
Telomere Targeting Approaches in Cancer: Beyond Length Maintenance


Abstract
:1. Introduction
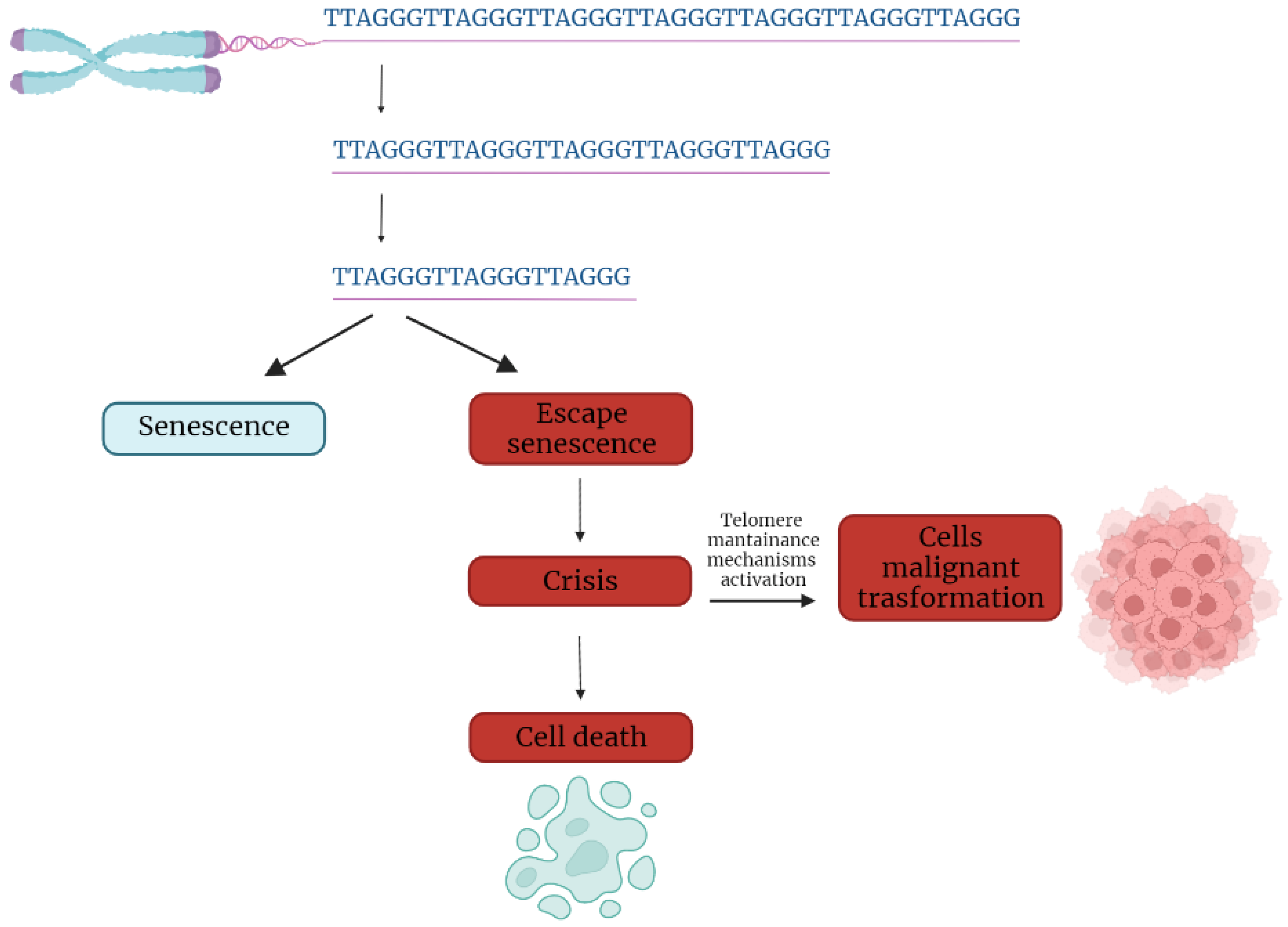
2. Telomere Evolution and Length Maintenance in Aging and Cancer
3. Telomere Structure
3.1. Protein Complexes at Telomeres
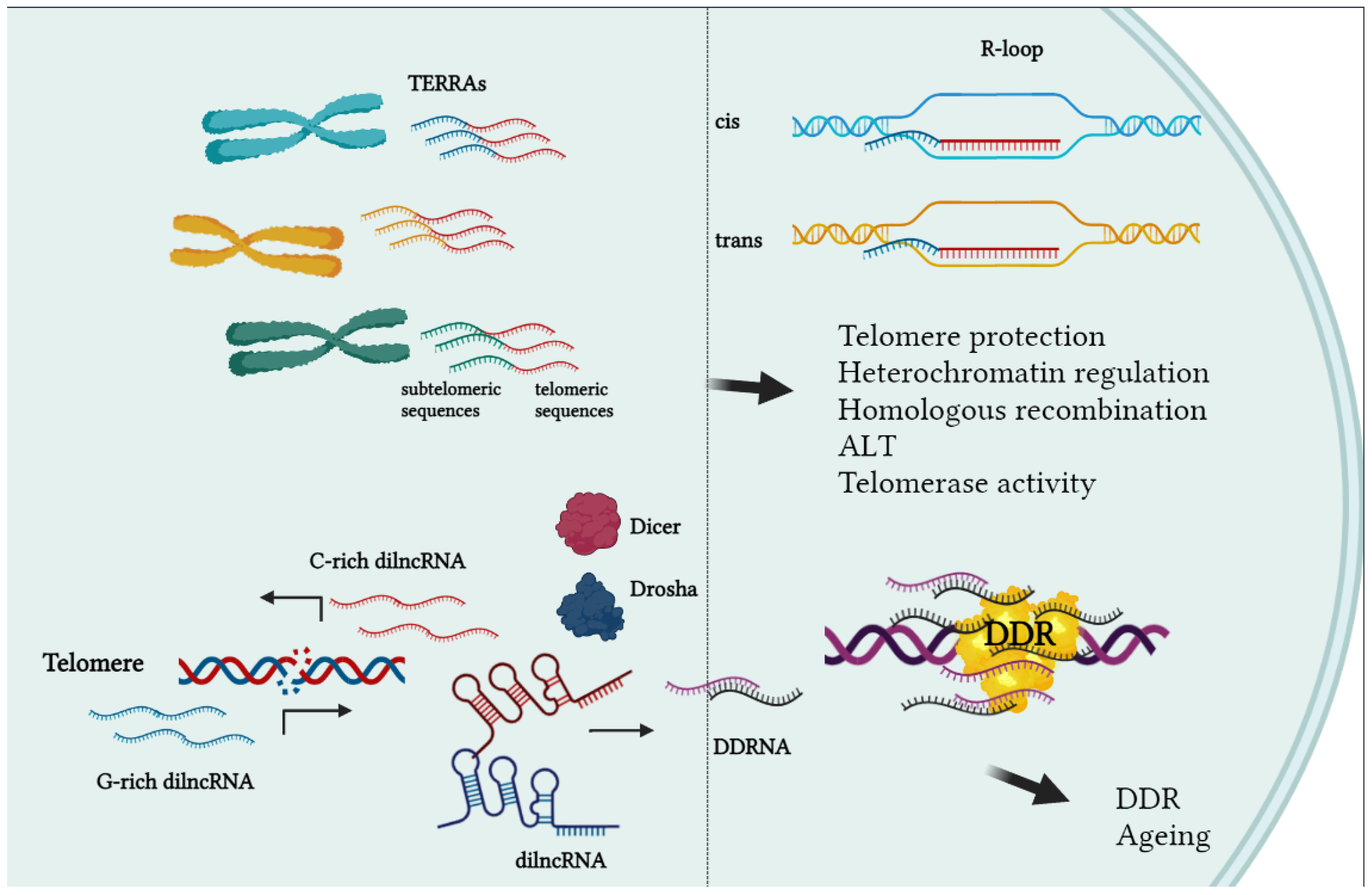
3.2. RNA Transcription at Telomeres
3.3. Telomeric DNA and Secondary Structures
4. Telomere Dysfunction in Cancer Initiation and Progression
5. Targeting Approaches against Telomere Components
5.1. Telomerase Targeting
5.2. Targeting Telomeric DNA Secondary Structures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Telomeric Targets | Mechanism | Synergism/Synthetic Lethality | Anticancer Effect | Refs. |
|---|---|---|---|---|---|
| RHPS4 and derivatives | G-quadruplex i-motifs | TRF2 POT1 delocalization Replication perturbation DDR activation | Camptotecins PARPi Ionizing radiation | ALT cells Glioblastoma Colorectal cancer | [86,97] |
| BRACO19 | G-quadruplex i-motifs | T-loop disassembly POT1 downregulation DDR activation | Cis platinum | Lung cancer Breast cancer Glioblastoma | [98,99] |
| Telomestatin | G-quadruplex | POT1 and TRF2 displacement G-overhang loss DDR activation | Imatinib Vincristin Ionizing radiation | Glioma Neuroblastoma Sarcoma ALT cells Leukemia | [100,101] |
| Naphthalene diimides | G-quadruplex | DDR activation | Ionizing radiation | Glioma Pancreatic cancer | [102,103] |
| Pyridostatin | G-quadruplex i-motifs | DDR activation | BRCA1/2 mut | Colon cancer Renal cancer | [88,89] |
| Perilene coronene derivatives | G-quadruplex | DDR activation | N.A. | Colorectal cancer | [104,105] |
| AKT inhibitors | TRF1 | TRF1 downregulation | N.A. | Glioblastoma | [106,107] |
| APO D41 peptides | TRF2 | DDR activation | N.A. | N.A. | [108] |
| Curcusone C | TRF2 | DDR induction | N.A. | Ovarian cancer Endometrial cancer | [109] |
| Quindoline derivatives | G-quadruplex | TRF2 delocalization TERRA downregulation | N.A. | N.A. | [110] |
| BPBA | G-quadruplex | TERRA stabilization | N.A. | ALT cells | [111] |
5.3. Targeting Shelterins
5.4. Targeting TERRAs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Counter, C.M.; Avilion, A.A.; LeFeuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar] [CrossRef]
- Boutelle, A.M.; Attardi, L.D. p53 and Tumor Suppression: It Takes a Network. Trends Cell Biol. 2021, 31, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Cleal, K.; Baird, D.M. Catastrophic Endgames: Emerging Mechanisms of Telomere-Driven Genomic Instability. Trends Genet. 2020, 36, 347–359. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Protection of mammalian telomeres. Oncogene 2002, 21, 532–540. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.M.; Pendlebury, D.F.; Nandakumar, J. Structural biology of telomeres and telomerase. Cell. Mol. Life Sci. 2020, 77, 61–79. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. T-loops and the origin of telomeres. Nat. Rev. Mol. Cell Biol. 2004, 5, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Liu, D.; Wan, M.; Safari, A.; Kim, H.; Sun, W.; O’Connor, M.S.; Songyang, Z. TPP1 is a homologue of ciliate TEBP-β and interacts with POT1 to recruit telomerase. Nature 2007, 445, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Wellinger, R.J. In the End, What’s the Problem? Mol. Cell 2014, 53, 855–856. [Google Scholar] [CrossRef] [Green Version]
- Aubert, G.; Lansdorp, P.M. Telomeres and Aging. Physiol. Rev. 2008, 88, 557–579. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, D.; Labella, K.A.; Depinho, R.A. Telomeres: History, health, and hallmarks of aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Demanelis, K.; Jasmine, F.; Chen, L.S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; et al. Determinants of telomere length across human tissues. Science 2020, 369, eaaz6876. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, S.; Nagadoi, A.; Nishimura, Y. Comparison between TRF2 and TRF1 of their telomeric DNA-bound structures and DNA-binding activities. Protein Sci. 2009, 14, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Collins, K.; Mitchell, J.R. Telomerase in the human organism. Oncogene 2002, 21, 564–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roake, C.M.; Artandi, S.E. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J. Telomeres and Cell Senescence—Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zvereva, M.I.; Shcherbakova, D.M.; Dontsova, O.A. Telomerase: Structure, functions, and activity regulation. Biochemistry 2010, 75, 1563–1583. [Google Scholar] [CrossRef]
- Gomez, D.E.; Armando, R.G.; Farina, H.G.; Menna, P.L.; Cerrudo, C.S.; Ghiringhelli, P.D.; Alonso, D.F. Telomere structure and telomerase in health and disease. Int. J. Oncol. 2012, 41, 1561–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smogorzewska, A.; de Lange, T. Regulation of Telomerase by Telomeric Proteins. Annu. Rev. Biochem. 2004, 73, 177–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podlevsky, J.D.; Chen, J.J.-L. It all comes together at the ends: Telomerase structure, function, and biogenesis. Mutat. Res. Mol. Mech. Mutagen. 2012, 730, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angrisani, A.; Vicidomini, R.; Turano, M.; Furia, M. Human dyskerin: Beyond telomeres. Biol. Chem. 2014, 395, 593–610. [Google Scholar] [CrossRef]
- Pompili, L.; Leonetti, C.; Biroccio, A.; Salvati, E. Diagnosis and treatment of ALT tumors: Is Trabectedin a new therapeutic option? J. Exp. Clin. Cancer Res. 2017, 36, 189. [Google Scholar] [CrossRef] [Green Version]
- Dagg, R.A.; Pickett, H.A.; Neumann, A.A.; Napier, C.E.; Henson, J.; Teber, E.T.; Arthur, J.W.; Reynolds, C.P.; Murray, J.; Haber, M.; et al. Extensive Proliferation of Human Cancer Cells with Ever-Shorter Telomeres. Cell Rep. 2017, 19, 2544–2556. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; De Lange, T. Removal of Shelterin Reveals the Telomere End-Protection Problem. Science 2012, 336, 593–597. [Google Scholar] [CrossRef] [Green Version]
- Tommerup, H.; Dousmanis, A.; de Lange, T. Unusual chromatin in human telomeres. Mol. Cell. Biol. 1994, 14, 5777–5785. [Google Scholar] [CrossRef]
- Soman, A.; Liew, C.W.; Teo, H.L.; Berezhnoy, N.V.; Olieric, V.; Korolev, N.; Rhodes, D.; Nordenskiöld, L. The human telomeric nucleosome displays distinct structural and dynamic properties. Nucleic Acids Res. 2020, 48, 5383–5396. [Google Scholar] [CrossRef]
- Mechelli, R.; Anselmi, C.; Cacchione, S.; De Santis, P.; Savino, M. Organization of telomeric nucleosomes: Atomic force microscopy imaging and theoretical modeling. FEBS Lett. 2004, 566, 131–135. [Google Scholar] [CrossRef]
- Cacchione, S.; Biroccio, A.; Rizzo, A. Emerging roles of telomeric chromatin alterations in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 21. [Google Scholar] [CrossRef]
- Lim, C.J.; Cech, T.R. Shaping human telomeres: From shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 2021, 22, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, C.M.; Reichenbach, P.; Khoriauli, L.; Giulotto, E.; Lingner, J. Telomeric Repeat–Containing RNA and RNA Surveillance Factors at Mammalian Chromosome Ends. Science 2007, 318, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Schoeftner, S.; Blasco, M.A. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 2008, 10, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Feretzaki, M.; Lingner, J. A practical qPCR approach to detect TERRA, the elusive telomeric repeat-containing RNA. Methods 2017, 114, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Barral, A.; Déjardin, J. Telomeric Chromatin and TERRA. J. Mol. Biol. 2020, 432, 4244–4256. [Google Scholar] [CrossRef]
- Bettin, N.; Oss Pegorar, C.; Cusanelli, E. The Emerging Roles of TERRA in Telomere Maintenance and Genome Stability. Cells 2019, 8, 246. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Komiyama, M. Structure, function and targeting of human telomere RNA. Methods 2012, 57, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Rossiello, F.; Aguado, J.; Sepe, S.; Iannelli, F.; Nguyen, Q.; Pitchiaya, S.; Carninci, P.; Di Fagagna, F.D. DNA damage response inhibition at dysfunctional telomeres by modulation of telomeric DNA damage response RNAs. Nat. Commun. 2017, 8, 13980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelini, F.; Pitchiaya, S.; Vitelli, V.; Sharma, S.; Gioia, U.; Pessina, F.; Cabrini, M.; Wang, Y.; Capozzo, I.; Iannelli, F.; et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 2017, 19, 1400–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguado, J.; Sola-Carvajal, A.; Cancila, V.; Revêchon, G.; Ong, P.F.; Jones-Weinert, C.W.; Arzt, E.W.; Lattanzi, G.; Dreesen, O.; Tripodo, C.; et al. Inhibition of DNA damage response at telomeres improves the detrimental phenotypes of Hutchinson–Gilford Progeria Syndrome. Nat. Commun. 2019, 10, 4990. [Google Scholar] [CrossRef] [PubMed]
- Tomáška, L.; Cesare, A.J.; AlTurki, T.M.; Griffith, J.D. Twenty years of t-loops: A case study for the importance of collaboration in molecular biology. DNA Repair 2020, 94, 102901. [Google Scholar] [CrossRef]
- Ruis, P.; Boulton, S.J. The end protection problem—an unexpected twist in the tail. Genes Dev. 2021, 35, 1–21. [Google Scholar] [CrossRef]
- Kar, A.; Willcox, S.; Griffith, J.D. Transcription of telomeric DNA leads to high levels of homologous recombination and t-loops. Nucleic Acids Res. 2016, 44, 9369–9380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilson, E.; Géli, V. How telomeres are replicated. Nat. Rev. Mol. Cell Biol. 2007, 8, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Cusanelli, E.; Chartrand, P. Telomeric repeat-containing RNA TERRA: A noncoding RNA connecting telomere biology to genome integrity. Front. Genet. 2015, 6, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossiello, F.; Herbig, U.; Longhese, M.P.; Fumagalli, M.; di Fagagna, F.D. Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr. Opin. Genet. Dev. 2014, 26, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Noureen, N.; Wu, S.; Lv, Y.; Yang, J.; Yung, W.K.A.; Gelfond, J.; Wang, X.; Koul, D.; Ludlow, A.; Zheng, S. Integrated analysis of telomerase enzymatic activity unravels an association with cancer stemness and proliferation. Nat. Commun. 2021, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, S.G.; Dsouza, R.; Pandya, G.; Kirtonia, A.; Tergaonkar, V.; Lee, S.Y.; Garg, M.; Khattar, E. Role of Telomeres and Telomeric Proteins in Human Malignancies and Their Therapeutic Potential. Cancers 2020, 12, 1901. [Google Scholar] [CrossRef]
- Pestana, A.; Vinagre, J.; Sobrinho-Simões, M.; Soares, P. TERT biology and function in cancer: Beyond immortalisation. J. Mol. Endocrinol. 2017, 58, R129–R146. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Sfeir, A.J.; Shay, J.W.; E Wright, W.; De Lange, T. POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 2005, 24, 2667–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaires, J.B.; Gray, R.D.; Dean, W.L.; Monsen, R.; DeLeeuw, L.W.; Stribinskis, V.; O Trent, J. Human POT1 unfolds G-quadruplexes by conformational selection. Nucleic Acids Res. 2020, 48, 4976–4991. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Deng, Y.; Lin, Y.; Cosme-Blanco, W.; Chan, S.; He, H.; Yuan, G.; Brown, E.J.; Chang, S. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 2007, 26, 4709–4719. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Poulos, R.C.; Reddel, R.R. Role of POT1 in Human Cancer. Cancers 2020, 12, 2739. [Google Scholar] [CrossRef]
- Butler, K.; Hines, W.; Heaphy, C.M.; Griffith, J.K. Coordinate regulation between expression levels of telomere-binding proteins and telomere length in breast carcinomas. Cancer Med. 2012, 1, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, M.; Beswick, R.; Walne, A.J.; Hossain, U.; Casimir, C.; Vulliamy, T.; Dokal, I. Dyskeratosis congenita and the DNA damage response. Br. J. Haematol. 2011, 153, 634–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marión, R.M.; de Silanes, I.L.; Mosteiro, L.; Gamache, B.; Abad, M.; Guerra, C.; Megias, D.; Serrano, M.; Blasco, M.A. Common Telomere Changes during In Vivo Reprogramming and Early Stages of Tumorigenesis. Stem Cell Rep. 2017, 8, 460–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, H.; Qureshi, A.A.; Prescott, J.; De Vivo, I.; Han, J. Genetic variants in telomere-maintaining genes and skin cancer risk. Qual. Life Res. 2010, 129, 247–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zizza, P.; Dinami, R.; Porru, M.; Cingolani, C.; Salvati, E.; Rizzo, A.; D’Angelo, C.; Petti, E.; Amoreo, C.A.; Mottolese, M.; et al. TRF2 positively regulates SULF2 expression increasing VEGF-A release and activity in tumor microenvironment. Nucleic Acids Res. 2019, 47, 3365–3382. [Google Scholar] [CrossRef] [PubMed]
- EL Maï, M.; Wagner, K.-D.; Michiels, J.-F.; Ambrosetti, D.; Borderie, A.; Destree, S.; Renault, V.; Djerbi, N.; Giraud-Panis, M.-J.; Gilson, E.; et al. The Telomeric Protein TRF2 Regulates Angiogenesis by Binding and Activating the PDGFRβ Promoter. Cell Rep. 2014, 9, 1047–1060. [Google Scholar] [CrossRef] [Green Version]
- Pan-Cancer Analyses Reveal Regulation and Clinical Outcome Association of the Shelterin Complex in Cancer. Available online: https://pubmed.ncbi.nlm.nih.gov/33497432/ (accessed on 5 February 2021).
- Adishesh, M.; Alnafakh, R.; Baird, D.M.; Jones, R.E.; Simon, S.; Button, L.; Kamal, A.M.; Kirwan, J.; Decruze, S.B.; Drury, J.; et al. Human Endometrial Carcinogenesis Is Associated with Significant Reduction in Long Non-Coding RNA, TERRA. Int. J. Mol. Sci. 2020, 21, 8686. [Google Scholar] [CrossRef] [PubMed]
- Shea-Herbert, B.; Pongracz, K.; Shay, J.W.; Gryaznov, S.M. Oligonucleotide N3′ → P5′ phosphoramidates as efficient telomerase inhibitors. Oncogene 2002, 21, 638–642. [Google Scholar] [CrossRef] [Green Version]
- A Novel Telomerase Template Antagonist (GRN163) as a Potential Anticancer Agent. Available online: https://pubmed.ncbi.nlm.nih.gov/12873987/ (accessed on 15 December 2021).
- Tefferi, A.; Lasho, T.L.; Begna, K.H.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A. Telomerase Inhibitor Imetelstat in Essential Thrombocythemia and Myelofibrosis. N. Engl. J. Med. 2015, 373, 2579–2581. [Google Scholar] [CrossRef] [Green Version]
- Armanios, M.; Greider, C.W. Treating Myeloproliferation—On Target or Off? N. Engl. J. Med. 2015, 373, 965–966. [Google Scholar] [CrossRef]
- Sengupta, S.; Sobo, M.; Lee, K.; Kumar, S.S.; White, A.R.; Mender, I.; Fuller, C.; Chow, L.M.; Fouladi, M.; Shay, J.W.; et al. Induced Telomere Damage to Treat Telomerase Expressing Therapy-Resistant Pediatric Brain Tumors. Mol. Cancer Ther. 2018, 17, 1504–1514. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Uribe, P.; Adrianzen-Ruesta, M.P.; Deng, Z.; Echevarría-Vargas, I.; Mender, I.; Saheb, S.; Liu, Q.; Altieri, D.C.; Murphy, M.E.; Shay, J.W.; et al. Exploiting TERT dependency as a therapeutic strategy for NRAS-mutant melanoma. Oncogene 2018, 37, 4058–4072. [Google Scholar] [CrossRef]
- Zeng, X.; Hernandez-Sanchez, W.; Xu, M.; Whited, T.L.; Baus, D.; Zhang, J.; Berdis, A.J.; Taylor, D.J. Administration of a Nucleoside Analog Promotes Cancer Cell Death in a Telomerase-Dependent Manner. Cell Rep. 2018, 23, 3031–3041. [Google Scholar] [CrossRef]
- Seenisamy, J.; Bashyam, S.; Gokhale, V.; Vankayalapati, H.; Sun, D.; Siddiqui-Jain, A.; Streiner, N.; Shin-Ya, K.; White, E.; Wilson, A.W.D.; et al. Design and Synthesis of an Expanded Porphyrin That Has Selectivity for the c-MYC G-Quadruplex Structure. J. Am. Chem. Soc. 2005, 127, 2944–2959. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. A second chance for telomerase reverse transcriptase in anticancer immunotherapy. Nat. Rev. Clin. Oncol. 2017, 14, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Duperret, E.K.; Wise, M.C.; Trautz, A.; Villarreal, D.O.; Ferraro, B.; Walters, J.; Yan, J.; Khan, A.; Masteller, E.; Humeau, L.; et al. Synergy of Immune Checkpoint Blockade with a Novel Synthetic Consensus DNA Vaccine Targeting TERT. Mol. Ther. 2018, 26, 435–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Tannahill, D.; Balasubramanian, S. An Intramolecular G-Quadruplex Structure Is Required for Binding of Telomeric Repeat-Containing RNA to the Telomeric Protein TRF2. J. Am. Chem. Soc. 2012, 134, 11974–11976. [Google Scholar] [CrossRef] [PubMed]
- Moye, A.L.; Porter, K.C.; Cohen, S.B.; Phan, T.; Zyner, K.; Sasaki, N.; Lovrecz, G.; Beck, J.; Bryan, T.M. Telomeric G-quadruplexes are a substrate and site of localization for human telomerase. Nat. Commun. 2015, 6, 7643. [Google Scholar] [CrossRef] [Green Version]
- Gomez, D.; Aouali, N.; Renaud, A.; Douarre, C.; Shin-Ya, K.; Tazi, J.; Martinez, S.; Trentesaux, C.; Morjani, H.; Riou, J.-F. Resistance to senescence induction and telomere shortening by a G-quadruplex ligand inhibitor of telomerase. Cancer Res. 2003, 63, 6149–6153. [Google Scholar] [PubMed]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvati, E.; Leonetti, C.; Rizzo, A.; Scarsella, M.; Mottolese, M.; Galati, R.; Sperduti, I.; Stevens, M.F.; D’Incalci, M.; Blasco, M.A.; et al. Telomere damage induced by the G-quadruplex ligand RHPS4 has an antitumor effect. J. Clin. Investig. 2007, 117, 3236–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miglietta, G.; Russo, M.; Capranico, G. G-quadruplex–R-loop interactions and the mechanism of anticancer G-quadruplex binders. Nucleic Acids Res. 2020, 48, 11942–11957. [Google Scholar] [CrossRef]
- Salvati, E.; Scarsella, M.; Porru, M.; Rizzo, A.; Iachettini, S.; Tentori, L.; Graziani, G.; D’Incalci, M.; Stevens, M.F.G.; Orlandi, A.; et al. PARP1 is activated at telomeres upon G4 stabilization: Possible target for telomere-based therapy. Oncogene 2010, 29, 6280–6293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardinelli, F.; Siteni, S.; Tanzarella, C.; Stevens, M.; Sgura, A.; Antoccia, A. The G-quadruplex-stabilising agent RHPS4 induces telomeric dysfunction and enhances radiosensitivity in glioblastoma cells. DNA Repair 2015, 25, 104–115. [Google Scholar] [CrossRef]
- Berardinelli, F.; Tanori, M.; Muoio, D.; Buccarelli, M.; Di Masi, A.; Leone, S.; Ricci-Vitiani, L.; Pallini, R.; Mancuso, M.; Antoccia, A. G-quadruplex ligand RHPS4 radiosensitizes glioblastoma xenograft in vivo through a differential targeting of bulky differentiated- and stem-cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 311. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.; Dos Santos, N.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Teng, F.-Y.; Jiang, Z.-Z.; Guo, M.; Tan, X.-Z.; Chen, F.; Xi, X.-G.; Xu, Y. G-quadruplex DNA: A novel target for drug design. Cell. Mol. Life Sci. 2021, 78, 6557–6583. [Google Scholar] [CrossRef]
- Müller, S.; Sanders, D.A.; Di Antonio, M.; Matsis, S.; Riou, J.-F.; Rodriguez, R.; Balasubramanian, S. Pyridostatin analogues promote telomere dysfunction and long-term growth inhibition in human cancer cells. Org. Biomol. Chem. 2012, 10, 6537–6546. [Google Scholar] [CrossRef]
- Zimmer, J.; Tacconi, E.; Folio, C.; Badie, S.; Porru, M.; Klare, K.; Tumiati, M.; Markkanen, E.; Halder, S.; Ryan, A.; et al. Targeting BRCA1 and BRCA2 Deficiencies with G-Quadruplex-Interacting Compounds. Mol. Cell 2016, 61, 449–460. [Google Scholar] [CrossRef] [Green Version]
- Groelly, F.J.; Porru, M.; Zimmer, J.; Benainous, H.; De Visser, Y.; A Kosova, A.; Di Vito, S.; Serra, V.; Ryan, A.; Leonetti, C.; et al. Anti-tumoural activity of the G-quadruplex ligand pyridostatin against BRCA1/2-deficient tumours. EMBO Mol. Med. 2022, 14, e14501. [Google Scholar] [CrossRef]
- Pagano, B.; Amato, J.; Iaccarino, N.; Cingolani, C.; Zizza, P.; Biroccio, A.; Novellino, E.; Randazzo, A. Looking for Efficient G-Quadruplex Ligands: Evidence for Selective Stabilizing Properties and Telomere Damage by Drug-Like Molecules. ChemMedChem 2015, 10, 640–649. [Google Scholar] [CrossRef]
- Pagano, B.; Margarucci, L.; Zizza, P.; Amato, J.; Iaccarino, N.; Cassiano, C.; Salvati, E.; Novellino, E.; Biroccio, A.; Casapullo, A.; et al. Identification of novel interactors of human telomeric G-quadruplex DNA. Chem. Commun. 2015, 51, 2964–2967. [Google Scholar] [CrossRef]
- Pagano, A.; Iaccarino, N.; Abdelhamid, M.; Brancaccio, D.; Garzarella, E.U.; Di Porzio, A.; Novellino, E.; Waller, Z.; Pagano, B.; Amato, J.; et al. Common G-Quadruplex Binding Agents Found to Interact With i-Motif-Forming DNA: Unexpected Multi-Target-Directed Compounds. Front. Chem. 2018, 6. [Google Scholar] [CrossRef]
- Fedoroff, O.Y.; Rangan, A.; Chemeris, V.V.; Hurley, L.H. Cationic Porphyrins Promote the Formation of i-Motif DNA and Bind Peripherally by a Nonintercalative Mechanism. Biochemistry 2000, 39, 15083–15090. [Google Scholar] [CrossRef]
- Musumeci, D.; Amato, J.; Zizza, P.; Platella, C.; Cosconati, S.; Cingolani, C.; Biroccio, A.; Novellino, E.; Randazzo, A.; Giancola, C.; et al. Tandem application of ligand-based virtual screening and G4-OAS assay to identify novel G-quadruplex-targeting chemotypes. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2017, 1861 Pt B, 1341–1352. [Google Scholar] [CrossRef]
- Shu, B.; Cao, J.; Kuang, G.; Qiu, J.; Zhang, M.; Zhang, Y.; Wang, M.; Li, X.; Kang, S.; Ou, T.-M.; et al. Syntheses and evaluation of new acridone derivatives for selective binding of oncogene c-myc promoter i-motifs in gene transcriptional regulation. Chem. Commun. 2018, 54, 2036–2039. [Google Scholar] [CrossRef]
- Leonetti, C.; Scarsella, M.; Riggio, G.; Rizzo, A.; Salvati, E.; D’Incalci, M.; Staszewsky, L.; Frapolli, R.; Stevens, M.F.; Stoppacciaro, A.; et al. G-Quadruplex Ligand RHPS4 Potentiates the Antitumor Activity of Camptothecins in Preclinical Models of Solid Tumors. Clin. Cancer Res. 2008, 14, 7284–7291. [Google Scholar] [CrossRef] [Green Version]
- Gunaratnam, M.; Green, C.; Moreira, J.B.; Moorhouse, A.D.; Kelland, L.R.; Moses, J.E.; Neidle, S. G-quadruplex compounds and cis-platin act synergistically to inhibit cancer cell growth in vitro and in vivo. Biochem. Pharmacol. 2009, 78, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.-T.; Keppler, B.R.; Soares, J.; Jarstfer, M.B. BRACO19 analog dimers with improved inhibition of telomerase and hPot 1. Bioorganic Med. Chem. 2009, 17, 2030–2037. [Google Scholar] [CrossRef]
- Hasegawa, D.; Okabe, S.; Okamoto, K.; Nakano, I.; Shin-Ya, K.; Seimiya, H. G-quadruplex ligand-induced DNA damage response coupled with telomere dysfunction and replication stress in glioma stem cells. Biochem. Biophys. Res. Commun. 2016, 471, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Tauchi, T.; Ohyashiki, J.H.; Ohyashiki, K. Telomerase Inhibition Combined with Other Chemotherapeutic Reagents to Enhance Anti-Cancer Effect. Adv. Struct. Saf. Stud. 2007, 405, 181–189. [Google Scholar] [CrossRef]
- Muoio, D.; Berardinelli, F.; Leone, S.; Coluzzi, E.; Di Masi, A.; Doria, F.; Freccero, M.; Sgura, A.; Folini, M.; Antoccia, A. Naphthalene diimide-derivatives G-quadruplex ligands induce cell proliferation inhibition, mild telomeric dysfunction and cell cycle perturbation in U251MG glioma cells. FEBS J. 2018, 285, 3769–3785. [Google Scholar] [CrossRef] [PubMed]
- Berardinelli, F.; Coluzzi, E.; Sgura, A.; Antoccia, A. Targeting telomerase and telomeres to enhance ionizing radiation effects in in vitro and in vivo cancer models. Mutat. Res. Rev. Mutat. Res. 2017, 773, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, V.; Salvati, E.; Alvino, A.; Bianco, A.; Ciammaichella, A.; D’Angelo, C.; Ginnari-Satriani, L.; Serrilli, A.M.; Iachettini, S.; Leonetti, C.; et al. N-Cyclic Bay-Substituted Perylene G-Quadruplex Ligands Have Selective Antiproliferative Effects on Cancer Cells and Induce Telomere Damage. J. Med. Chem. 2011, 54, 1140–1156. [Google Scholar] [CrossRef] [PubMed]
- Franceschin, M.; Rizzo, A.; Casagrande, V.; Salvati, E.; Alvino, A.; Altieri, A.; Ciammaichella, A.; Iachettini, S.; Leonetti, C.; Ortaggi, G.; et al. Aromatic Core Extension in the Series of N-Cyclic Bay-Substituted Perylene G-Quadruplex Ligands: Increased Telomere Damage, Antitumor Activity, and Strong Selectivity for Neoplastic over Healthy Cells. ChemMedChem 2012, 7, 2144–2154. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Pertuz, M.; Martínez, P.; Blanco-Aparicio, C.; Gómez-Casero, E.; Garcia, A.B.; Martinez-Torrecuadrada, J.L.; Palafox, M.; Cortés, J.; Serra, V.; Pastor, J.; et al. Modulation of telomere protection by the PI3K/AKT pathway. Nat. Commun. 2017, 8, 1278. [Google Scholar] [CrossRef]
- Bejarano, L.; Bosso, G.; Louzame, J.; Serrano, R.; Gómez-Casero, E.; Martinez-Torrecuadrada, J.L.; Martínez, S.; Blanco-Aparicio, C.; Pastor, J.; A Blasco, M. Multiple cancer pathways regulate telomere protection. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Di Maro, S.; Zizza, P.; Salvati, E.; De Luca, V.; Capasso, C.; Fotticchia, I.; Pagano, B.; Marinelli, L.; Gilson, E.; Novellino, E.; et al. Shading the TRF2 Recruiting Function: A New Horizon in Drug Development. J. Am. Chem. Soc. 2014, 136, 16708–16711. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, J.; Zhu, J.-Y.; Qiu, J.; Zhang, Y.; Shu, B.; Ou, T.-M.; Tan, J.-H.; Gu, L.-Q.; Huang, Z.-S.; et al. Curcusone C induces telomeric DNA-damage response in cancer cells through inhibition of telomeric repeat factor 2. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2017, 1865, 1372–1382. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, D.; Cao, J.; Wang, M.; Shu, B.; Kuang, G.; Ou, T.-M.; Tan, J.-H.; Gu, L.-Q.; Huang, Z.-S.; et al. Interaction of Quindoline derivative with telomeric repeat–containing RNA induces telomeric DNA-damage response in cancer cells through inhibition of telomeric repeat factor 2. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2017, 1861, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Marzano, S.; Pagano, B.; Iaccarino, N.; Di Porzio, A.; De Tito, S.; Vertecchi, E.; Salvati, E.; Randazzo, A.; Amato, J. Targeting of Telomeric Repeat-Containing RNA G-Quadruplexes: From Screening to Biophysical and Biological Characterization of a New Hit Compound. Int. J. Mol. Sci. 2021, 22, 10315. [Google Scholar] [CrossRef]
- Bejarano, L.; Schuhmacher, A.J.; Méndez, M.; Megias, D.; Blanco-Aparicio, C.; Martínez, S.; Pastor, J.; Squatrito, M.; Blasco, M.A. Inhibition of TRF1 Telomere Protein Impairs Tumor Initiation and Progression in Glioblastoma Mouse Models and Patient-Derived Xenografts. Cancer Cell 2017, 32, 590–607.e4. [Google Scholar] [CrossRef] [Green Version]
- García-Beccaria, M.; Martínez, P.; Méndez-Pertuz, M.; Martínez, S.; Blanco-Aparicio, C.; Cañamero, M.; Mulero, F.; Ambrogio, C.; Flores, J.M.; Megias, D.; et al. Therapeutic inhibition of TRF 1 impairs the growth of p53-deficient K-Ras G12V-induced lung cancer by induction of telomeric DNA damage. EMBO Mol. Med. 2015, 7, 930–949. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, L.; Louzame, J.; Montero, J.J.; Megias, D.; Flores, J.M.; Blasco, M.A. Safety of Whole-Body Abrogation of the TRF1 Shelterin Protein in Wild-Type and Cancer-Prone Mouse Models. iScience 2019, 19, 572–585. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Vázquez, R.; Martínez, P.; Blasco, M.A. AKT-dependent signaling of extracellular cues through telomeres impact on tumorigenesis. PLoS Genet. 2021, 17, e1009410. [Google Scholar] [CrossRef]
- Biroccio, A.; Cherfils-Vicini, J.; Augereau, A.; Pinte, S.; Bauwens, S.; Ye, J.; Simonet, T.; Horard, B.; Jamet, K.; Cervera, L.; et al. TRF2 inhibits a cell-extrinsic pathway through which natural killer cells eliminate cancer cells. Nat. Cell Biol. 2013, 15, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Cherfils-Vicini, J.; Iltis, C.; Cervera, L.; Pisano, S.; Croce, O.; Sadouni, N.; Győrffy, B.; Collet, R.; Renault, V.M.; Rey-Millet, M.; et al. Cancer cells induce immune escape via glycocalyx changes controlled by the telomeric protein TRF 2. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Aramburu, T.; Plucinsky, S.; Skordalakes, E. POT1-TPP1 telomere length regulation and disease. Comput. Struct. Biotechnol. J. 2020, 18, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, L.; Chen, Y.; Yang, Y.; Yang, C.-Y.; Guo, T.; Lei, M.; Sun, H.; Wang, S. Cyclic Peptidic Mimetics of Apollo Peptides Targeting Telomeric Repeat Binding Factor 2 (TRF2) and Apollo Interaction. ACS Med. Chem. Lett. 2018, 9, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Sarek, G.; Kotsantis, P.; Ruis, P.; Van Ly, D.; Margalef, P.; Borel, V.; Zheng, X.-F.; Flynn, H.; Snijders, B.; Chowdhury, D.; et al. CDK phosphorylation of TRF2 controls t-loop dynamics during the cell cycle. Nature 2019, 575, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, F.; Giraud-Panis, M.-J.; Jaco, I.; Amé, J.-C.; Schultz, I.; Blasco, M.; Koering, C.-E.; Gilson, E.; Menissier-de Murcia, J.; de Murcia, G.; et al. Functional Interaction between Poly(ADP-Ribose) Polymerase 2 (PARP-2) and TRF2: PARP Activity Negatively Regulates TRF2. Mol. Cell. Biol. 2004, 24, 1595–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picco, V.; Coste, I.; Giraud-Panis, M.-J.; Renno, T.; Gilson, E.; Pagès, G. ERK1/2/MAPK pathway-dependent regulation of the telomeric factor TRF2. Oncotarget 2016, 7, 46615–46627. [Google Scholar] [CrossRef] [Green Version]
- El Maï, M.; Hreich, S.J.D.; Gaggioli, C.; Roisin, A.; Wagner, N.; Ye, J.; Jalinot, P.; Cherfils-Vicini, J.; Gilson, E. A Novel Screen for Expression Regulators of the Telomeric Protein TRF2 Identified Small Molecules That Impair TRF2 Dependent Immunosuppression and Tumor Growth. Cancers 2021, 13, 2998. [Google Scholar] [CrossRef]
- Diala, I.; Wagner, N.; Magdinier, F.; Shkreli, M.; Sirakov, M.; Bauwens, S.; Schluth-Bolard, C.; Simonet, T.; Renault, V.M.; Ye, J.; et al. Telomere protection and TRF2 expression are enhanced by the canonical Wnt signalling pathway. EMBO Rep. 2013, 14, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Li, Y.; Ma, C.; Song, Y.; Xu, H.; Yu, H.; Xu, S.; Mu, Q.; Li, H.; Chen, Y.; et al. Arsenic trioxide inhibits glioma cell growth through induction of telomerase displacement and telomere dysfunction. Oncotarget 2016, 7, 12682–12692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, A.; Iachettini, S.; Salvati, E.; Zizza, P.; Maresca, C.; D’Angelo, C.; Benarroch-Popivker, D.; Capolupo, A.; Del Gaudio, F.; Cosconati, S.; et al. SIRT6 interacts with TRF2 and promotes its degradation in response to DNA damage. Nucleic Acids Res. 2017, 45, 1820–1834. [Google Scholar] [CrossRef]
- Altschuler, S.E.; Croy, J.E.; Wuttke, D.S. A Small Molecule Inhibitor of Pot1 Binding to Telomeric DNA. Biochemistry 2012, 51, 7833–7845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, M.; Mohammad, T.; Prasad, K.; Hasan, G.M.; Kumar, V.; Dohare, R.; Islam, A.; Ahmad, F.; Hassan, I. Virtual high-throughput screening of natural compounds in-search of potential inhibitors for protection of telomeres 1 (POT1). J. Biomol. Struct. Dyn. 2019, 38, 4625–4634. [Google Scholar] [CrossRef]
- Dinami, R.; Ercolani, C.; Petti, E.; Piazza, S.; Ciani, Y.; Sestito, R.; Sacconi, A.; Biagioni, F.; Le Sage, C.; Agami, R.; et al. miR-155 Drives Telomere Fragility in Human Breast Cancer by Targeting TRF1. Cancer Res. 2014, 74, 4145–4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Feng, X.; Wang, H.; XuYang, F.; Zhao, Y.; Ma, W.; Jiang, S.; Liu, D.; Huang, J.; Songyang, Z. Mir-23a induces telomere dysfunction and cellular senescence by inhibiting TRF 2 expression. Aging Cell 2015, 14, 391–399. [Google Scholar] [CrossRef]
- Li, T.; Luo, Z.; Lin, S.; Li, C.; Dai, S.; Wang, H.; Huang, J.; Ma, W.; Songyang, Z.; Huang, Y. MiR-185 targets POT1 to induce telomere dysfunction and cellular senescence. Aging 2020, 12, 14791–14807. [Google Scholar] [CrossRef]
- Cao, H.; Zhai, Y.; Ji, X.; Wang, Y.; Zhao, J.; Xing, J.; An, J.; Ren, T. Noncoding telomeric repeat-containing RNA inhibits the progression of hepatocellular carcinoma by regulating telomerase-mediated telomere length. Cancer Sci. 2020, 111, 2789–2802. [Google Scholar] [CrossRef]
- Kang, S.; Cao, J.; Zhang, M.; Li, X.; Guo, Q.-L.; Zeng, H.; Wei, Z.; Gong, X.; Wang, J.; Liu, B.; et al. Transcriptional regulation of telomeric repeat-containing RNA by acridine derivatives. RNA Biol. 2021, 18, 2261–2277. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kaminaga, K.; Komiyama, M. G-Quadruplex Formation by Human Telomeric Repeats-Containing RNA in Na + Solution. J. Am. Chem. Soc. 2008, 130, 11179–11184. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ishizuka, T.; Yang, J.; Ito, K.; Katada, H.; Komiyama, M.; Hayashi, T. Oligonucleotide Models of Telomeric DNA and RNA Form a Hybrid G-quadruplex Structure as a Potential Component of Telomeres. J. Biol. Chem. 2012, 287, 41787–41796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, A.T. Human telomeric G-quadruplex: Structures of DNA and RNA sequences. FEBS J. 2010, 277, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Salvati, E.; Rizzo, A.; Iachettini, S.; Zizza, P.; Cingolani, C.; D’Angelo, C.; Porru, M.; Mondello, C.; Aiello, A.; Farsetti, A.; et al. A basal level of DNA damage and telomere deprotection increases the sensitivity of cancer cells to G-quadruplex interactive compounds. Nucleic Acids Res. 2015, 43, 1759–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vertecchi, E.; Rizzo, A.; Salvati, E. Telomere Targeting Approaches in Cancer: Beyond Length Maintenance. Int. J. Mol. Sci. 2022, 23, 3784. https://doi.org/10.3390/ijms23073784
Vertecchi E, Rizzo A, Salvati E. Telomere Targeting Approaches in Cancer: Beyond Length Maintenance. International Journal of Molecular Sciences. 2022; 23(7):3784. https://doi.org/10.3390/ijms23073784
Chicago/Turabian StyleVertecchi, Eleonora, Angela Rizzo, and Erica Salvati. 2022. "Telomere Targeting Approaches in Cancer: Beyond Length Maintenance" International Journal of Molecular Sciences 23, no. 7: 3784. https://doi.org/10.3390/ijms23073784