
The Expanding Role of Extracellular Traps in Inflammation and Autoimmunity: The New Players in Casting Dark Webs



Abstract
:1. A Brief History of the Discovery of NETs
2. Pathways to NETosis
2.1. Vital NETosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of NETosis | Vital | Mitochondrial | Suicidal | Caspase Dependent |
|---|---|---|---|---|
| Stimuli | LPS [65,66] | GM-CSF [78], C5a [78], RNP ICs [79] | PMA [4,5,60], LPS [5], viral glycoproteins [69,70,71], C. albicans [80], C5a [68], IL-8 [5], crystalline particles [61], activated platelets [62], ANCA [63], TNFα [67] | Cytosolic LPS [75] |
| Receptors | TLR2 [65] | TLR7 [81] | TLR4 [70], TLR7 [69,71], TLR8 [69,71], C5aR1 [68], TNFαR | |
| Adaptors | C3 [65] | PKC [72] GSDMD [59], MLKL/RIPK3 (stimulus dependent) [60,61,62,63] | Caspase-11, GSDMD [75] | |
| Cascades | Raf-MEK-ERK [73], ERK and p38 MAPK [70], AKT [82] | |||
| Oxidant reliance | ROS independent [66] | ROS dependent [78,79] | ROS production [56,57,70,83] | |
| Components released | DNA, histones and dense granules [65,66] | Mitochondrial DNA | DNA, histones and proteins from primary [5,74,84,85], secondary and tertiary granules [5] | DNA and histones [75] |
| Downstream pathways activated | TLR9 [86], NFκB [87], cGAS-STING, Type I IFN [79]; | Complement cascade [63], IL-17 |
2.2. Suicidal NETosis
2.3. Caspase-Dependent NETosis
3. NETs—Friend or Foe in Human Disease?
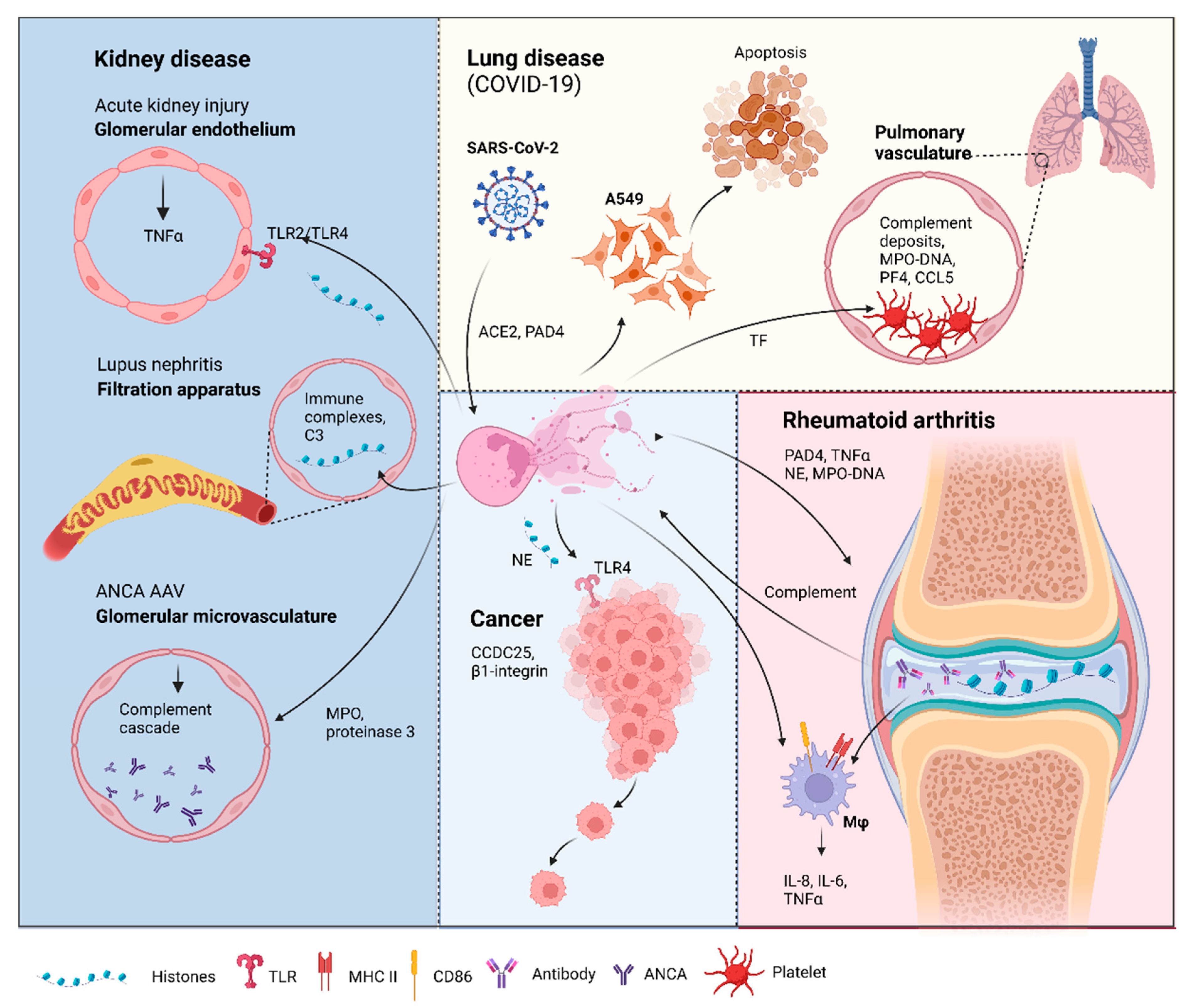
3.1. Cancer
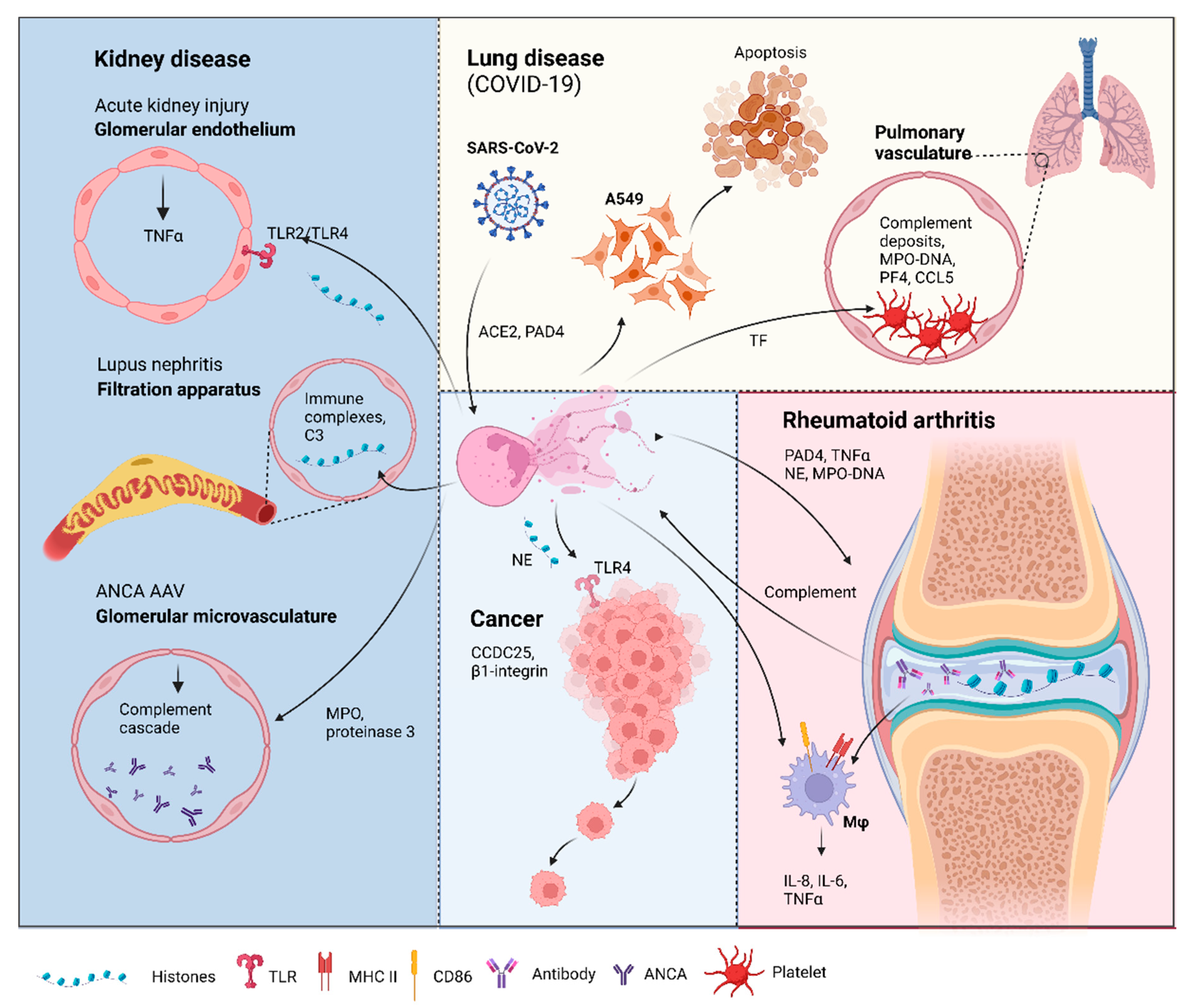
3.2. Multiple Sclerosis
3.3. Kidney Disease
3.3.1. Acute Kidney Injury (AKI)
3.3.2. Lupus Nephritis
3.3.3. ANCA-Associated Vasculitis
3.3.4. Goodpastures Disease (Anti-Glomerular Basement Membrane Disease)
3.3.5. Vasculitis
3.4. Rheumatoid Arthritis
3.5. Lung Disease
4. Role of NETs in Inducing T Cell Responses
5. Lymphocyte Extracellular Traps
B Cells
6. Extracellular Traps Are No Longer the Strict Domain of Neutrophils
6.1. Eosinophils
6.1.1. Vital EETs
6.1.2. Suicidal EETs
6.1.3. Are EETs Protective or Pathogenic?
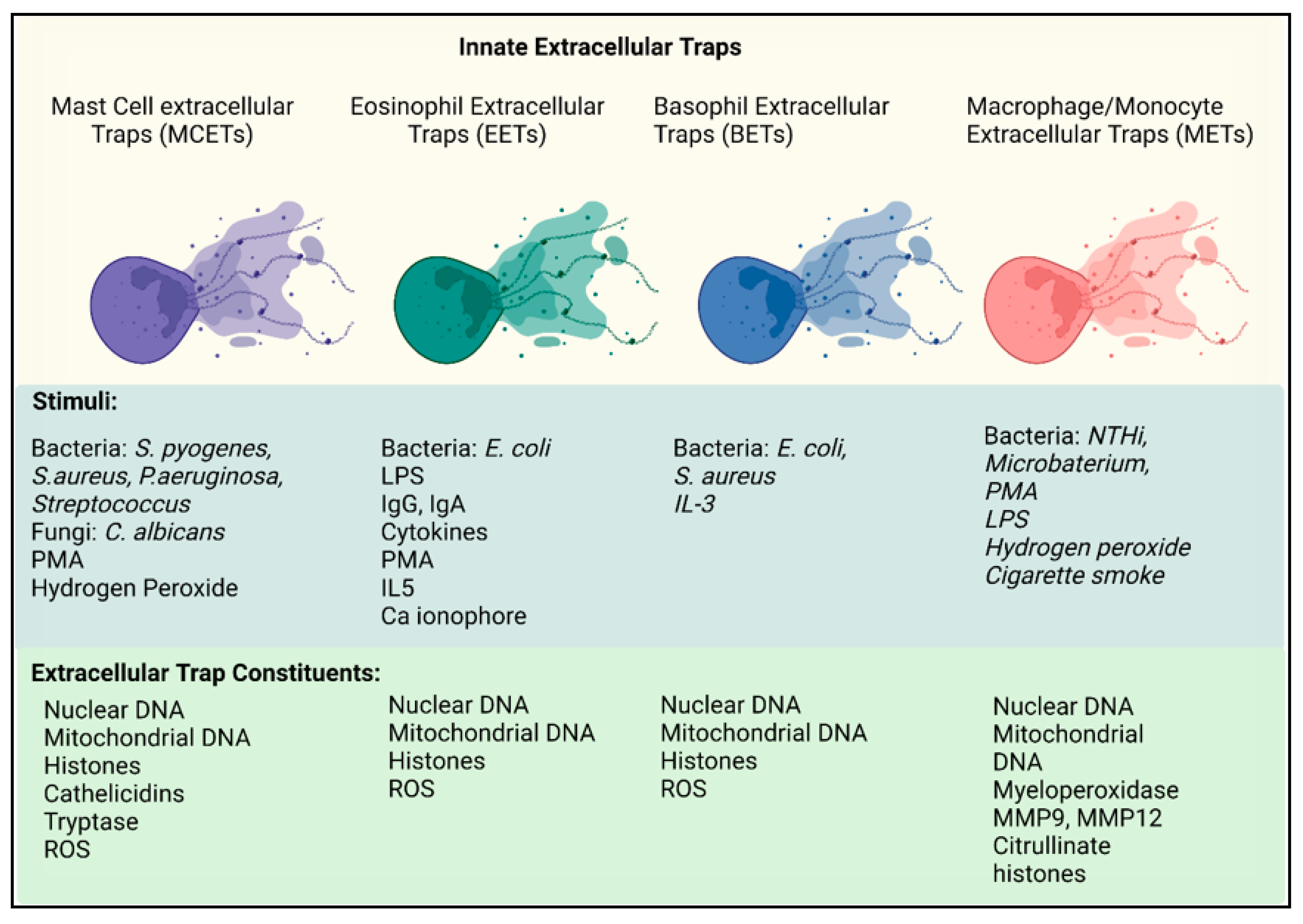
6.2. Mast Cells
Mast Cell Extracellular Traps
6.3. Basophils
6.4. Monocytes/Macrophages
Macrophage Extracellular Traps (METs)
6.5. Dendritic Cells
7. Potential Therapy Targeting the Injurious Functions of Extracellular Traps
7.1. Therapy That Targets Extracellular Traps after Formation
DNase I
7.2. Therapeutic Targeting of Critical Enzymes Required for ET Formation
7.2.1. Peptidyl Arginase Deiminases
7.2.2. Neutrophil Elastase Inhibition
7.2.3. Gasdermin
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anker, P.; Stroun, M.; Maurice, P.A. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res. 1975, 35, 2375–2382. [Google Scholar] [PubMed]
- Rogers, J.C. Identification of an intracellular precursor to DNA excreted by human lymphocytes. Proc. Natl. Acad. Sci. USA 1976, 73, 3211–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jachertz, D.; Anker, P.; Maurice, P.A.; Stroun, M. Information carried by the DNA released by antigen-stimulated lymphocytes. Immunology 1979, 37, 753–763. [Google Scholar] [PubMed]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Ingelsson, B.; Söderberg, D.; Strid, T.; Söderberg, A.; Bergh, A.-C.; Loitto, V.; Lotfi, K.; Segelmark, M.; Spyrou, G.; Rosén, A. Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of class C. Proc. Natl. Acad. Sci. USA 2018, 115, E478–E487. [Google Scholar] [CrossRef] [Green Version]
- Arrieta, Y.C.R.; Rojas, M.; Vasquez, G.; López, M.R. The Lymphocytes Stimulation Induced DNA Release, a Phenomenon Similar to NETosis. Scand. J. Immunol. 2017, 86, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Costanza, M.; Poliani, P.L.; Portararo, P.; Cappetti, B.; Musio, S.; Pagani, F.; Steinman, L.; Colombo, M.P.; Pedotti, R.; Sangaletti, S. DNA threads released by activated CD4+ T lymphocytes provide autocrine costimulation. Proc. Natl. Acad. Sci. USA 2019, 116, 8985–8994. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.C.; Wardini, A.B.; Vieira, M.; Passos, L.S.A.; Martinelli, P.M.; Neves, E.G.A.; Antonelli, L.R.D.V.; Barbosa, D.F.; Velikkakam, T.; Gutseit, E.; et al. Human CD8+ T Cells Release Extracellular Traps Co-Localized With Cytotoxic Vesicles That Are Associated With Lesion Progression and Severity in Human Leishmaniasis. Front. Immunol. 2020, 11, 594581. [Google Scholar] [CrossRef]
- Dworski, R.; Simon, H.-U.; Hoskins, A.; Yousefi, S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J. Allergy Clin. Immunol. 2011, 127, 1260–1266. [Google Scholar] [CrossRef] [Green Version]
- Aulik, N.A.; Hellenbrand, K.M.; Czuprynski, C.J. Mannheimia haemolytica and Its Leukotoxin Cause Macrophage Extracellular Trap Formation by Bovine Macrophages. Infect. Immun. 2012, 80, 1923–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, O.A.; Von Köckritz-Blickwede, M.; Bright, A.T.; Hensler, M.E.; Zinkernagel, A.; Cogen, A.L.; Gallo, R.; Monestier, M.; Wang, Y.; Glass, C.K.; et al. Statins Enhance Formation of Phagocyte Extracellular Traps. Cell Host Microbe 2010, 8, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, S.; Morshed, M.; Amini, P.; Stojkov, D.; Simon, D.; von Gunten, S.; Kaufmann, T.; Simon, H.-U. Basophils exhibit antibacterial activity through extracellular trap formation. Allergy 2015, 70, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Swain, D.K.; Kushwah, M.S.; Kaur, M.; Patbandha, T.K.; Mohanty, A.K.; Dang, A. Formation of NET, phagocytic activity, surface architecture, apoptosis and expression of toll like receptors 2 and 4 (TLR2 and TLR4) in neutrophils of mastitic cows. Vet. Res. Commun. 2014, 38, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Gondaira, S.; Higuchi, H.; Nishi, K.; Iwano, H.; Nagahata, H. Mycoplasma bovis escapes bovine neutrophil extracellular traps. Vet. Microbiol. 2017, 199, 68–73. [Google Scholar] [CrossRef]
- Caro, T.M.; Hermosilla, C.; Silva, L.M.R.; Cortes, H.; Taubert, A. Neutrophil Extracellular Traps as Innate Immune Reaction against the Emerging Apicomplexan Parasite Besnoitia besnoiti. PLoS ONE 2014, 9, e91415. [Google Scholar] [CrossRef]
- Aulik, N.A.; Hellenbrand, K.M.; Klos, H.; Czuprynski, C.J. Mannheimia haemolytica and Its Leukotoxin Cause Neutrophil Extracellular Trap Formation by Bovine Neutrophils. Infect. Immun. 2010, 78, 4454–4466. [Google Scholar] [CrossRef] [Green Version]
- Grinberg, N.; Elazar, S.; Rosenshine, I.; Shpigel, N.Y. β-Hydroxybutyrate Abrogates Formation of Bovine Neutrophil Extracellular Traps and Bactericidal Activity against Mammary Pathogenic Escherichia coli. Infect. Immun. 2008, 76, 2802–2807. [Google Scholar] [CrossRef] [Green Version]
- Lippolis, J.D.; Reinhardt, T.A.; Goff, J.P.; Horst, R.L. Neutrophil extracellular trap formation by bovine neutrophils is not inhibited by milk. Vet. Immunol. Immunopathol. 2006, 113, 248–255. [Google Scholar] [CrossRef]
- Behrendt, J.H.; Ruiz, A.; Zahner, H.; Taubert, A.; Hermosilla, C. Neutrophil extracellular trap formation as innate immune reactions against the apicomplexan parasite Eimeria bovis. Vet. Immunol. Immunopathol. 2010, 133, 1–8. [Google Scholar] [CrossRef]
- Brea, D.; Meurens, F.; Dubois, A.V.; Gaillard, J.; Chevaleyre, C.; Jourdan, M.-L.; Winter, N.; Arbeille, B.; Si-Tahar, M.; Gauthier, F.; et al. The pig as a model for investigating the role of neutrophil serine proteases in human inflammatory lung diseases. Biochem. J. 2012, 447, 363–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Buhr, N.; Neumann, A.; Jerjomiceva, N.; Von Köckritz-Blickwede, M.; Baums, C.G. Streptococcus suis DNase SsnA contributes to degradation of neutrophil extracellular traps (NETs) and evasion of NET-mediated antimicrobial activity. Microbiology 2014, 160, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loving, C.L.; Kehrli, M.E.; Brockmeier, S.L.; Bayles, D.O.; Michael, D.D.; Schlink, S.N.; Lager, K.M. Porcine granulocyte-colony stimulating factor (G-CSF) delivered via replication-defective adenovirus induces a sustained increase in circulating peripheral blood neutrophils. Biologicals 2013, 41, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Rebordão, M.; Carneiro, C.; Alexandre-Pires, G.; Brito, P.; Pereira, C.; Nunes, T.; Galvão, A.; Leitão, A.; Vilela, C.; Ferreira-Dias, G. Neutrophil extracellular traps formation by bacteria causing endometritis in the mare. J. Reprod. Immunol. 2014, 106, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.S.; Foster, D.N. Seminal DNase Frees Spermatozoa Entangled in Neutrophil Extracellular Traps. Biol. Reprod. 2005, 73, 1174–1181. [Google Scholar] [CrossRef]
- Herteman, N.; Vargas, A.; Lavoie, J.-P. Characterization of Circulating Low-Density Neutrophils Intrinsic Properties in Healthy and Asthmatic Horses. Sci. Rep. 2017, 7, 7743. [Google Scholar] [CrossRef] [Green Version]
- Chuammitri, P.; Redmond, S.B.; Kimura, K.; Andreasen, C.B.; Lamont, S.J.; Palić, D. Heterophil functional responses to dietary immunomodulators vary in genetically distinct chicken lines. Vet. Immunol. Immunopathol. 2011, 142, 219–227. [Google Scholar] [CrossRef]
- Chuammitri, P.; Ostojić, J.; Andreasen, C.B.; Redmond, S.B.; Lamont, S.J.; Palić, D. Chicken heterophil extracellular traps (HETs): Novel defense mechanism of chicken heterophils. Vet. Immunol. Immunopathol. 2009, 129, 126–131. [Google Scholar] [CrossRef]
- Palić, D.; Andreasen, C.B.; Ostojić, J.; Tell, R.M.; Roth, J.A. Zebrafish (Danio rerio) whole kidney assays to measure neutrophil extracellular trap release and degranulation of primary granules. J. Immunol. Methods 2007, 319, 87–97. [Google Scholar] [CrossRef]
- Gratacap, R.L.; Scherer, A.K.; Seman, B.; Wheeler, R.T. Control of Mucosal Candidiasis in the Zebrafish Swim Bladder Depends on Neutrophils That Block Filament Invasion and Drive Extracellular-Trap Production. Infect. Immun. 2017, 85, e00276-17. [Google Scholar] [CrossRef] [Green Version]
- Brogden, G.; von Köckritz-Blickwede, M.; Adamek, M.; Reuner, F.; Jung-Schroers, V.; Naim, H.Y.; Steinhagen, D. β-Glucan protects neutrophil extracellular traps against degradation by Aeromonas hydrophila in carp (Cyprinus carpio). Fish Shellfish Immunol. 2012, 33, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Pijanowski, L.; Golbach, L.; Kolaczkowska, E.; Scheer, M.; Kemenade, B.V.-V.; Chadzinska, M. Carp neutrophilic granulocytes form extracellular traps via ROS-dependent and independent pathways. Fish Shellfish Immunol. 2013, 34, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Sun, L. Neutrophils of Scophthalmus maximus produce extracellular traps that capture bacteria and inhibit bacterial infection. Dev. Comp. Immunol. 2016, 56, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-L.; Chi, H.; Sun, L. Neutrophil Extracellular Traps of Cynoglossus semilaevis: Production Characteristics and Antibacterial Effect. Front. Immunol. 2017, 8, 290. [Google Scholar] [CrossRef] [Green Version]
- Robb, C.T.; Dyrynda, E.A.; Gray, R.; Rossi, A.G.; Smith, V.J. Invertebrate extracellular phagocyte traps show that chromatin is an ancient defence weapon. Nat. Commun. 2014, 5, 4627. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.H.; Chang, S.-H.; Wu, M.-H.; Wang, H.-C. Shrimp hemocytes release extracellular traps that kill bacteria. Dev. Comp. Immunol. 2013, 41, 644–651. [Google Scholar] [CrossRef]
- Ng, T.H.; Wu, M.-H.; Chang, S.-H.; Aoki, T.; Wang, H.-C. The DNA fibers of shrimp hemocyte extracellular traps are essential for the clearance of Escherichia coli. Dev. Comp. Immunol. 2015, 48, 229–233. [Google Scholar] [CrossRef]
- Lange, M.K.; Penagos-Tabares, F.; Muñoz-Caro, T.; Gärtner, U.; Mejer, H.; Schaper, R.; Hermosilla, C.; Taubert, A. Gastropod-derived haemocyte extracellular traps entrap metastrongyloid larval stages of Angiostrongylus vasorum, Aelurostrongylus abstrusus and Troglostrongylus brevior. Parasites Vectors 2017, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Poirier, A.C.; Schmitt, P.; Rosa, R.D.; Vanhove, A.S.; Kieffer-Jaquinod, S.; Rubio, T.P.; Charrière, G.M.; Destoumieux-Garzón, D. Antimicrobial Histones and DNA Traps in Invertebrate Immunity. J. Biol. Chem. 2014, 289, 24821–24831. [Google Scholar] [CrossRef] [Green Version]
- Wen, F.; White, G.J.; VanEtten, H.D.; Xiong, Z.; Hawes, M.C. Extracellular DNA Is Required for Root Tip Resistance to Fungal Infection. Plant Physiol. 2009, 151, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.M.; MacIntyre, A.; Hawes, M.; Allen, C. Escaping Underground Nets: Extracellular DNases Degrade Plant Extracellular Traps and Contribute to Virulence of the Plant Pathogenic Bacterium Ralstonia solanacearum. PLoS Pathog. 2016, 12, e1005686. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martínez, E.; Hernández-González, L.; Ramos-Martínez, I.; Mayoral, L.P.-C.; López-Cortés, G.I.; Pérez-Campos, E.; Andrade, G.M.; Hernández-Huerta, M.T.; José, M.V. Multiple Origins of Extracellular DNA Traps. Front. Immunol. 2021, 12, 621311. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, J.T.; Simpson, A.J.; Aziz, R.; Liu, G.Y.; Kristian, S.A.; Kotb, M.; Feramisco, J.; Nizet, V. DNase Expression Allows the Pathogen Group A Streptococcus to Escape Killing in Neutrophil Extracellular Traps. Curr. Biol. 2006, 16, 396–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beiter, K.; Wartha, F.; Albiger, B.; Normark, S.; Zychlinsky, A.; Henriques-Normark, B. An Endonuclease Allows Streptococcus pneumoniae to Escape from Neutrophil Extracellular Traps. Curr. Biol. 2006, 16, 401–407. [Google Scholar] [CrossRef] [Green Version]
- Walker, M.; Hollands, A.; Sanderson-Smith, M.; Cole, J.N.; Kirk, J.K.; Henningham, A.; McArthur, J.D.; Dinkla, K.; Aziz, R.; Kansal, R.G.; et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat. Med. 2007, 13, 981–985. [Google Scholar] [CrossRef]
- Tonello, S.; Rizzi, M.; Migliario, M.; Rocchetti, V.; Renò, F. Low concentrations of neutrophil extracellular traps induce proliferation in human keratinocytes via NF-kB activation. J. Dermatol. Sci. 2017, 88, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Hofbauer, T.M.; Mangold, A.; Scherz, T.; Seidl, V.; Panzenböck, A.; Ondracek, A.S.; Müller, J.; Schneider, M.; Binder, T.; Hell, L.; et al. Neutrophil extracellular traps and fibrocytes in ST-segment elevation myocardial infarction. Basic Res. Cardiol. 2019, 114, 33. [Google Scholar] [CrossRef] [Green Version]
- Arampatzioglou, A.; Papazoglou, D.; Konstantinidis, T.; Chrysanthopoulou, A.; Mitsios, A.; Angelidou, I.; Maroulakou, I.; Ritis, K.; Skendros, P. Clarithromycin Enhances the Antibacterial Activity and Wound Healing Capacity in Type 2 Diabetes Mellitus by Increasing LL-37 Load on Neutrophil Extracellular Traps. Front. Immunol. 2018, 9, 2064. [Google Scholar] [CrossRef]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [Green Version]
- Njeim, R.; Azar, W.S.; Fares, A.H.; Azar, S.T.; Kassouf, H.K.; Eid, A.A. NETosis contributes to the pathogenesis of diabetes and its complications. J. Mol. Endocrinol. 2020, 65, R65–R76. [Google Scholar] [CrossRef]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttocao, A.; Ciciliot, S.; Mammano, F.; Ciubotaru, C.D.; Brocco, E.; et al. NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Bitschar, K.; Staudenmaier, L.; Klink, L.; Focken, J.; Sauer, B.; Fehrenbacher, B.; Herster, F.; Bittner, Z.; Bleul, L.; Schaller, M.; et al. Staphylococcus aureus Skin Colonization Is Enhanced by the Interaction of Neutrophil Extracellular Traps with Keratinocytes. J. Investig. Dermatol. 2020, 140, 1054–1065.e4. [Google Scholar] [CrossRef] [PubMed]
- Neeli, I.; Khan, S.; Radic, M. Histone Deimination As a Response to Inflammatory Stimuli in Neutrophils. J. Immunol. 2008, 180, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Stadler, S.C.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Remijsen, Q.; Berghe, T.V.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef] [Green Version]
- Kenny, E.F.; Herzig, A.; Krüger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; Von Bernuth, H.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife 2017, 6, e24437. [Google Scholar] [CrossRef]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [Green Version]
- Desai, J.; Kumar, S.V.; Mulay, S.R.; Konrad, L.; Romoli, S.; Schauer, C.; Herrmann, M.; Bilyy, R.; Müller, S.; Popper, B.; et al. PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur. J. Immunol. 2015, 46, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Desai, J.; Foresto-Neto, O.; Honarpisheh, M.; Steiger, S.; Nakazawa, D.; Popper, B.; Buhl, E.M.; Boor, P.; Mulay, S.R.; Anders, H.-J. Particles of different sizes and shapes induce neutrophil necroptosis followed by the release of neutrophil extracellular trap-like chromatin. Sci. Rep. 2017, 7, 15003. [Google Scholar] [CrossRef]
- Nakazawa, D.; Desai, J.; Steiger, S.; Müller, S.; Devarapu, S.K.; Mulay, S.R.; Iwakura, T.; Anders, H.-J. Activated platelets induce MLKL-driven neutrophil necroptosis and release of neutrophil extracellular traps in venous thrombosis. Cell Death Discov. 2018, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Rousselle, A.; Becker, J.U.; von Mässenhausen, A.; Linkermann, A.; Kettritz, R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl. Acad. Sci. USA 2017, 114, E9618–E9625. [Google Scholar] [CrossRef] [Green Version]
- Amini, P.; Stojkov, D.; Wang, X.; Wicki, S.; Kaufmann, T.; Wong, W.W.; Simon, H.; Yousefi, S. NET formation can occur independently of RIPK3 and MLKL signaling. Eur. J. Immunol. 2016, 46, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PLoS ONE 2012, 7, e48111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, J.; Gan, P.-Y.; Ford, S.L.; Odobasic, D.; Alikhan, M.A.; Loosen, S.H.; Hall, P.; Westhorpe, C.L.; Li, A.; Ooi, J.D.; et al. C5a receptor 1 promotes autoimmunity, neutrophil dysfunction and injury in experimental anti-myeloperoxidase glomerulonephritis. Kidney Int. 2017, 93, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil Extracellular Traps Mediate a Host Defense Response to Human Immunodeficiency Virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funchal, G.A.; Jaeger, N.; Czepielewski, R.S.; Machado, M.S.; Muraro, S.P.; Stein, R.T.; Bonorino, C.B.C.; Porto, B.N. Respiratory Syncytial Virus Fusion Protein Promotes TLR-4–Dependent Neutrophil Extracellular Trap Formation by Human Neutrophils. PLoS ONE 2015, 10, e0124082. [Google Scholar] [CrossRef]
- Hiroki, C.H.; Toller-Kawahisa, J.E.; Fumagalli, M.J.; Colon, D.; Figueiredo, L.T.M.; Fonseca, B.A.L.D.; Franca, R.F.O.; Cunha, F.Q. Neutrophil Extracellular Traps Effectively Control Acute Chikungunya Virus Infection. Front. Immunol. 2020, 10, 3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, M.; McPhail, L.C.; Nasrallah, V.N.; Snyderman, R. Phorbol myristate acetate mediates redistribution of protein kinase C in human neutrophils: Potential role in the activation of the respiratory burst enzyme. J. Immunol. 1985, 135, 2057–2062. [Google Scholar] [PubMed]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D–dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [Green Version]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Hilbrands, L.B.; van der Vlag, J. Neutrophils Discriminate between Lipopolysaccharides of Different Bacterial Sources and Selectively Release Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 484. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2005, 8, 668–676. [Google Scholar] [CrossRef]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting Neutrophils Are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, ra20–ra73. [Google Scholar] [CrossRef] [Green Version]
- Dömer, D.; Walther, T.; Möller, S.; Behnen, M.; Laskay, T. Neutrophil Extracellular Traps Activate Proinflammatory Functions of Human Neutrophils. Front. Immunol. 2021, 12, 1190. [Google Scholar] [CrossRef]
- Ermert, D.; Zychlinsky, A.; Urban, C. Fungal and Bacterial Killing by Neutrophils. Inflammation 2009, 470, 293–312. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.-J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- O’Donoghue, A.J.; Jin, Y.; Knudsen, G.M.; Perera, N.C.; Jenne, D.E.; Murphy, J.E.; Craik, C.S.; Hermiston, T.W. Global Substrate Profiling of Proteases in Human Neutrophil Extracellular Traps Reveals Consensus Motif Predominantly Contributed by Elastase. PLoS ONE 2013, 8, e75141. [Google Scholar] [CrossRef] [Green Version]
- Itagaki, K.; Kaczmarek, E.; Lee, Y.T.; Tang, I.T.; Isal, B.; Adibnia, Y.; Sandler, N.; Grimm, M.J.; Segal, B.H.; Otterbein, L.E.; et al. Mitochondrial DNA Released by Trauma Induces Neutrophil Extracellular Traps. PLoS ONE 2015, 10, e0120549. [Google Scholar] [CrossRef]
- Arai, Y.; Nishinaka, Y.; Arai, T.; Morita, M.; Mizugishi, K.; Adachi, S.; Takaori-Kondo, A.; Watanabe, T.; Yamashita, K. Uric acid induces NADPH oxidase-independent neutrophil extracellular trap formation. Biochem. Biophys. Res. Commun. 2014, 443, 556–561. [Google Scholar] [CrossRef]
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; Von Köckritz-Blickwede, M. Nuclease Expression by Staphylococcus aureus Facilitates Escape from Neutrophil Extracellular Traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [Green Version]
- Sumby, P.; Barbian, K.D.; Gardner, D.J.; Whitney, A.R.; Welty, D.M.; Long, R.D.; Bailey, J.R.; Parnell, M.J.; Hoe, N.P.; Adams, G.G.; et al. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc. Natl. Acad. Sci. USA 2005, 102, 1679–1684. [Google Scholar] [CrossRef] [Green Version]
- McIlroy, D.J.; Jarnicki, A.G.; Au, G.G.; Lott, N.; Smith, D.W.; Hansbro, P.; Balogh, Z.J. Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J. Crit. Care 2014, 29, 1133.e1–1133.e5. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Fuchs, T.A.; Nauseef, W.M.; Reumaux, D.; Roesler, J.; Schulze, I.; Wahn, V.; Papayannopoulos, V.; Zychlinsky, A. Myeloperoxidase is required for neutrophil extracellular trap formation: Implications for innate immunity. Blood 2011, 117, 953–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.-C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Ospina, F.E.; Betancur, J.F.; Suso, J.P.; Muñoz-Buitron, E.; Cañas, C.A.; Tobón, G.J. Role of the cytokine BAFF in autoimmune diseases: Physiopathology and therapeutic targets. Rev. Colomb. Reumatol. (Engl. Ed.) 2016, 23, 177–194. [Google Scholar] [CrossRef]
- Homa-Mlak, I.; Majdan, A.; Mlak, R.; Małecka-Massalska, T. Metastatic potential of NET in neoplastic disease. Postepy Hig. Med. Dosw. (Online) 2016, 70, 887–895. [Google Scholar] [CrossRef]
- Oklu, R.; Sheth, R.A.; Wong, K.H.K.; Jahromi, A.H.; Albadawi, H. Neutrophil extracellular traps are increased in cancer patients but does not associate with venous thrombosis. Cardiovasc. Diagn. Ther. 2017, 7 (Suppl. 3), S140–S149. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Gan, T.; Zhou, J.; Hu, F.; Hao, N.; Yuan, B.; Chen, Y.; Zhang, M. Extracellular RNAs from lung cancer cells activate epithelial cells and induce neutrophil extracellular traps. Int. J. Oncol. 2019, 55, 69–80. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil Extracellular Traps Directly Induce Epithelial and Endothelial Cell Death: A Predominant Role of Histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Cedervall, J.; Hamidi, A.; Olsson, A.-K. Platelets, NETs and cancer. Thromb. Res. 2018, 164 (Suppl. 1), S148–S152. [Google Scholar] [CrossRef]
- Schedel, F.; Mayer-Hain, S.; Pappelbaum, K.I.; Metze, D.; Stock, M.; Goerge, T.; Loser, K.; Sunderkötter, C.; Luger, T.A.; Weishaupt, C. Evidence and impact of neutrophil extracellular traps in malignant melanoma. Pigment Cell Melanoma Res. 2020, 33, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Najmeh, S.; Cools-Lartigue, J.; Rayes, R.F.; Gowing, S.; Vourtzoumis, P.; Bourdeau, F.; Giannias, B.; Berube, J.; Rousseau, S.; Ferri, L.E.; et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int. J. Cancer 2017, 140, 2321–2330. [Google Scholar] [CrossRef] [Green Version]
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749. [Google Scholar] [CrossRef]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef]
- Zhu, T.; Zou, X.; Yang, C.; Li, L.; Wang, B.; Li, R.; Li, H.; Xu, Z.; Huang, D.; Wu, Q. Neutrophil extracellular traps promote gastric cancer metastasis by inducing epithelial-mesenchymal transition. Int. J. Mol. Med. 2021, 48, 127. [Google Scholar] [CrossRef]
- Kumar, S.V.; Kulkarni, O.P.; Mulay, S.R.; Darisipudi, M.N.; Romoli, S.; Thomasova, D.; Scherbaum, C.R.; Hohenstein, B.; Hugo, C.; Müller, S.; et al. Neutrophil Extracellular Trap-Related Extracellular Histones Cause Vascular Necrosis in Severe GN. J. Am. Soc. Nephrol. 2015, 26, 2399–2413. [Google Scholar] [CrossRef] [Green Version]
- Mistry, P.; Kaplan, M.J. Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin. Immunol. 2017, 185, 59–73. [Google Scholar] [CrossRef]
- Rother, N.; Pieterse, E.; Lubbers, J.; Hilbrands, L.; Van Der Vlag, J. Acetylated Histones in Apoptotic Microparticles Drive the Formation of Neutrophil Extracellular Traps in Active Lupus Nephritis. Front. Immunol. 2017, 8, 1136. [Google Scholar] [CrossRef] [Green Version]
- Söderberg, D.; Segelmark, M. Neutrophil Extracellular Traps in ANCA-Associated Vasculitis. Front. Immunol. 2016, 7, 256. [Google Scholar] [CrossRef] [Green Version]
- Kitching, A.R.; Anders, H.-J.; Basu, N.; Brouwer, E.; Gordon, J.; Jayne, D.R.; Kullman, J.; Lyons, P.A.; Merkel, P.A.; Savage, C.O.S.; et al. ANCA-associated vasculitis. Nat. Rev. Dis. Prim. 2020, 6, 71. [Google Scholar] [CrossRef]
- Radford, D.J.; Savage, C.O.S.; Nash, G.B. Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheumatol. 2000, 43, 1337–1345. [Google Scholar] [CrossRef]
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative Complement Pathway in the Pathogenesis of Disease Mediated by Anti-Neutrophil Cytoplasmic Autoantibodies. Am. J. Pathol. 2007, 170, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2–triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.; Skendros, P.; Konstantinides, S.; Ritis, K. Traps N’ Clots: NET-Mediated Thrombosis and Related Diseases. Thromb. Haemost. 2020, 120, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef]
- Clavel, C.; Nogueira, L.; Laurent, L.; Iobagiu, C.; Vincent, C.; Sebbag, M.; Serre, G. Induction of macrophage secretion of tumor necrosis factor α through Fcγ receptor IIa engagement by rheumatoid arthritis–specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Care Res. 2008, 58, 678–688. [Google Scholar] [CrossRef]
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Care Res. 2010, 63, 53–62. [Google Scholar] [CrossRef]
- Yazdani, H.O.; Roy, E.; Comerci, A.J.; van der Windt, D.J.; Zhang, H.; Huang, H.; Loughran, P.; Shiva, S.; Geller, D.A.; Bartlett, D.L.; et al. Neutrophil Extracellular Traps Drive Mitochondrial Homeostasis in Tumors to Augment Growth. Cancer Res. 2019, 79, 5626–5639. [Google Scholar] [CrossRef] [Green Version]
- Moscarello, M.A.; Wood, D.D.; Ackerley, C.; Boulias, C. Myelin in multiple sclerosis is developmentally immature. J. Clin. Investig. 1994, 94, 146–154. [Google Scholar] [CrossRef]
- Pritzker, L.B.; Joshi, S.; Gowan, J.J.; Harauz, G.; Moscarello, M.A. Deimination of Myelin Basic Protein. 1. Effect of Deimination of Arginyl Residues of Myelin Basic Protein on Its Structure and Susceptibility to Digestion by Cathepsin D. Biochemistry 2000, 39, 5374–5381. [Google Scholar] [CrossRef]
- Boggs, J.M.; Rangaraj, G.; Koshy, K.M.; Ackerley, C.; Wood, D.D.; Moscarello, M.A. Highly deiminated isoform of myelin basic protein from multiple sclerosis brain causes fragmentation of lipid vesicles. J. Neurosci. Res. 1999, 57, 529–535. [Google Scholar] [CrossRef]
- Harauz, G.; Musse, A.A. A Tale of Two Citrullines—Structural and Functional Aspects of Myelin Basic Protein Deimination in Health and Disease. Neurochem. Res. 2007, 32, 137–158. [Google Scholar] [CrossRef] [PubMed]
- Opdenakker, G.; Proost, P.; Van Damme, J. Microbiomic and Posttranslational Modifications as Preludes to Autoimmune Diseases. Trends Mol. Med. 2016, 22, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Caprariello, A.V.; Rogers, J.A.; Morgan, M.L.; Hoghooghi, V.; Plemel, J.; Koebel, A.; Tsutsui, S.; Dunn, J.F.; Kotra, L.P.; Ousman, S.S.; et al. Biochemically altered myelin triggers autoimmune demyelination. Proc. Natl. Acad. Sci. USA 2018, 115, 5528–5533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastronardi, F.G.; Wood, D.D.; Mei, J.; Raijmakers, R.; Tseveleki, V.; Dosch, H.-M.; Probert, L.; Casaccia-Bonnefil, P.; Moscarello, M.A. Increased Citrullination of Histone H3 in Multiple Sclerosis Brain and Animal Models of Demyelination: A Role for Tumor Necrosis Factor-Induced Peptidylarginine Deiminase 4 Translocation. J. Neurosci. 2006, 26, 11387–11396. [Google Scholar] [CrossRef]
- Naegele, M.; Tillack, K.; Reinhardt, S.; Schippling, S.; Martin, R.; Sospedra, M. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J. Neuroimmunol. 2012, 242, 60–71. [Google Scholar] [CrossRef]
- Rumble, J.M.; Huber, A.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-related factors as biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Tillack, K.; Naegele, M.; Haueis, C.; Schippling, S.; Wandinger, K.-P.; Martin, R.; Sospedra, M. Gender differences in circulating levels of neutrophil extracellular traps in serum of multiple sclerosis patients. J. Neuroimmunol. 2013, 261, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Paryzhak, S.; Dumych, T.; Mahorivska, I.; Boichuk, M.; Bila, G.; Peshkova, S.; Nehrych, T.; Bilyy, R. Neutrophil-released enzymes can influence composition of circulating immune complexes in multiple sclerosis. Autoimmunity 2018, 51, 297–303. [Google Scholar] [CrossRef]
- Zhang, H.; Ray, A.; Miller, N.M.; Hartwig, D.; Pritchard, K.A., Jr.; Dittel, B.N. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J. Neurochem. 2015, 136, 826–836. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Zheng, S.; Zhang, H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces experimental autoimmune encephalomyelitis-induced injury and promotes oligodendrocyte regeneration and neurogenesis in a murine model of progressive multiple sclerosis. NeuroReport 2018, 29, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.A.; Noel, S.; Sadasivam, M.; Hamad, A.R.; Rabb, H. Role of Immune Cells in Acute Kidney Injury and Repair. Nephron Exp. Nephrol. 2017, 137, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, S.; Agarwal, A. Neutrophils in acute kidney injury: Not neutral any more. Kidney Int. 2009, 75, 674–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonavia, A.; Singbartl, K. A review of the role of immune cells in acute kidney injury. Pediatr. Nephrol. 2018, 33, 1629–1639. [Google Scholar] [CrossRef]
- Tecklenborg, J.; Clayton, D.; Siebert, S.; Coley, S.M. The role of the immune system in kidney disease. Clin. Exp. Immunol. 2018, 192, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Mulay, S.R.; Linkermann, A.; Anders, H.-J. Necroinflammation in Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, D.; Kumar, S.V.; Marschner, J.; Desai, J.; Holderied, A.; Rath, L.; Kraft, F.; Lei, Y.; Fukasawa, Y.; Moeckel, G.; et al. Histones and Neutrophil Extracellular Traps Enhance Tubular Necrosis and Remote Organ Injury in Ischemic AKI. J. Am. Soc. Nephrol. 2017, 28, 1753–1768. [Google Scholar] [CrossRef] [Green Version]
- Nawroth, P.; Handley, D.; Matsueda, G.; De Waal, R.; Gerlach, H.; Blohm, D.; Stern, D. Tumor necrosis factor/cachectin-induced intravascular fibrin formation in meth A fibrosarcomas. J. Exp. Med. 1988, 168, 637–647. [Google Scholar] [CrossRef] [Green Version]
- Hertig, A.; Rondeau, E. Role of the coagulation/fibrinolysis system in fibrin-associated glomerular injury. J. Am. Soc. Nephrol. 2004, 15, 844–853. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Kanjanabuch, T.; Kittikowit, W.; Eiam-Ong, S. An update on acute postinfectious glomerulonephritis worldwide. Nat. Rev. Nephrol. 2009, 5, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Tesař, V.; Hruskova, Z. Treatment of proliferative lupus nephritis: A slowly changing landscape. Nat. Rev. Nephrol. 2010, 7, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Menzi, C.P.; Bucher, B.S.; Bianchetti, M.G.; Ardissino, G.; Simonetti, G.D. Management and outcomes of childhood Goodpasture’s disease. Pediatr. Res. 2018, 83, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Lu, X.; Shu, X.; Tian, X.; Yang, H.; Yang, W.; Zhang, Y.; Wang, G. Elevated Plasma cfDNA May be Associated with Active Lupus Nephritis and Partially Attributed to Abnormal Regulation of Neutrophil Extracellular Traps (NETs) in Patients with Systemic Lupus Erythematosus. Intern. Med. 2014, 53, 2763–2771. [Google Scholar] [CrossRef] [Green Version]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [Green Version]
- Kallenberg, C.G.M. Pathogenesis of ANCA-associated vasculitides. Ann. Rheum. Dis. 2011, 70 (Suppl. 1), i59–i63. [Google Scholar] [CrossRef]
- O’Sullivan, K.M.; Lo, C.; Summers, S.A.; Elgass, K.D.; McMillan, P.; Longano, A.; Ford, S.L.; Gan, P.Y.; Kerr, P.G.; Kitching, A.R.; et al. Renal participation of myeloperoxidase in antineutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Kidney Int. 2015, 88, 1030–1046. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Yamada, M.; Sudo, Y.; Kojima, T.; Tomiyasu, T.; Yoshikawa, N.; Oda, T.; Yamada, M. Myeloperoxidase anti-neutrophil cytoplasmic antibody affinity is associated with the formation of neutrophil extracellular traps in the kidney and vasculitis activity in myeloperoxidase anti-neutrophil cytoplasmic antibody-associated microscopic polyangiitis. Nephrology 2016, 21, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Gadola, S.D.; Gross, W.L. The renaissance of granulomatous inflammation in AAV. Nat. Rev. Rheumatol. 2012, 8, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Shida, H.; Tomaru, U.; Yoshida, M.; Nishio, S.; Atsumi, T.; Ishizu, A. Enhanced Formation and Disordered Regulation of NETs in Myeloperoxidase-ANCA–Associated Microscopic Polyangiitis. J. Am. Soc. Nephrol. 2014, 25, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Kraaij, T.; Kamerling, S.; Bakker, J.; Brunini, F.; Pusey, C.; Scherer, H.U.; Toes, R.E.; Rabelink, T.; van Kooten, C.; Teng, O. NET-inducing capacity is a biomarker in ANCA-associated vasculitis independent of ANCA antibodies. Arthritis Rheumatol. 2016, 68 (Suppl. 10), 1–15. [Google Scholar]
- Nakazawa, D.; Marschner, J.A.; Platen, L.; Anders, H.-J. Extracellular traps in kidney disease. Kidney Int. 2018, 94, 1087–1098. [Google Scholar] [CrossRef]
- Chen, K.; Nishi, H.; Travers, R.; Tsuboi, N.; Martinod, K.; Wagner, D.D.; Stan, R.; Croce, K.; Mayadas, T.N. Endocytosis of soluble immune complexes leads to their clearance by FcγRIIIB but induces neutrophil extracellular traps via FcγRIIA in vivo. Blood 2012, 120, 4421–4431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, H.; Furuhashi, K.; Cullere, X.; Saggu, G.; Miller, M.J.; Chen, Y.; Rosetti, F.; Hamilton, S.L.; Yang, L.; Pittman, S.P.; et al. Neutrophil FcγRIIA promotes IgG-mediated glomerular neutrophil capture via Abl/Src kinases. J. Clin. Investig. 2017, 127, 3810–3826. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Herter, J.M.; Rosetti, F.; Campbell, A.; Nishi, H.; Kashgarian, M.; Bastacky, S.I.; Marinov, A.; Nickerson, K.; Mayadas, T.N.; et al. Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight 2017, 2, e92926. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Shinagawa, S.; Mikage, H. Vasculitis syndrome-diagnosis and therapy. J. Gen. Fam. Med. 2017, 18, 72–78. [Google Scholar] [CrossRef]
- Yates, M.; Watts, R. ANCA-associated vasculitis. Clin. Med. 2017, 17, 60–64. [Google Scholar] [CrossRef]
- Yoshida, M.; Sasaki, M.; Sugisaki, K.; Yamaguchi, Y.; Yamada, M. Neutrophil extracellular trap components in fibrinoid necrosis of the kidney with myeloperoxidase-ANCA-associated vasculitis. Clin. Kidney J. 2013, 6, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Nishide, M.; Nojima, S.; Ito, D.; Takamatsu, H.; Koyama, S.; Kang, S.; Kimura, T.; Morimoto, K.; Hosokawa, T.; Hayama, Y.; et al. Semaphorin 4D inhibits neutrophil activation and is involved in the pathogenesis of neutrophil-mediated autoimmune vasculitis. Ann. Rheum. Dis. 2017, 76, 1440–1448. [Google Scholar] [CrossRef] [PubMed]
- Sha, L.-L.; Wang, H.; Wang, C.; Peng, H.-Y.; Chen, M.; Zhao, M.-H. Autophagy is induced by anti-neutrophil cytoplasmic Abs and promotes neutrophil extracellular traps formation. Innate Immun. 2016, 22, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Ohkawa, K.; Hosui, A.; Hiramatsu, N.; Kanto, T.; Ueda, K.; Takehara, T.; Hayashi, N. Involvement of p38 signaling pathway in interferon-α-mediated antiviral activity toward hepatitis C virus. Biochem. Biophys. Res. Commun. 2004, 321, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-H.; Ma, T.-T.; Wang, C.; Wang, H.; Chang, D.-Y.; Chen, M.; Zhao, M.-H. High-mobility group box 1 potentiates antineutrophil cytoplasmic antibody-inducing neutrophil extracellular traps formation. Arthritis Res. Ther. 2016, 18, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shida, H.; Nakazawa, D.; Tateyama, Y.; Miyoshi, A.; Kusunoki, Y.; Hattanda, F.; Masuda, S.; Tomaru, U.; Kawakami, T.; Atsumi, T.; et al. The Presence of Anti-Lactoferrin Antibodies in a Subgroup of Eosinophilic Granulomatosis with Polyangiitis Patients and Their Possible Contribution to Enhancement of Neutrophil Extracellular Trap Formation. Front. Immunol. 2016, 7, 636. [Google Scholar] [CrossRef] [Green Version]
- Kettritz, R.; Jennette, J.C.; Falk, R.J. Crosslinking of ANCA-antigens stimulates superoxide release by human neutrophils. J. Am. Soc. Nephrol. 1997, 8, 386–394. [Google Scholar] [CrossRef]
- Shingu, M.; Nonaka, S.; Nishimukai, H.; Nobunaga, M.; Kitamura, H.; Tomo-Oka, K. Activation of complement in normal serum by hydrogen peroxide and hydrogen peroxide-related oxygen radicals produced by activated neutrophils. Clin. Exp. Immunol. 1992, 90, 72–78. [Google Scholar] [CrossRef]
- Vogt, W. Complement Activation by Myeloperoxidase Products Released from Stimulated Human Polymorphonuclear Leukocytes. Immunobiology 1996, 195, 334–346. [Google Scholar] [CrossRef]
- Wirthmueller, U.; Dewald, B.; Thelen, M.; Schäfer, M.K.; Stover, C.; Whaley, K.; North, J.; Eggleton, P.; Reid, K.B.; Schwaeble, W.J. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J. Immunol. 1997, 158, 4444–4451. [Google Scholar]
- Jayne, D.R.W.; Bruchfeld, A.N.; Harper, L.; Schaier, M.; Venning, M.C.; Hamilton, P.; Burst, V.; Grundmann, F.; Jadoul, M.; Szombati, I.; et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2017, 28, 2756–2767. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, K.; Nijenhuis, S.; Vossenaar, E.R.; Palmblad, K.; Van Venrooij, W.J.; Klareskog, L.; Zendman, A.J.W.; Harris, H.E. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res. Ther. 2005, 7, R458–R467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holers, V.M.; Banda, N.K. Complement in the Initiation and Evolution of Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1057. [Google Scholar] [CrossRef]
- Chowdhury, C.S.; Giaglis, S.; Walker, U.A.; Buser, A.; Hahn, S.; Hasler, P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: Analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res. Ther. 2014, 16, R122. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Akahoshi, T.; Takagishi, K.; Kashiwazaki, S.; Matsushima, K. Elevation of interleukin-8 (IL-8) levels in joint fluids of patients with rheumatoid arthritis and the induction by IL-8 of leukocyte infiltration and synovitis in rabbit joints. Lymphokine Cytokine Res. 1991, 10, 245–252. [Google Scholar] [PubMed]
- Kaneko, S.; Satoh, T.; Chiba, J.; Ju, C.; Inoue, K.; Kagawa, J. Interleukin–6 and interleukin–8 levels in serum and synovial fluid of patients with osteoarthritis. Cytokines Cell. Mol. Ther. 2000, 6, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, M.; Baron, B. The role of TNF-α in rheumatoid arthritis: A focus on regulatory T cells. J. Clin. Transl. Res. 2016, 2, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Banik, S. Pharmacotherapy Options in Rheumatoid Arthritis. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2013, 6, 35–43. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.N.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; on behalf of the HLH across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Ritis, K.; Doumas, M.; Mastellos, D.; Micheli, A.; Giaglis, S.; Magotti, P.; Rafail, S.; Kartalis, G.; Sideras, P.; Lambris, J. A Novel C5a Receptor-Tissue Factor Cross-Talk in Neutrophils Links Innate Immunity to Coagulation Pathways. J. Immunol. 2006, 177, 4794–4802. [Google Scholar] [CrossRef]
- Redecha, P.; Tilley, R.; Tencati, M.; Salmon, J.E.; Kirchhofer, D.; Mackman, N.; Girardi, G. Tissue factor: A link between C5a and neutrophil activation in antiphospholipid antibody–induced fetal injury. Blood 2007, 110, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Kourtzelis, I.; Markiewski, M.M.; Doumas, M.; Rafail, S.; Kambas, K.; Mitroulis, I.; Panagoutsos, S.; Passadakis, P.; Vargemezis, V.; Magotti, P.; et al. Complement anaphylatoxin C5a contributes to hemodialysis-associated thrombosis. Blood 2010, 116, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.H.; Park, H.; Dong, C. Act1 Adaptor Protein Is an Immediate and Essential Signaling Component of Interleukin-17 Receptor. J. Biol. Chem. 2006, 281, 35603–35607. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Liu, C.; Hartupee, J.; Altuntas, C.Z.; Gulen, M.F.; Jane-Wit, D.; Xiao, J.; Lu, Y.; Giltiay, N.V.; Liu, J.; et al. The adaptor Act1 is required for interleukin 17–dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 2007, 8, 247–256. [Google Scholar] [CrossRef]
- Sønder, S.U.; Saret, S.; Tang, W.; Sturdevant, D.E.; Porcella, S.F.; Siebenlist, U. IL-17-induced NF-κB Activation via CIKS/Act1: Physiologic significance and signaling mechanisms. J. Biol. Chem. 2011, 286, 12881–12890. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Qian, W.; Qian, Y.; Giltiay, N.V.; Lu, Y.; Swaidani, S.; Misra, S.; Deng, L.; Chen, Z.J.; Li, X. Act1, a U-box E3 Ubiquitin Ligase for IL-17 Signaling. Sci. Signal. 2009, 2, ra63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Q.; Sun, Y.; Liu, W.; Qian, C.; Jin, B.; Tao, F.; Gu, Y.; Wu, X.; Shen, Y.; Xu, Q. A Novel Disease-Modifying Antirheumatic Drug, Iguratimod, Ameliorates Murine Arthritis by Blocking IL-17 Signaling, Distinct from Methotrexate and Leflunomide. J. Immunol. 2013, 191, 4969–4978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinghaus, E.; Ellinghaus, D.; Stuart, P.E.; Nair, R.P.; Debrus, S.; Raelson, J.V.; Belouchi, M.; Fournier, H.; Reinhard, C.; Ding, J.; et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 2010, 42, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Hüffmeier, U.; Uebe, S.; Ekici, A.B.; Bowes, J.; Giardina, E.; Korendowych, E.; Juneblad, K.; Apel, M.; McManus, R.; Ho, P.; et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 2010, 42, 996–999. [Google Scholar] [CrossRef] [Green Version]
- Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010, 42, 985–990. [CrossRef] [Green Version]
- Stuart, P.E.; Nair, R.P.; Tsoi, L.C.; Tejasvi, T.; Das, S.; Kang, H.M.; Ellinghaus, E.; Chandran, V.; Callis-Duffin, K.; Ike, R.; et al. Genome-wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture. Am. J. Hum. Genet. 2015, 97, 816–836. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wu, L.; Bulek, K.; Martin, B.; Zepp, J.A.; Kang, Z.; Liu, C.; Herjan, T.; Misra, S.; Carman, J.A.; et al. The psoriasis-associated D10N variant of the adaptor Act1 with impaired regulation by the molecular chaperone hsp90. Nat. Immunol. 2012, 14, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.C.-S.; Yu, H.-S.; Yen, F.-L.; Lin, C.-L.; Chen, G.-S.; Lan, C.-C.E. Neutrophil extracellular trap formation is increased in psoriasis and induces human β-defensin-2 production in epidermal keratinocytes. Sci. Rep. 2016, 6, 31119. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.M.; Rubin, C.J.; Khandpur, R.; Wang, J.Y.; Riblett, M.; Yalavarthi, S.; Villanueva, E.C.; Shah, P.; Kaplan, M.J.; Bruce, A.T. Mast Cells and Neutrophils Release IL-17 through Extracellular Trap Formation in Psoriasis. J. Immunol. 2011, 187, 490–500. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.; Hambro, C.A.; Johnston, A.; Stuart, P.E.; Tsoi, L.C.; Nair, R.P.; Elder, J.T. Neutrophil Extracellular Traps Induce Human Th17 Cells: Effect of Psoriasis-Associated TRAF3IP2 Genotype. J. Investig. Dermatol. 2019, 139, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Chen, H.; Yang, Z.-T.; Mao, E.-Q.; Chen, Y.; Chen, E.-Z. Free fatty acids-induced neutrophil extracellular traps lead to dendritic cells activation and T cell differentiation in acute lung injury. Aging 2021, 13, 26148–26160. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qiu, S.-L.; Tang, Q.-Y.; Zhou, X.; Zhang, J.-Q.; He, Z.-Y.; Bai, J.; Li, M.-H.; Deng, J.-M.; Liang, Y.; et al. Erythromycin suppresses neutrophil extracellular traps in smoking-related chronic pulmonary inflammation. Cell Death Dis. 2019, 10, 678. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, N.; Douda, D.N.; Brüggemann, T.R.; Ricklefs, I.; Duvall, M.G.; Abdulnour, R.-E.E.; Martinod, K.; Tavares, L.; Wang, X.; Cernadas, M.; et al. Neutrophil cytoplasts induce T H 17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci. Immunol. 2018, 3, eaao4747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.S.; Randall, K.L.; Pettitt, J.A.; Ellyard, J.I.; Blumenthal, A.; Enders, A.; Quah, B.J.; Bopp, T.; Parish, C.R.; Brüstle, A. Neutrophil extracellular traps and their histones promote Th17 cell differentiation directly via TLR2. Nat. Commun. 2022, 13, 528. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.F. STAT3 Regulates the Generation of Th17 Cells. Sci. STKE 2007, 2007, tw113. [Google Scholar] [CrossRef]
- Komiyama, Y.; Nakae, S.; Matsuki, T.; Nambu, A.; Ishigame, H.; Kakuta, S.; Sudo, K.; Iwakura, Y. IL-17 Plays an Important Role in the Development of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2006, 177, 566–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, D.R.; Gollob, K.J.; Barbosa, J.; Schriefer, A.; Machado, P.R.L.; Lessa, H.; Carvalho, L.P.; Romano-Silva, M.A.; de Jesus, A.R.; Carvalho, E.M.; et al. Decreased In Situ Expression of Interleukin-10 Receptor Is Correlated with the Exacerbated Inflammatory and Cytotoxic Responses Observed in Mucosal Leishmaniasis. Infect. Immun. 2005, 73, 7853–7859. [Google Scholar] [CrossRef] [Green Version]
- Faria, D.R.; Souza, P.E.A.; Durães, F.V.; Carvalho, E.M.; Gollob, K.J.; Machado, P.R.; Dutra, W.O. Recruitment of CD8+T cells expressing granzyme A is associated with lesion progression in human cutaneous leishmaniasis. Parasite Immunol. 2009, 31, 432–439. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, T.M.; Machado, A.; Costa, D.L.; Carvalho, L.P.; Queiroz, A.; Machado, P.; Scott, P.; Carvalho, E.M.; Bacellar, O. Protective and Pathological Functions of CD8+ T Cells in Leishmania braziliensis Infection. Infect. Immun. 2015, 83, 898–906. [Google Scholar] [CrossRef] [Green Version]
- Guimarães-Costa, A.B.; Nascimento, M.T.C.; Froment, G.S.; Soares, R.P.P.; Morgado, F.N.; Conceição-Silva, F.; Saraiva, E.M. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. USA 2009, 106, 6748–6753. [Google Scholar] [CrossRef] [Green Version]
- Morgado, F.; Nascimento, M.T.C.; Saraiva, E.M.; De Oliveira-Ribeiro, C.; Madeira, M.D.F.; Da Costa-Santos, M.; Vasconcellos, E.C.F.; Pimentel, M.I.F.; Lyra, M.R.; Schubach, A.D.O.; et al. Are Neutrophil Extracellular Traps Playing a Role in the Parasite Control in Active American Tegumentary Leishmaniasis Lesions? PLoS ONE 2015, 10, e0133063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muniz, V.S.; Weller, P.F.; Neves, J.S. Eosinophil crystalloid granules: Structure, function, and beyond. J. Leukoc. Biol. 2012, 92, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, S.; Gold, J.; Andina, N.; Lee, J.J.; Kelly, A.M.; Kozlowski, E.; Schmid, I.; Straumann, A.; Reichenbach, J.; Gleich, G.J.; et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008, 14, 949–953. [Google Scholar] [CrossRef]
- Ueki, S.; Melo, R.; Ghiran, I.; Spencer, L.A.; Dvorak, A.M.; Weller, P.F. Eosinophil extracellular DNA trap cell death mediates lytic release of free secretion-competent eosinophil granules in humans. Blood 2013, 121, 2074–2083. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, I.; Ceulemans, M.; Wauters, L.; Breynaert, C.; Vermeire, S.; Verstockt, B.; Vanuytsel, T. Role of Eosinophils in Intestinal Inflammation and Fibrosis in Inflammatory Bowel Disease: An Overlooked Villain? Front. Immunol. 2021, 12, 754413. [Google Scholar] [CrossRef]
- Choi, Y.; Le Pham, D.; Lee, D.-H.; Lee, S.-H.; Kim, S.-H.; Park, H.-S. Biological function of eosinophil extracellular traps in patients with severe eosinophilic asthma. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Cunha, A.A.; Nuñez, N.K.; de Souza, R.G.; Vargas, M.H.M.; Silveira, J.S.; Antunes, G.L.; Durante, L.D.S.; Porto, B.N.; Marczak, E.S.; Jones, M.H.; et al. Recombinant human deoxyribonuclease therapy improves airway resistance and reduces DNA extracellular traps in a murine acute asthma model. Exp. Lung Res. 2016, 42, 66–74. [Google Scholar] [CrossRef]
- Shak, S.; Capon, D.J.; Hellmiss, R.; Marsters, S.A.; Baker, C.L. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 1990, 87, 9188–9192. [Google Scholar] [CrossRef] [Green Version]
- Ueki, S.; Konno, Y.; Takeda, M.; Moritoki, Y.; Hirokawa, M.; Matsuwaki, Y.; Honda, K.; Ohta, N.; Yamamoto, S.; Takagi, Y.; et al. Eosinophil extracellular trap cell death–derived DNA traps: Their presence in secretions and functional attributes. J. Allergy Clin. Immunol. 2016, 137, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Hwang, C.S.; Park, S.C.; Cho, H.-J.; Park, D.-J.; Yoon, J.-H.; Kim, C.-H. Eosinophil extracellular trap formation is closely associated with disease severity in chronic rhinosinusitis regardless of nasal polyp status. Sci. Rep. 2019, 9, 8061. [Google Scholar] [CrossRef] [Green Version]
- Gevaert, E.; Zhang, N.; Krysko, O.; Lan, F.; Holtappels, G.; De Ruyck, N.; Nauwynck, H.; Yousefi, S.; Simon, H.-U.; Bachert, C. Extracellular eosinophilic traps in association with Staphylococcus aureus at the site of epithelial barrier defects in patients with severe airway inflammation. J. Allergy Clin. Immunol. 2017, 139, 1849–1860.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, N.; Ueki, S.; Konno, Y.; Hirokawa, M.; Kubota, T.; Tomioka-Matsutani, S.; Suzuki, T.; Ishida, Y.; Kawano, T.; Miyasaka, T.; et al. ETosis-derived DNA trap production in middle ear effusion is a common feature of eosinophilic otitis media. Allergol. Int. 2018, 67, 414–416. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Ueki, S.; Kamide, Y.; Miyabe, Y.; Fukuchi, M.; Yokoyama, Y.; Furukawa, T.; Azuma, N.; Oka, N.; Takeuchi, H.; et al. Increased Circulating Cell-Free DNA in Eosinophilic Granulomatosis With Polyangiitis: Implications for Eosinophil Extracellular Traps and Immunothrombosis. Front. Immunol. 2021, 12, 801897. [Google Scholar] [CrossRef] [PubMed]
- Beaven, M.A. Our perception of the mast cell from Paul Ehrlich to now. Eur. J. Immunol. 2009, 39, 11–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, D.F.; Barrett, N.A.; Austen, K.F.; The Immunological Genome Project Consortium. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat. Immunol. 2016, 17, 878–887. [Google Scholar] [CrossRef]
- Pejler, G.; Åbrink, M.; Ringvall, M.; Wernersson, S. Mast Cell Proteases. Adv. Immunol. 2007, 95, 167–255. [Google Scholar] [CrossRef]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef]
- Kraft, S.; Kinet, J.-P.P. New developments in FcεRI regulation, function and inhibition. Nat. Rev. Immunol. 2007, 7, 365–378. [Google Scholar] [CrossRef]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Supajatura, V.; Ushio, H.; Nakao, A.; Akira, S.; Okumura, K.; Ra, C.; Ogawa, H. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J. Clin. Investig. 2002, 109, 1351–1359. [Google Scholar] [CrossRef]
- Gaudenzio, N.; Laurent, C.; Valitutti, S.; Espinosa, E. Human mast cells drive memory CD4+ T cells toward an inflammatory IL-22+ phenotype. J. Allergy Clin. Immunol. 2013, 131, 1400–1407.e11. [Google Scholar] [CrossRef] [PubMed]
- Nija, R.J.; Sanju, S.; Sidharthan, N.; Mony, U. Extracellular Trap by Blood Cells: Clinical Implications. Tissue Eng. Regen. Med. 2020, 17, 141–153. [Google Scholar] [CrossRef] [PubMed]
- von KöCkritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008, 111, 3070–3080. [Google Scholar] [CrossRef] [PubMed]
- Cregar, L.; Elrod, K.C.; Putnam, D.; Moore, W.R. Neutrophil Myeloperoxidase Is a Potent and Selective Inhibitor of Mast Cell Tryptase. Arch. Biochem. Biophys. 1999, 366, 125–130. [Google Scholar] [CrossRef]
- Lowy, F.D. Staphylococcus aureus Infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, K.M.; Bahri, R.; Sattentau, C.; Roberts, I.S.; Goenka, A.; Bulfone-Paus, S. Human mast cells exhibit an individualized pattern of antimicrobial responses. Immun. Inflamm. Dis. 2020, 8, 198–210. [Google Scholar] [CrossRef]
- Lauth, X.; Von Köckritz-Blickwede, M.; McNamara, C.W.; Myskowski, S.; Zinkernagel, A.S.; Beall, B.; Ghosh, P.; Gallo, R.L.; Nizet, V. M1 Protein Allows Group A Streptococcal Survival in Phagocyte Extracellular Traps through Cathelicidin Inhibition. J. Innate Immun. 2009, 1, 202–214. [Google Scholar] [CrossRef]
- Lopes, J.P.; Stylianou, M.; Nilsson, G.; Urban, C.F. Opportunistic pathogen Candida albicans elicits a temporal response in primary human mast cells. Sci. Rep. 2015, 5, 12287. [Google Scholar] [CrossRef] [Green Version]
- Pertiwi, K.R.; De Boer, O.J.; Mackaaij, C.; Pabittei, D.R.; De Winter, R.J.; Li, X.; van der Wal, A. Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis. J. Pathol. 2019, 247, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Morshed, M.; Hlushchuk, R.; Simon, D.; Walls, A.F.; Obata-Ninomiya, K.; Karasuyama, H.; Djonov, V.; Eggel, A.; Kaufmann, T.; Simon, H.-U.; et al. NADPH Oxidase–Independent Formation of Extracellular DNA Traps by Basophils. J. Immunol. 2014, 192, 5314–5323. [Google Scholar] [CrossRef] [Green Version]
- King, P.T.; Sharma, R.; O’Sullivan, K.M.; Selemidis, S.; Lim, S.; Radhakrishna, N.; Lo, C.; Prasad, J.; Callaghan, J.; McLaughlin, P.; et al. Nontypeable Haemophilus influenzae Induces Sustained Lung Oxidative Stress and Protease Expression. PLoS ONE 2015, 10, e0120371. [Google Scholar] [CrossRef] [Green Version]
- King, P.T.; Sharma, R.; O’Sullivan, K.M.; Callaghan, J.; Dousha, L.; Thomas, B.; Ruwanpura, S.; Lim, S.; Farmer, M.W.; Jennings, B.R.; et al. Deoxyribonuclease 1 reduces pathogenic effects of cigarette smoke exposure in the lung. Sci. Rep. 2017, 7, 12128. [Google Scholar] [CrossRef] [PubMed]
- King, P.T.; Dousha, L.; Clarke, N.; Schaefer, J.; Carzino, R.; Sharma, R.; Wan, K.L.; Anantharajah, A.; O’Sullivan, K.; Lu, Z.X.; et al. Phagocyte extracellular traps in children with neutrophilic airway inflammation. ERJ Open Res. 2021, 7, 00883-2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wu, X.; Liao, C.; Liu, X.; Du, J.; Shi, H.; Wang, X.; Bai, X.; Peng, P.; Yu, L.; et al. Escherichia coli and Candida albicans Induced Macrophage Extracellular Trap-Like Structures with Limited Microbicidal Activity. PLoS ONE 2014, 9, e90042. [Google Scholar] [CrossRef]
- Loures, F.V.; Röhm, M.; Lee, C.K.; Santos, E.; Wang, J.P.; Specht, C.A.; Calich, V.L.G.; Urban, C.F.; Levitz, S.M. Recognition of Aspergillus fumigatus Hyphae by Human Plasmacytoid Dendritic Cells Is Mediated by Dectin-2 and Results in Formation of Extracellular Traps. PLoS Pathog. 2015, 11, e1004643. [Google Scholar] [CrossRef]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Chamardani, T.M.; Amiritavassoli, S. Inhibition of NETosis for treatment purposes: Friend or foe? Mol. Cell. Biochem. 2022, 477, 673–688. [Google Scholar] [CrossRef]
- O’Sullivan, K.M.; Holdsworth, S.R. Neutrophil Extracellular Traps: A Potential Therapeutic Target in MPO-ANCA Associated Vasculitis? Front. Immunol. 2021, 12, 635188. [Google Scholar] [CrossRef]
- Davis, J.C.; Manzi, S.; Yarboro, C.; Rairie, J.; McInnes, I.; Averthelyi, D.; Sinicropi, D.; Hale, V.G.; Balow, J.; Austin, H.; et al. Recombinant human Dnase I (rhDNase) in patients with lupus nephritis. Lupus 1999, 8, 68–76. [Google Scholar] [CrossRef]
- Shiokawa, D.; Tanuma, S.-I. Characterization of Human DNase I Family Endonucleases and Activation of DNase γ during Apoptosis. Biochemistry 2000, 40, 143–152. [Google Scholar] [CrossRef]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, J.S.; Zhao, W.; Luo, W.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Hodgin, J.B.; Eitzman, D.T.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Investig. 2013, 123, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Biron, B.; Chung, C.-S.; O’Brien, X.M.; Chen, Y.; Reichner, J.S.; Ayala, A. Cl-Amidine Prevents Histone 3 Citrullination and Neutrophil Extracellular Trap Formation, and Improves Survival in a Murine Sepsis Model. J. Innate Immun. 2017, 9, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine Deiminase Inhibition Reduces Vascular Damage and Modulates Innate Immune Responses in Murine Models of Atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Curran, A.M.; Naik, P.; Giles, J.T.; Darrah, E. PAD enzymes in rheumatoid arthritis: Pathogenic effectors and autoimmune targets. Nat. Rev. Rheumatol. 2020, 16, 301–315. [Google Scholar] [CrossRef]
- Elborn, J.S.; Perrett, J.; Forsman-Semb, K.; Marks-Konczalik, J.; Gunawardena, K.; Entwistle, N. Efficacy, safety and effect on biomarkers of AZD9668 in cystic fibrosis. Eur. Respir. J. 2012, 40, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Kuna, P.; Jenkins, M.; O’Brien, C.D.; Fahy, W.A. AZD9668, a neutrophil elastase inhibitor, plus ongoing budesonide/formoterol in patients with COPD. Respir. Med. 2012, 106, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Stockley, R.; de Soyza, A.; Gunawardena, K.; Perrett, J.; Forsman-Semb, K.; Entwistle, N.; Snell, N. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir. Med. 2013, 107, 524–533. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhou, X.; Tan, H.; Hu, Y.; Zhang, L.; Liu, S.; Dai, M.; Li, Y.; Li, Q.; Mao, Z.; et al. Neutrophil extracellular traps contribute to the pathogenesis of acid-aspiration-induced ALI/ARDS. Oncotarget 2018, 9, 1772–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Adrover, J.M.; Carrau, L.; Daßler-Plenker, J.; Bram, Y.; Chandar, V.; Houghton, S.; Redmond, D.; Merrill, J.R.; Shevik, M.; Tenoever, B.R.; et al. Disulfiram inhibits neutrophil extracellular trap formation and protects rodents from acute lung injury and SARS-CoV-2 infection. JCI Insight 2022, 7, e157342. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.U.-S.; O’Sullivan, K.M. The Expanding Role of Extracellular Traps in Inflammation and Autoimmunity: The New Players in Casting Dark Webs. Int. J. Mol. Sci. 2022, 23, 3793. https://doi.org/10.3390/ijms23073793
Huang SU-S, O’Sullivan KM. The Expanding Role of Extracellular Traps in Inflammation and Autoimmunity: The New Players in Casting Dark Webs. International Journal of Molecular Sciences. 2022; 23(7):3793. https://doi.org/10.3390/ijms23073793
Chicago/Turabian StyleHuang, Stephanie U-Shane, and Kim Maree O’Sullivan. 2022. "The Expanding Role of Extracellular Traps in Inflammation and Autoimmunity: The New Players in Casting Dark Webs" International Journal of Molecular Sciences 23, no. 7: 3793. https://doi.org/10.3390/ijms23073793
APA StyleHuang, S. U. -S., & O’Sullivan, K. M. (2022). The Expanding Role of Extracellular Traps in Inflammation and Autoimmunity: The New Players in Casting Dark Webs. International Journal of Molecular Sciences, 23(7), 3793. https://doi.org/10.3390/ijms23073793