Dodhia, V.R.; Fantuzzi, A.; Gilardi, G. Engineering Human Cytochrome P450 Enzymes into Catalytically Self-Sufficient Chimeras Using Molecular Lego. J. Biol. Inorg. Chem. 2006, 11, 903–916. [Google Scholar] [CrossRef]
- Degregorio, D.; Sadeghi, S.J.; Di Nardo, G.; Gilardi, G.; Solinas, S.P. Understanding Uncoupling in the Multiredox Centre P450 3A4–BMR Model System. J. Biol. Inorg. Chem. 2011, 16, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Degregorio, D.; D’Avino, S.; Castrignanò, S.; Di Nardo, G.; Sadeghi, S.J.; Catucci, G.; Gilardi, G. Human Cytochrome P450 3A4 as a Biocatalyst: Effects of the Engineered Linker in Modulation of Coupling Efficiency in 3A4-BMR Chimeras. Front. Pharmacol. 2017, 8, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castrignanò, S.; D’Avino, S.; Di Nardo, G.; Catucci, G.; Sadeghi, S.J.; Gilardi, G. Modulation of the Interaction between Human P450 3A4 and B. Megaterium Reductase via Engineered Loops. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2018, 1866, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Castrignanò, S.; Di Nardo, G.; Sadeghi, S.J.; Gilardi, G. Influence of Inter-Domain Dynamics and Surrounding Environment Flexibility on the Direct Electrochemistry and Electrocatalysis of Self-Sufficient Cytochrome P450 3A4-BMR Chimeras. J. Inorg. Biochem. 2018, 188, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, G.; Breitner, M.; Sadeghi, S.J.; Castrignanò, S.; Mei, G.; Di Venere, A.; Nicolai, E.; Allegra, P.; Gilardi, G. Dynamics and Flexibility of Human Aromatase Probed by FTIR and Time Resolved Fluorescence Spectroscopy. PLoS ONE 2013, 8, e82118. [Google Scholar] [CrossRef] [Green Version]
- Hui Bon Hoa, G.; Di Primo, C.; Dondaine, I.; Sligar, S.G.; Gunsalus, I.C.; Douzou, P. Conformational Changes of Cytochromes P-450cam and P-450lin Induced by High Pressure. Biochemistry 1989, 28, 651–656. [Google Scholar] [CrossRef]
- Davydov, D.R.; Deprez, E.; Hoa, G.H.; Knyushko, T.V.; Kuznetsova, G.P.; Koen, Y.M.; Archakov, A.I. High-Pressure-Induced Transitions in Microsomal Cytochrome P450 2B4 in Solution: Evidence for Conformational Inhomogeneity in the Oligomers. Arch. Biochem. Biophys. 1995, 320, 330–344. [Google Scholar] [CrossRef]
- Martinis, S.A.; Blanke, S.R.; Hager, L.P.; Sligar, S.G.; Hoa, G.H.; Rux, J.J.; Dawson, J.H. Probing the Heme Iron Coordination Structure of Pressure-Induced Cytochrome P420cam. Biochemistry 1996, 35, 14530–14536. [Google Scholar] [CrossRef]
- Davydov, D.R.; Hui Bon Hoa, G.; Peterson, J.A. Dynamics of Protein-Bound Water in the Heme Domain of P450BM3 Studied by High-Pressure Spectroscopy: Comparison with P450cam and P450 2B4. Biochemistry 1999, 38, 751–761. [Google Scholar] [CrossRef]
- Tschirret-Guth, R.A.; Koo, L.S.; Hui Bon Hoa, G.; Ortiz de Montellano, P.R. Reversible Pressure Deformation of A Thermophilic Cytochrome P450 Enzyme (CYP119) and Its Active-Site Mutants. J. Am. Chem. Soc. 2001, 123, 3412–3417. [Google Scholar] [CrossRef]
- Di Nardo, G.; Breitner, M.; Bandino, A.; Ghosh, D.; Jennings, G.K.; Hackett, J.C.; Gilardi, G. Evidence for an Elevated Aspartate PK(a) in the Active Site of Human Aromatase. J. Biol. Chem. 2015, 290, 1186–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baravalle, R.; Di Nardo, G.; Bandino, A.; Barone, I.; Catalano, S.; Andò, S.; Gilardi, G. Impact of R264C and R264H Polymorphisms in Human Aromatase Function. J. Steroid Biochem. Mol. Biol. 2017, 167, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Baravalle, R.; Ciaramella, A.; Baj, F.; Di Nardo, G.; Gilardi, G. Identification of Endocrine Disrupting Chemicals Acting on Human Aromatase. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, V.; Choi, J.H.; Giacomini, K.M.; Miller, W.L. Substrate-Specific Modulation of CYP3A4 Activity by Genetic Variants of Cytochrome P450 Oxidoreductase. Pharm. Genom. 2010, 20, 611–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nardo, G.; Cimicata, G.; Baravalle, R.; Dell’Angelo, V.; Ciaramella, A.; Catucci, G.; Ugliengo, P.; Gilardi, G. Working at the Membrane Interface: Ligand-Induced Changes in Dynamic Conformation and Oligomeric Structure in Human Aromatase: Ligand-Induced Structural Changes in Aromatase. Biotechnol. Appl. Biochem. 2018, 65, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Farber, P.; Darmawan, H.; Sprules, T.; Mittermaier, A. Analyzing Protein Folding Cooperativity by Differential Scanning Calorimetry and NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 6214–6222. [Google Scholar] [CrossRef]
- Treuheit, N.A.; Redhair, M.; Kwon, H.; McClary, W.D.; Guttman, M.; Sumida, J.P.; Atkins, W.M. Membrane Interactions, Ligand-Dependent Dynamics, and Stability of Cytochrome P4503A4 in Lipid Nanodiscs. Biochemistry 2016, 55, 1058–1069. [Google Scholar] [CrossRef] [Green Version]
- Ebrecht, A.C.; van der Bergh, N.; Harrison, S.T.L.; Smit, M.S.; Sewell, B.T.; Opperman, D.J. Biochemical and Structural Insights into the Cytochrome P450 Reductase from Candida Tropicalis. Sci. Rep. 2019, 9, 20088. [Google Scholar] [CrossRef]
- Morlock, L.K.; Böttcher, D.; Bornscheuer, U.T. Simultaneous Detection of NADPH Consumption and H2O2 Production Using the AmplifluTM Red Assay for Screening of P450 Activities and Uncoupling. Appl. Microbiol. Biotechnol. 2018, 102, 985–994. [Google Scholar] [CrossRef]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms That Regulate Production of Reactive Oxygen Species by Cytochrome P450. Toxicol. Appl. Pharmacol. 2004, 199, 316–331. [Google Scholar] [CrossRef]
- Sadeghi, S.J.; Ferrero, S.; Di Nardo, G.; Gilardi, G. Drug–Drug Interactions and Cooperative Effects Detected in Electrochemically Driven Human Cytochrome P450 3A4. Bioelectrochemistry 2012, 86, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Catucci, G.; Di Nardo, G.; Gilardi, G. Effector Role of Cytochrome P450 Reductase for Androstenedione Binding to Human Aromatase. Int. J. Biol. Macromol. 2020, 164, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Lo, J.; Morton, D.; Valette, D.; Xi, J.; Griswold, J.; Hubbell, S.; Egbuta, C.; Jiang, W.; An, J.; et al. Novel Aromatase Inhibitors by Structure-Guided Design. J. Med. Chem. 2012, 55, 8464–8476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistrato, A.; Sgrignani, J.; Krause, R.; Cavalli, A. Single or Multiple Access Channels to the CYP450s Active Site? An Answer from Free Energy Simulations of the Human Aromatase Enzyme. J. Phys. Chem. Lett. 2017, 8, 2036–2042. [Google Scholar] [CrossRef]
- Zárate-Pérez, F.; Hackett, J.C. Conformational Selection Is Present in Ligand Binding to Cytochrome P450 19A1 Lipoprotein Nanodiscs. J. Inorg. Biochem. 2020, 209, 111120. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Wilkey, C.J.; Phan, T.T.N. Human Cytochrome P450 Enzymes Bind Drugs and Other Substrates Mainly through Conformational-Selection Modes. J. Biol. Chem. 2019, 294, 10928–10941. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Wilkey, C.J.; Glass, S.M.; Reddish, M.J. Conformational Selection Dominates Binding of Steroids to Human Cytochrome P450 17A1. J. Biol. Chem. 2019, 294, 10028–10041. [Google Scholar] [CrossRef] [Green Version]
- Otyepka, M.; Berka, K.; Anzenbacher, P. Is There a Relationship Between the Substrate Preferences and Structural Flexibility of Cytochromes P450? CDM 2012, 13, 130–142. [Google Scholar] [CrossRef]
- Hendrychová, T.; Anzenbacherová, E.; Hudeček, J.; Skopalík, J.; Lange, R.; Hildebrandt, P.; Otyepka, M.; Anzenbacher, P. Flexibility of Human Cytochrome P450 Enzymes: Molecular Dynamics and Spectroscopy Reveal Important Function-Related Variations. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2011, 1814, 58–68. [Google Scholar] [CrossRef]
- Skopalík, J.; Anzenbacher, P.; Otyepka, M. Flexibility of Human Cytochromes P450: Molecular Dynamics Reveals Differences between CYPs 3A4, 2C9, and 2A6, Which Correlate with Their Substrate Preferences. J. Phys. Chem. B 2008, 112, 8165–8173. [Google Scholar] [CrossRef]
- Foti, R.S.; Honaker, M.; Nath, A.; Pearson, J.T.; Buttrick, B.; Isoherranen, N.; Atkins, W.M. Catalytic versus Inhibitory Promiscuity in Cytochrome P450s: Implications for Evolution of New Function. Biochemistry 2011, 50, 2387–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedison, T.M.; Scrutton, N.S. Tripping the Light Fantastic in Membrane Redox Biology: Linking Dynamic Structures to Function in ER Electron Transfer Chains. FEBS J. 2019, 286, 2004–2017. [Google Scholar] [CrossRef] [PubMed]
- Tsotsou, G.E.; Cass, A.E.G.; Gilardi, G. High Throughput Assay for Cytochrome P450 BM3 for Screening Libraries of Substrates and Combinatorial Mutants. Biosens. Bioelectron. 2002, 17, 119–131. [Google Scholar] [CrossRef]
- Omura, T.; Sato, R. THE CARBON MONOXIDE-BINDING PIGMENT OF LIVER MICROSOMES. I. EVIDENCE FOR ITS HEMOPROTEIN NATURE. J. Biol. Chem. 1964, 239, 2370–2378. [Google Scholar] [CrossRef]
- Gao, C.; Catucci, G.; Castrignanò, S.; Gilardi, G.; Sadeghi, S.J. Inactivation Mechanism of N61S Mutant of Human FMO3 towards Trimethylamine. Sci. Rep. 2017, 7, 14668. [Google Scholar] [CrossRef] [Green Version]
- Catucci, G.; Aramini, D.; Sadeghi, S.J.; Gilardi, G. Ligand Stabilization and Effect on Unfolding by Polymorphism in Human Flavin-Containing Monooxygenase 3. Int. J. Biol. Macromol. 2020, 162, 1484–1493. [Google Scholar] [CrossRef]
- Henderson, C.J.; McLaughlin, L.A.; Scheer, N.; Stanley, L.A.; Wolf, C.R. Cytochrome B5 Is a Major Determinant of Human Cytochrome P450 CYP2D6 and CYP3A4 Activity in Vivo. Mol. Pharmacol. 2015, 87, 733–739. [Google Scholar] [CrossRef] [Green Version]
- Kitazume, T.; Haines, D.C.; Estabrook, R.W.; Chen, B.; Peterson, J.A. Obligatory Intermolecular Electron-Transfer from FAD to FMN in Dimeric P450BM-3. Biochemistry 2007, 46, 11892–11901. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | kcat (min−1) | Enzyme | kcat (min−1) | Refs. |
|---|---|---|---|---|
| ARO | 1.9 ± 0.1 | CYP3A4 | 2.74 ± 0.26 | [44,46] |
| ARO-BMR | 0.0021 ± 0.0002 | 3A4-BMR | 0.120 ± 0.004 | [35] |
| ARO-BMR-3GLY | 0.0023 ± 0.0001 | 3A4-BMR-3GLY | 0.156 ± 0.003 | [35] |
| ARO-BMR-5GLY | 0.0026 ± 0.0002 | 3A4-BMR-5GLY | 0.216 ± 0.007 | [35] |
| Activity measurements are based on estrone formation for ARO and ARO chimeras and 6β-hydroxylation of testosterone for 3A4 and 3A4 chimeras. | ||||
| Tm Melting T/°C | ΔH kcal/mol/°C | |||||
|---|---|---|---|---|---|---|
| HEME DOMAIN | FAD DOMAIN | FMN DOMAIN | HEME DOMAIN | FAD DOMAIN | FMN DOMAIN | |
| ARO-BMR | 45.66 | 49.48 | 56.37 | 181.14 | 324.89 | 33.97 |
| ARO-BMR-3GLY | 44.08 | 48.54 | 55.18 | 146.75 | 390.04 | 84.76 |
| ARO-BMR-5GLY | 46.23 | 49.61 | 56.36 | 464.29 | 549.98 | 324.47 |
| 3A4-BMR | 44.38 | 48.47 | 54.04 | 67.52 | 266.94 | 109.35 |
| 3A4-BMR-3GLY | 45.25 | 48.78 | 54.70 | 99.97 | 239.57 | 95.76 |
| 3A4-BMR-5GLY | 45.71 | 49.18 | 54.50 | 143.01 | 438.18 | 90.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catucci, G.; Ciaramella, A.; Di Nardo, G.; Zhang, C.; Castrignanò, S.; Gilardi, G. Molecular Lego of Human Cytochrome P450: The Key Role of Heme Domain Flexibility for the Activity of the Chimeric Proteins. Int. J. Mol. Sci. 2022, 23, 3618. https://doi.org/10.3390/ijms23073618
Catucci G, Ciaramella A, Di Nardo G, Zhang C, Castrignanò S, Gilardi G. Molecular Lego of Human Cytochrome P450: The Key Role of Heme Domain Flexibility for the Activity of the Chimeric Proteins. International Journal of Molecular Sciences. 2022; 23(7):3618. https://doi.org/10.3390/ijms23073618
Chicago/Turabian StyleCatucci, Gianluca, Alberto Ciaramella, Giovanna Di Nardo, Chao Zhang, Silvia Castrignanò, and Gianfranco Gilardi. 2022. "Molecular Lego of Human Cytochrome P450: The Key Role of Heme Domain Flexibility for the Activity of the Chimeric Proteins" International Journal of Molecular Sciences 23, no. 7: 3618. https://doi.org/10.3390/ijms23073618
APA StyleCatucci, G., Ciaramella, A., Di Nardo, G., Zhang, C., Castrignanò, S., & Gilardi, G. (2022). Molecular Lego of Human Cytochrome P450: The Key Role of Heme Domain Flexibility for the Activity of the Chimeric Proteins. International Journal of Molecular Sciences, 23(7), 3618. https://doi.org/10.3390/ijms23073618