
Molecular Effects of Auto-Antibodies on Angiotensin II Type 1 Receptor Signaling and Cell Proliferation

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Endothelial Cell Proliferation Is Induced by AT1R-Abs-Mediated Gq/11 Activation and Is Inhibited by Basal Gi Signaling
2.2. Auto-Antibodies Attenuate Ang II-Mediated Internalization of AT1R
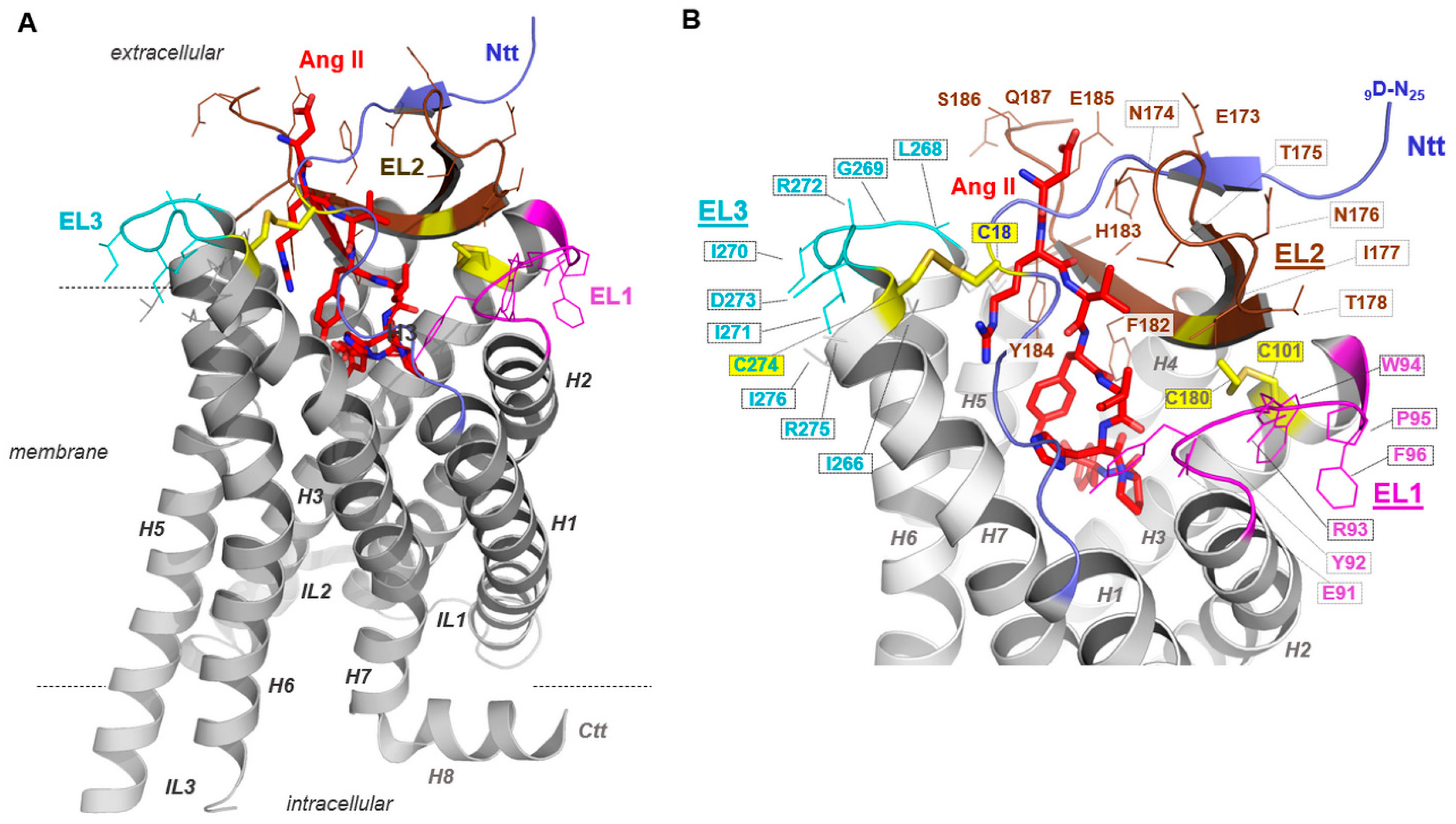
2.3. The Second Extracellular Loop Is Involved in AT1R-Mediated Signaling Induced by AT1R Abs
3. Discussion
3.1. AT1R Auto-Antibodies Inhibit Ang II-Mediated Receptor Internalization
3.2. Cell Proliferation Is Mediated by Gq/11 Activation and Inhibited by Basal Gi Signaling
3.3. EL2 Contributes to Antibody-Mediated Signaling in Systemic Sclerosis
3.4. Conclusions
4. Materials and Methods
4.1. Clinical Samples of Patients with Systemic Sclerosis
4.2. Expression of Constructs and Site-Directed Mutagenesis
4.3. Yeast Culture and Transformation
4.4. Cell Culture and Transfection
4.5. GPCR-Induced Yeast-Growth Assay
4.6. Luciferase Reporter Assay
4.7. EdU Proliferation Assay
4.8. Receptor Expression at the Cell Surface
4.9. Receptor Internalization after Ligand Exposure
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACEI | angiotensin converting enzyme I |
| ACEi | angiotensin converting enzyme inhibitors |
| Ang II | angiotensin II |
| AP1 | Activator Protein 1 |
| ARB | Angiotensin Receptor Blockers |
| AT1R | angiotensin II type 1 receptor |
| AT2R | angiotensin II type 2 receptor |
| AT1R-Abs | angiotensin II type 1 receptor auto-antibodies |
| EL | extracellular loop |
| ERK1/2 | extracellular signal-regulated kinases 1/2 |
| ETAR | endothelin-1 type A receptor |
| ETBR | endothelin-1 type B receptor |
| FCS | fetal calf serum |
| GPCR | G-protein coupled receptor |
| HEK293T | human embryonic kidney 293T cells |
| HMEC-1 | human microvascular endothelial cells |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| HUVEC | human umbilical vascular endothelial cells |
| IgG | immunoglobulin G |
| MAPK | mitogen activated protein kinase |
| NFAT | nuclear factor of activated T-cells |
| NFκB | Nuclear Factor of “κappa-light-chain-enhancer” of activated B-cells |
| Ntt | N-terminal tail |
| PTx | pertussis toxin |
| RAAS | renin-angiotensin-aldosterone system |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus-2 |
| SSc | systemic sclerosis |
| SRC | scleroderma renal crisis |
| TM | transmembrane helix |
| TSHR | thyroid stimulating hormone receptor |
| VSMC | vascular smooth muscle cells |
| VEGF | vascular endothelial growth factor |
| WT | wild-type |
References
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, K.P.; Scheerer, P.; Hildebrand, P.W.; Choe, H.W.; Park, J.H.; Heck, M.; Ernst, O.P. A G protein-coupled receptor at work: The rhodopsin model. Trends Biochem. Sci. 2009, 34, 540–552. [Google Scholar] [CrossRef]
- Allen, A.M.; Zhuo, J.; Mendelsohn, F.A. Localization of angiotensin AT1 and AT2 receptors. J. Am. Soc. Nephrol. 1999, 10 (Suppl. S11), S23–S29. [Google Scholar] [PubMed]
- Abe, K.; Nakashima, H.; Ishida, M.; Miho, N.; Sawano, M.; Soe, N.N.; Kurabayashi, M.; Chayama, K.; Yoshizumi, M.; Ishida, T. Angiotensin II-induced osteopontin expression in vascular smooth muscle cells involves Gq/11, Ras, ERK, Src and Ets-1. Hypertens Res. 2008, 31, 987–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunyady, L.; Catt, K.J. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol. Endocrinol. 2006, 20, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Miserey-Lenkei, S.; Lenkei, Z.; Parnot, C.; Corvol, P.; Clauser, E. A functional enhanced green fluorescent protein (EGFP)-tagged angiotensin II at(1a) receptor recruits the endogenous Galphaq/11 protein to the membrane and induces its specific internalization independently of receptor-g protein coupling in HEK-293 cells. Mol. Endocrinol. 2001, 15, 294–307. [Google Scholar] [CrossRef] [Green Version]
- Ohtsu, H.; Higuchi, S.; Shirai, H.; Eguchi, K.; Suzuki, H.; Hinoki, A.; Brailoiu, E.; Eckhart, A.D.; Frank, G.D.; Eguchi, S. Central role of Gq in the hypertrophic signal transduction of angiotensin II in vascular smooth muscle cells. Endocrinology 2008, 149, 3569–3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laragh, J.H.; Angers, M.; Kelly, W.G.; Lieberman, S. Hypotensive agents and pressor substances. The effect of epinephrine, norepinephrine, angiotensin II, and others on the secretory rate of aldosterone in man. JAMA 1960, 174, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Mouw, D.; Bonjour, J.P.; Malvin, R.L.; Vander, A. Central action of angiotensin in stimulating ADH release. Am. J. Physiol. 1971, 220, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallukat, G.; Homuth, V.; Fischer, T.; Lindschau, C.; Horstkamp, B.; Jupner, A.; Baur, E.; Nissen, E.; Vetter, K.; Neichel, D.; et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J. Clin. Invest. 1999, 103, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Lefaucheur, C.; Viglietti, D.; Bouatou, Y.; Philippe, A.; Pievani, D.; Aubert, O.; Duong Van Huyen, J.P.; Taupin, J.L.; Glotz, D.; Legendre, C.; et al. Non-HLA agonistic anti-angiotensin II type 1 receptor antibodies induce a distinctive phenotype of antibody-mediated rejection in kidney transplant recipients. Kidney. Int. 2019, 96, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Sas-Strozik, A.; Donizy, P.; Koscielska-Kasprzak, K.; Kaminska, D.; Gawlik, K.; Mazanowska, O.; Madziarska, K.; Halon, A.; Krajewska, M.; Banasik, M. Angiotensin II type 1 receptor expression in renal transplant biopsies and anti-AT1R antibodies in serum indicates the risk of transplant loss. Transplant. Proc. 2020, 52, 2299–2304. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, L.J.; Hickey, M.J.; Chan, A.P.; Venick, R.S.; Farmer, D.G.; Busuttil, R.W.; Reed, E.F.; McDiarmid, S.V. Angiotensin II type-1 receptor antibodies are associated with active allograft dysfunction following pediatric liver transplantation. Transplantation 2020, 104, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Dragun, D.; Muller, D.N.; Brasen, J.H.; Fritsche, L.; Nieminen-Kelha, M.; Dechend, R.; Kintscher, U.; Rudolph, B.; Hoebeke, J.; Eckert, D.; et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N. Engl. J. Med. 2005, 352, 558–569. [Google Scholar] [CrossRef] [Green Version]
- Hinchcliff, M.; Varga, J. Obliterative vasculopathy in systemic sclerosis: Endothelial precursor cells as novel targets for therapy. Expert. Rev. Clin. Immunol. 2007, 3, 11–15. [Google Scholar] [CrossRef]
- Kuwana, M.; Kaburaki, J.; Okazaki, Y.; Yasuoka, H.; Kawakami, Y.; Ikeda, Y. Increase in circulating endothelial precursors by atorvastatin in patients with systemic sclerosis. Arthritis. Rheum. 2006, 54, 1946–1951. [Google Scholar] [CrossRef]
- Dragun, D.; Catar, R.; Philippe, A. Non-HLA antibodies against endothelial targets bridging allo- and autoimmunity. Kidney Int. 2016, 90, 280–288. [Google Scholar] [CrossRef]
- Sas-Strozik, A.; Krajewska, M.; Banasik, M. The significance of angiotensin II type 1 receptor (AT1 receptor) in renal transplant injury. Adv. Clin. Exp. Med. 2020, 29, 629–633. [Google Scholar] [CrossRef]
- Sorohan, B.M.; Ismail, G.; Leca, N.; Tacu, D.; Obrisca, B.; Constantinescu, I.; Baston, C.; Sinescu, I. Angiotensin II type 1 receptor antibodies in kidney transplantation: An evidence-based comprehensive review. Transplant. Rev. 2020, 34, 100573. [Google Scholar] [CrossRef]
- Zhang, X.; Reinsmoen, N.L. Angiotensin II type I receptor antibodies in thoracic transplantation. Hum. Immunol. 2019, 80, 579–582. [Google Scholar] [CrossRef]
- Zhang, X.; Reinsmoen, N.L. Impact and production of Non-HLA-specific antibodies in solid organ transplantation. Int. J. Immunogenet. 2020, 47, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, Y.X.; Wang, X.F.; Zheng, Y.Q.; Jin, Z.; Zhi, J.M. Role of agonistic autoantibodies against type-1 angiotensin II receptor in the pathogenesis of retinopathy in preeclampsia. Sci. Rep. 2016, 6, 29036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, A.H.; Irani, R.A.; Zhang, W.; Wang, W.; Blackwell, S.C.; Kellems, R.E.; Xia, Y. Angiotensin receptor agonistic autoantibody-mediated soluble fms-like tyrosine kinase-1 induction contributes to impaired adrenal vasculature and decreased aldosterone production in preeclampsia. Hypertension 2013, 61, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.C.; Ahmad, S.; Mi, T.; Abbasi, S.; Xia, L.; Day, M.C.; Ramin, S.M.; Ahmed, A.; Kellems, R.E.; Xia, Y. Autoantibody from women with preeclampsia induces soluble Fms-like tyrosine kinase-1 production via angiotensin type 1 receptor and calcineurin/nuclear factor of activated T-cells signaling. Hypertension 2008, 51, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Catar, R.; Herse-Naether, M.; Zhu, N.; Wagner, P.; Wischnewski, O.; Kusch, A.; Kamhieh-Milz, J.; Eisenreich, A.; Rauch, U.; Hegner, B.; et al. Autoantibodies targeting AT1- and ETA-receptors link endothelial proliferation and coagulation via Ets-1 transcription factor. Int. J. Mol. Sci. 2021, 23, 244. [Google Scholar] [CrossRef]
- Riemekasten, G.; Philippe, A.; Nather, M.; Slowinski, T.; Muller, D.N.; Heidecke, H.; Matucci-Cerinic, M.; Czirjak, L.; Lukitsch, I.; Becker, M.; et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.J.; Fortune, A.; Phalipon, A.; Marcel-Peyre, V.; Simenel, C.; Imberty, A.; Delepierre, M.; Mulard, L.A. Toward a better understanding of the basis of the molecular mimicry of polysaccharide antigens by peptides: The example of Shigella flexneri 5a. J. Biol. Chem. 2006, 281, 2317–2332. [Google Scholar] [CrossRef] [Green Version]
- Fillion, D.; Cabana, J.; Guillemette, G.; Leduc, R.; Lavigne, P.; Escher, E. Structure of the human angiotensin II type 1 (AT1) receptor bound to angiotensin II from multiple chemoselective photoprobe contacts reveals a unique peptide binding mode. J. Biol. Chem. 2013, 288, 8187–8197. [Google Scholar] [CrossRef] [Green Version]
- Laporte, S.A.; Boucard, A.A.; Servant, G.; Guillemette, G.; Leduc, R.; Escher, E. Determination of peptide contact points in the human angiotensin II type I receptor (AT1) with photosensitive analogs of angiotensin II. Mol. Endocrinol. 1999, 13, 578–586. [Google Scholar] [CrossRef]
- Unal, H.; Jagannathan, R.; Bhat, M.B.; Karnik, S.S. Ligand-specific conformation of extracellular loop-2 in the angiotensin II type 1 receptor. J. Biol. Chem. 2010, 285, 16341–16350. [Google Scholar] [CrossRef] [Green Version]
- Unal, H.; Jagannathan, R.; Bhatnagar, A.; Tirupula, K.; Desnoyer, R.; Karnik, S.S. Long range effect of mutations on specific conformational changes in the extracellular loop 2 of angiotensin II type 1 receptor. J. Biol. Chem. 2013, 288, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.W.; Suomivuori, C.M.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020, 367, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Stamatelopoulos, K.; Terpos, E.; Tsitsilonis, O.E.; Aivalioti, E.; Paraskevis, D.; Kastritis, E.; Pavlakis, G.N.; Dimopoulos, M.A. Insights to SARS-CoV-2 life cycle, pathophysiology, and rationalized treatments that target COVID-19 clinical complications. J. Biomed. Sci. 2021, 28, 9. [Google Scholar] [CrossRef] [PubMed]
- Bellis, A.; Mauro, C.; Barbato, E.; Trimarco, B.; Morisco, C. The rationale for angiotensin receptor neprilysin inhibitors in a multi-targeted therapeutic approach to COVID-19. Int. J. Mol. Sci. 2020, 21, 8612. [Google Scholar] [CrossRef]
- Sharma, T.; Mehan, S. Possible therapeutic interventions in COVID-19 induced ARDS by cotinine as an ACE-2 promoter and AT-1R blocker. Infect. Disord. Drug Targets. 2020, 21, e170721189261. [Google Scholar] [CrossRef]
- Manglik, A.; Wingler, L.M.; Rockman, H.A.; Lefkowitz, R.J. Beta-arrestin-biased angiotensin II receptor agonists for COVID-19. Circulation 2020, 142, 318–320. [Google Scholar] [CrossRef]
- Kowal-Bielecka, O.; Fransen, J.; Avouac, J.; Becker, M.; Kulak, A.; Allanore, Y.; Distler, O.; Clements, P.; Cutolo, M.; Czirjak, L.; et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja, V. Management of scleroderma renal crisis. Curr. Opin. Rheumatol. 2019, 31, 223–230. [Google Scholar] [CrossRef]
- Bruni, C.; Cometi, L.; Gigante, A.; Rosato, E.; Matucci-Cerinic, M. Prediction and primary prevention of major vascular complications in systemic sclerosis. Eur. J. Intern. Med. 2021, 87, 51–58. [Google Scholar] [CrossRef]
- Gordon, S.M.; Hughes, J.B.; Nee, R.; Stitt, R.S.; Bailey, W.T.; Little, D.J.; Edison, J.D.; Olson, S.W. Systemic sclerosis medications and risk of scleroderma renal crisis. BMC Nephrol. 2019, 20, 279. [Google Scholar] [CrossRef]
- Butikofer, L.; Varisco, P.A.; Distler, O.; Kowal-Bielecka, O.; Allanore, Y.; Riemekasten, G.; Villiger, P.M.; Adler, S.; Collaborators, E. ACE inhibitors in SSc patients display a risk factor for scleroderma renal crisis-a EUSTAR analysis. Arthritis. Res. Ther. 2020, 22, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez Sandoval, Y.H.; Levesque, L.O.; Anand-Srivastava, M.B. Contribution of epidermal growth factor receptor transactivation in angiotensin II-induced enhanced expression of Gi protein and proliferation in A10 vascular smooth muscle cells. Can. J. Physiol. Pharmacol. 2009, 87, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Boss, V.; Talpade, D.J.; Murphy, T.J. Induction of NFAT-mediated transcription by Gq-coupled receptors in lymphoid and non-lymphoid cells. J. Biol. Chem. 1996, 271, 10429–10432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishita, R.; Ueda, H.; Ito, H.; Takasaki, J.; Nagata, K.; Asano, T. Involvement of Gq/11 in both integrin signal-dependent and -independent pathways regulating endothelin-induced neural progenitor proliferation. Neurosci. Res. 2007, 59, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, O.; Li, Y.; Anand-Srivastava, M.B. Nitric oxide attenuates overexpression of Gialpha proteins in vascular smooth muscle cells from SHR: Role of ROS and ROS-mediated signaling. PLoS ONE 2017, 12, e0179301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaraj, K.K.; Li, R.; Albarran-Juarez, J.; Wang, S.; Tischner, D.; Grimm, M.; Swiercz, J.M.; Offermanns, S.; Wettschureck, N. Endothelial Galphaq/11 is required for VEGF-induced vascular permeability and angiogenesis. Cardiovasc. Res. 2015, 108, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Hsia, J.A.; Moss, J.; Hewlett, E.L.; Vaughan, M. ADP-ribosylation of adenylate cyclase by pertussis toxin. Effects on inhibitory agonist binding. J. Biol. Chem. 1984, 259, 1086–1090. [Google Scholar] [CrossRef]
- Raymond, J.R. Multiple mechanisms of receptor-G protein signaling specificity. Am. J. Physiol. 1995, 269, F141–F158. [Google Scholar] [CrossRef]
- Thomas, W.G.; Thekkumkara, T.J.; Baker, K.M. Molecular mechanisms of angiotensin II (AT1a) receptor endocytosis. Clin. Exp. Pharmacol. Physiol. 1996, 23 (Suppl. S3), S74–S80. [Google Scholar] [CrossRef]
- Bian, J.; Lei, J.; Yin, X.; Wang, P.; Wu, Y.; Yang, X.; Wang, L.; Zhang, S.; Liu, H.; Fu, M.L.X. Limited AT1 receptor internalization is a novel mechanism underlying sustained vasoconstriction induced by AT1 receptor autoantibody from preeclampsia. J. Am. Heart Assoc. 2019, 8, e011179. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingler, L.M.; McMahon, C.; Staus, D.P.; Lefkowitz, R.J.; Kruse, A.C. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell 2019, 176, 479–490.e412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, H.; Pipolo, L.; Thomas, W.G. Association of beta-Arrestin 1 with the type 1A angiotensin II receptor involves phosphorylation of the receptor carboxyl terminus and correlates with receptor internalization. Mol. Endocrinol. 2001, 15, 1706–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad Nezhady, M.A.; Rivera, J.C.; Chemtob, S. Location bias as emerging paradigm in GPCR biology and drug discovery. iScience 2020, 23, 101643. [Google Scholar] [CrossRef]
- Catar, R.A.; Wischnewski, O.; Chen, L.; Heidecke, H.; Rutz, C.; Schulein, R.; Dragun, D.; Philippe, A.; Kusch, A. Non-HLA antibodies targeting angiotensin II type 1 receptors and endothelin-1 type A receptors impair endothelial repair via a beta2-arrestin link to the mTOR pathway. Kidney Int. 2021, 101, 498–509. [Google Scholar] [CrossRef]
- Violin, J.D.; DeWire, S.M.; Yamashita, D.; Rominger, D.H.; Nguyen, L.; Schiller, K.; Whalen, E.J.; Gowen, M.; Lark, M.W. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 2010, 335, 572–579. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Abraham, D.; Williams, B.; Violin, J.D.; Mao, L.; Rockman, H.A. beta-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1001–H1010. [Google Scholar] [CrossRef] [Green Version]
- Monasky, M.M.; Taglieri, D.M.; Henze, M.; Warren, C.M.; Utter, M.S.; Soergel, D.G.; Violin, J.D.; Solaro, R.J. The beta-arrestin-biased ligand TRV120023 inhibits angiotensin II-induced cardiac hypertrophy while preserving enhanced myofilament response to calcium. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H856–H866. [Google Scholar] [CrossRef] [Green Version]
- Tarigopula, M.; Davis, R.T., 3rd; Mungai, P.T.; Ryba, D.M.; Wieczorek, D.F.; Cowan, C.L.; Violin, J.D.; Wolska, B.M.; Solaro, R.J. Cardiac myosin light chain phosphorylation and inotropic effects of a biased ligand, TRV120023, in a dilated cardiomyopathy model. Cardiovasc. Res. 2015, 107, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Kill, A.; Tabeling, C.; Undeutsch, R.; Kuhl, A.A.; Gunther, J.; Radic, M.; Becker, M.O.; Heidecke, H.; Worm, M.; Witzenrath, M.; et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis. Res. Ther. 2014, 16, R29. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.O.; Kill, A.; Kutsche, M.; Guenther, J.; Rose, A.; Tabeling, C.; Witzenrath, M.; Kuhl, A.A.; Heidecke, H.; Ghofrani, H.A.; et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am. J. Respir. Crit. Care Med. 2014, 190, 808–817. [Google Scholar] [CrossRef]
- Zrein, A.; Bagher, A.M.; Young, A.P.; Denovan-Wright, E.M.; Kelly, M.E.M. Endothelin receptor heteromerization inhibits beta-arrestin function in HEK293 cells. Can. J. Physiol. Pharmacol. 2020, 98, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Banasik, M.; Boratynska, M.; Koscielska-Kasprzak, K.; Kaminska, D.; Zmonarski, S.; Mazanowska, O.; Krajewska, M.; Bartoszek, D.; Zabinska, M.; Myszka, M.; et al. Non-HLA antibodies: Angiotensin II type 1 receptor (anti-AT1R) and endothelin-1 type A receptor (anti-ETAR) are associated with renal allograft injury and graft loss. Transplant. Proc. 2014, 46, 2618–2621. [Google Scholar] [CrossRef]
- Buttrup Larsen, S.; Wallukat, G.; Schimke, I.; Sandager, A.; Tvilum Christensen, T.; Uldbjerg, N.; Torring, N. Functional autoantibodies against Endothelin-1 receptor type A and Angiotensin II receptor type 1 in patients with preeclampsia. Pregnancy Hypertens 2018, 14, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Hiemann, N.E.; Meyer, R.; Wellnhofer, E.; Schoenemann, C.; Heidecke, H.; Lachmann, N.; Hetzer, R.; Dragun, D. Non-HLA antibodies targeting vascular receptors enhance alloimmune response and microvasculopathy after heart transplantation. Transplantation 2012, 94, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Boursier, M.E.; Levin, S.; Zimmerman, K.; Machleidt, T.; Hurst, R.; Butler, B.L.; Eggers, C.T.; Kirkland, T.A.; Wood, K.V.; Friedman Ohana, R. The luminescent HiBiT peptide enables selective quantitation of G protein-coupled receptor ligand engagement and internalization in living cells. J. Biol. Chem. 2020, 295, 5124–5135. [Google Scholar] [CrossRef]
- Murphy, T.J.; Alexander, R.W.; Griendling, K.K.; Runge, M.S.; Bernstein, K.E. Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature 1991, 351, 233–236. [Google Scholar] [CrossRef]
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Keys, J.R.; Greene, E.A.; Koch, W.J.; Eckhart, A.D. Gq-coupled receptor agonists mediate cardiac hypertrophy via the vasculature. Hypertension 2002, 40, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Herr, D.; Rodewald, M.; Fraser, H.M.; Hack, G.; Konrad, R.; Kreienberg, R.; Wulff, C. Regulation of endothelial proliferation by the renin-angiotensin system in human umbilical vein endothelial cells. Reproduction 2008, 136, 125–130. [Google Scholar] [CrossRef] [Green Version]
- AbdAlla, S.; Lother, H.; Quitterer, U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature 2000, 407, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Anton, E.L.; Fernandes, D.; Assreuy, J.; da Silva-Santos, J.E. Bradykinin increases BP in endotoxemic rat: Functional and biochemical evidence of angiotensin II AT1 /bradykinin B2 receptor heterodimerization. Br. J. Pharmacol. 2019, 176, 2608–2626. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.C.; Lee, M.H.; Appleton, K.M.; El-Shewy, H.M.; Morinelli, T.A.; Peterson, Y.K.; Luttrell, L.M.; Jaffa, A.A. The arrestin-selective angiotensin AT1 receptor agonist [Sar1,Ile4,Ile8]-AngII negatively regulates bradykinin B2 receptor signaling via AT1-B2 receptor heterodimers. J. Biol. Chem. 2013, 288, 18872–18884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, M.; Hutchinson, H.G.; Fujinaga, M.; Hayashida, W.; Morishita, R.; Zhang, L.; Horiuchi, M.; Pratt, R.E.; Dzau, V.J. The angiotensin II type 2 (AT2) receptor antagonizes the growth effects of the AT1 receptor: Gain-of-function study using gene transfer. Proc. Natl. Acad. Sci. USA 1995, 92, 10663–10667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bou Daou, G.; Li, Y.; Anand-Srivastava, M.B. Enhanced expression of Gialpha proteins contributes to the hyperproliferation of vascular smooth muscle cells from spontaneously hypertensive rats via MAP kinase- and PI3 kinase-independent pathways. Can. J. Physiol. Pharmacol. 2016, 94, 49–58. [Google Scholar] [CrossRef]
- Hossain, E.; Li, Y.; Anand-Srivastava, M.B. Angiotensin II-induced overexpression of sirtuin 1 contributes to enhanced expression of Gialpha proteins and hyperproliferation of vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H496–H508. [Google Scholar] [CrossRef]
- Li, Y.; Hossain, E.; Arifen, N.; Srivastava, A.K.; Anand-Srivastava, M.B. Sirtuin1 contributes to the overexpression of Gialpha proteins and hyperproliferation of vascular smooth muscle cells from spontaneously hypertensive rats. J. Hypertens 2022, 40, 117–127. [Google Scholar] [CrossRef]
- Lyu, C.; Ye, Y.; Lensing, M.M.; Wagner, K.U.; Weigel, R.J.; Chen, S. Targeting Gi/o protein-coupled receptor signaling blocks HER2-induced breast cancer development and enhances HER2-targeted therapy. JCI Insight 2021, 6, e150532. [Google Scholar] [CrossRef] [PubMed]
- Massotte, D.; Kieffer, B.L. The second extracellular loop: A damper for G protein-coupled receptors? Nat. Struct. Mol. Biol. 2005, 12, 287–288. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.C.; van Westen, G.J.; Li, Q.; AP, I.J. Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol. Sci. 2011, 32, 35–42. [Google Scholar] [CrossRef]
- Nicastro, G.; Peri, F.; Franzoni, L.; de Chiara, C.; Sartor, G.; Spisni, A. Conformational features of a synthetic model of the first extracellular loop of the angiotensin II AT1A receptor. J. Pept. Sci. 2003, 9, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Olesnicky, N.S.; Brown, A.J.; Dowell, S.J.; Casselton, L.A. A constitutively active G-protein-coupled receptor causes mating self-compatibility in the mushroom Coprinus. EMBO J. 1999, 18, 2756–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.J.; Dyos, S.L.; Whiteway, M.S.; White, J.H.; Watson, M.A.; Marzioch, M.; Clare, J.J.; Cousens, D.J.; Paddon, C.; Plumpton, C.; et al. Functional coupling of mammalian receptors to the yeast mating pathway using novel yeast/mammalian G protein alpha-subunit chimeras. Yeast 2000, 16, 11–22. [Google Scholar] [CrossRef]
- Dowell, S.J.; Brown, A.J. Yeast assays for G protein-coupled receptors. Methods Mol. Biol. 2009, 552, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Beukers, M.W.; van Oppenraaij, J.; van der Hoorn, P.P.; Blad, C.C.; den Dulk, H.; Brouwer, J.; AP, I.J. Random mutagenesis of the human adenosine A2B receptor followed by growth selection in yeast. Identification of constitutively active and gain of function mutations. Mol. Pharmacol. 2004, 65, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, M.C.; Wisse, L.E.; Dinaj, A.; Vroling, B.; Vriend, G.; Ijzerman, A.P. The role of the second and third extracellular loops of the adenosine A1 receptor in activation and allosteric modulation. Biochem. Pharmacol. 2012, 84, 76–87. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| polyA265-276 | AGCTGCCGCTGCAGCTGCA GATATTGTGGACACGGCC | GCGGCGGCTGCTGCAGCCGC TACATCCAGAAAAGTGAATATTTG |
| polyA173-178 | GCTGCTGCCGTTTGTGCTTTCCATTATGAG | GGCGGCCGCAATGAAAAATACATTTCGATGGATTATAG |
| polyA182-187 | TGCGGCCGCAAATTCAACCCTTCCGATAG | GCAGCGGCAGCACAAACTGTAATATTGGTGTTC |
| polyA92-97 | GCCGCTGCCAATTACCTATGTAAGATTGCTTC | CGCGGCGGCTTCCATAGCTGTGTAGAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Philippe, A.; Kleinau, G.; Gruner, J.J.; Wu, S.; Postpieszala, D.; Speck, D.; Heidecke, H.; Dowell, S.J.; Riemekasten, G.; Hildebrand, P.W.; et al. Molecular Effects of Auto-Antibodies on Angiotensin II Type 1 Receptor Signaling and Cell Proliferation. Int. J. Mol. Sci. 2022, 23, 3984. https://doi.org/10.3390/ijms23073984
Philippe A, Kleinau G, Gruner JJ, Wu S, Postpieszala D, Speck D, Heidecke H, Dowell SJ, Riemekasten G, Hildebrand PW, et al. Molecular Effects of Auto-Antibodies on Angiotensin II Type 1 Receptor Signaling and Cell Proliferation. International Journal of Molecular Sciences. 2022; 23(7):3984. https://doi.org/10.3390/ijms23073984
Chicago/Turabian StylePhilippe, Aurélie, Gunnar Kleinau, Jason Jannis Gruner, Sumin Wu, Daniel Postpieszala, David Speck, Harald Heidecke, Simon J. Dowell, Gabriela Riemekasten, Peter W. Hildebrand, and et al. 2022. "Molecular Effects of Auto-Antibodies on Angiotensin II Type 1 Receptor Signaling and Cell Proliferation" International Journal of Molecular Sciences 23, no. 7: 3984. https://doi.org/10.3390/ijms23073984
APA StylePhilippe, A., Kleinau, G., Gruner, J. J., Wu, S., Postpieszala, D., Speck, D., Heidecke, H., Dowell, S. J., Riemekasten, G., Hildebrand, P. W., Kamhieh-Milz, J., Catar, R., Szczepek, M., Dragun, D., & Scheerer, P. (2022). Molecular Effects of Auto-Antibodies on Angiotensin II Type 1 Receptor Signaling and Cell Proliferation. International Journal of Molecular Sciences, 23(7), 3984. https://doi.org/10.3390/ijms23073984