
Life Entrapped in a Network of Atavistic Attractors: How to Find a Rescue


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Theoretical Bases
2.1. Molecular Fundamentals of Unified Cell Bioenergetics and Bioenergetic Disturbances
2.2. Layered Model of Evolution of Cellular Functionalities
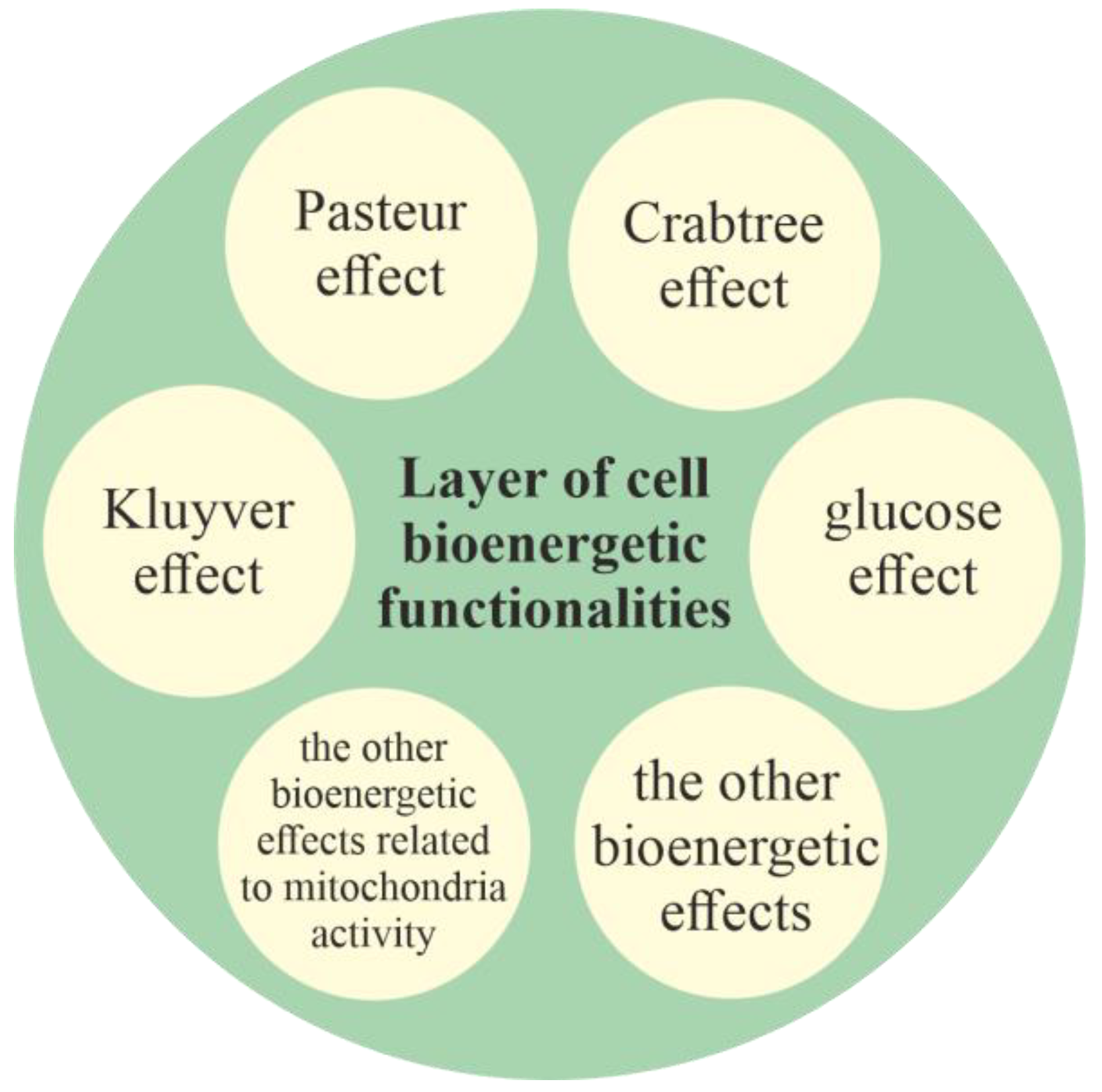
2.2.1. Layer of Cell Bioenergetic Functionalities, i.e., a Phylogenetic Memory Layer of Universal Cell Bioenergetic Functionalities
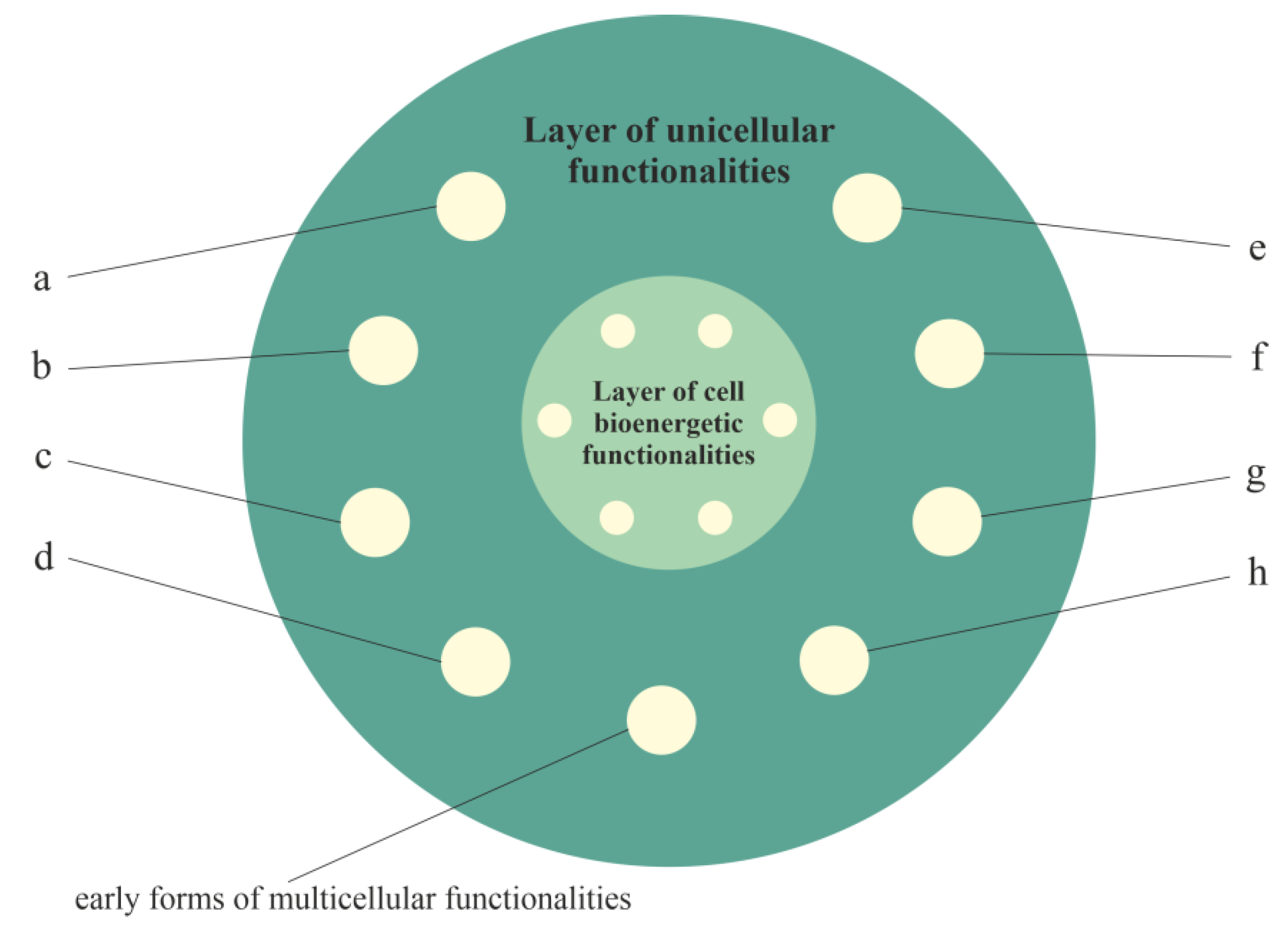
2.2.2. Layer of Unicellular Functionalities, i.e., a Phylogenetic Memory Layer of Atavistic Functionalities
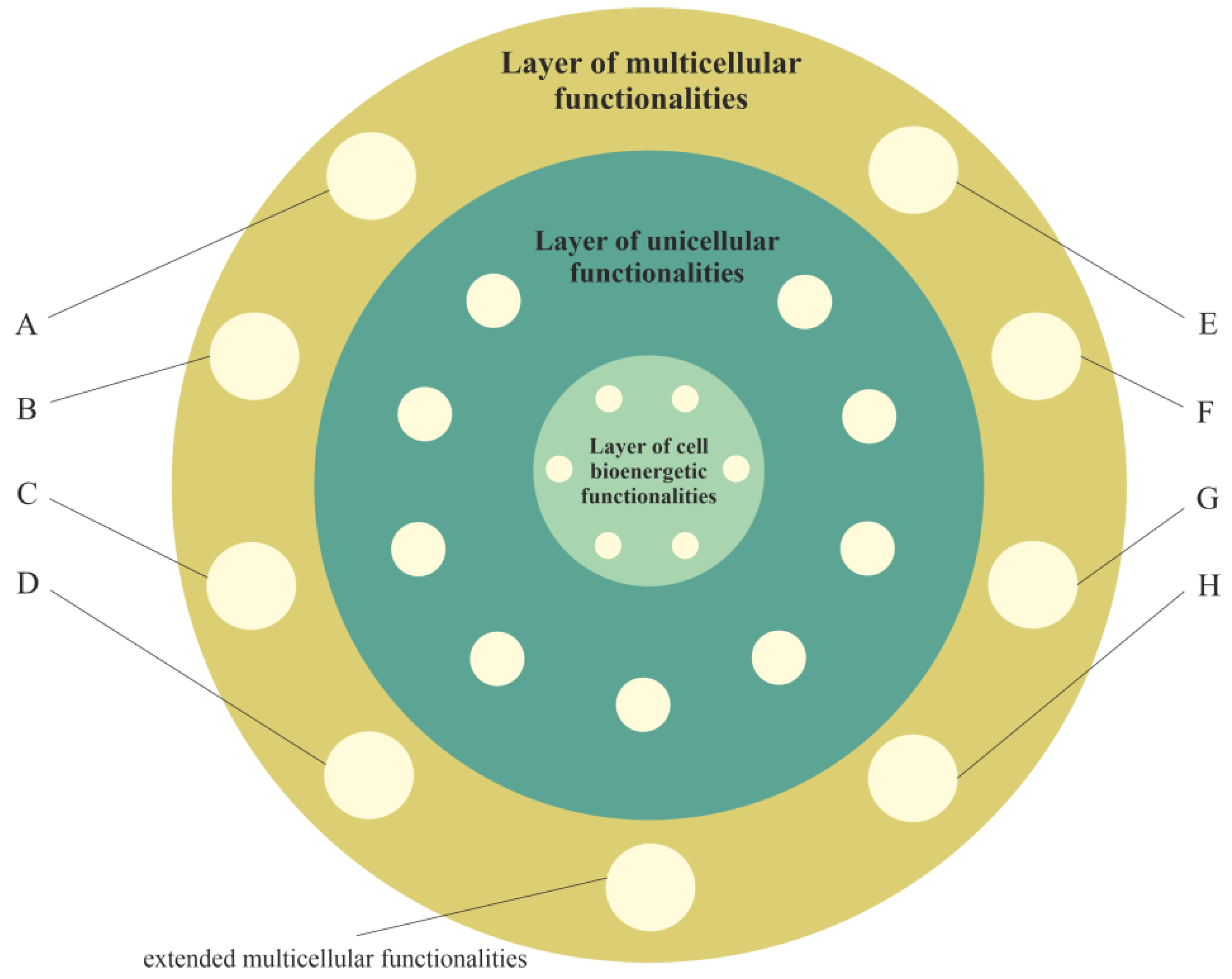
2.2.3. Layer of Multicellular Functionalities, i.e., a Phylogenetic Memory Layer of Multicellular Advanced Functionalities
3. Discussion on the Universal Model of Cancer Transformation and Development
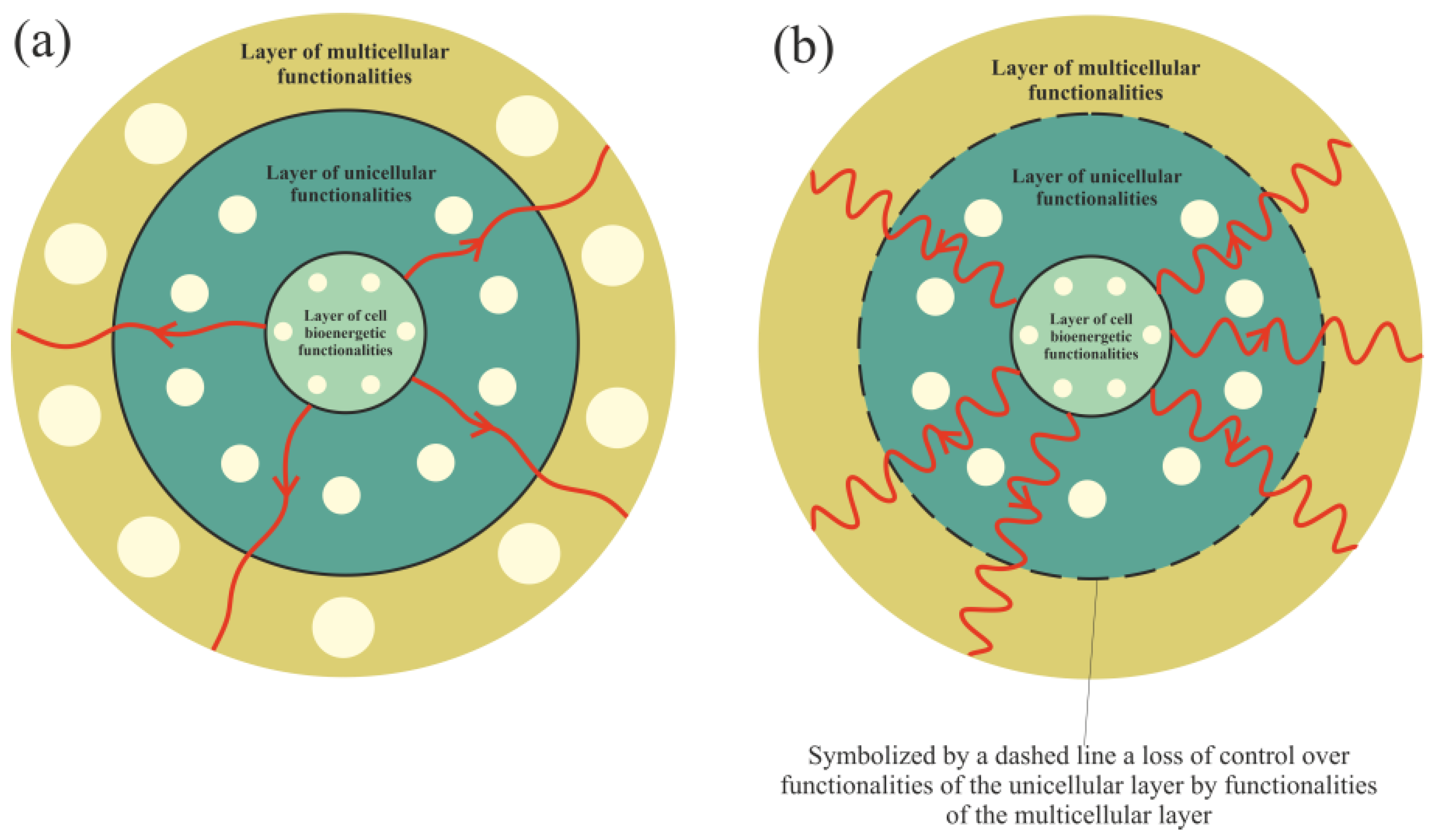
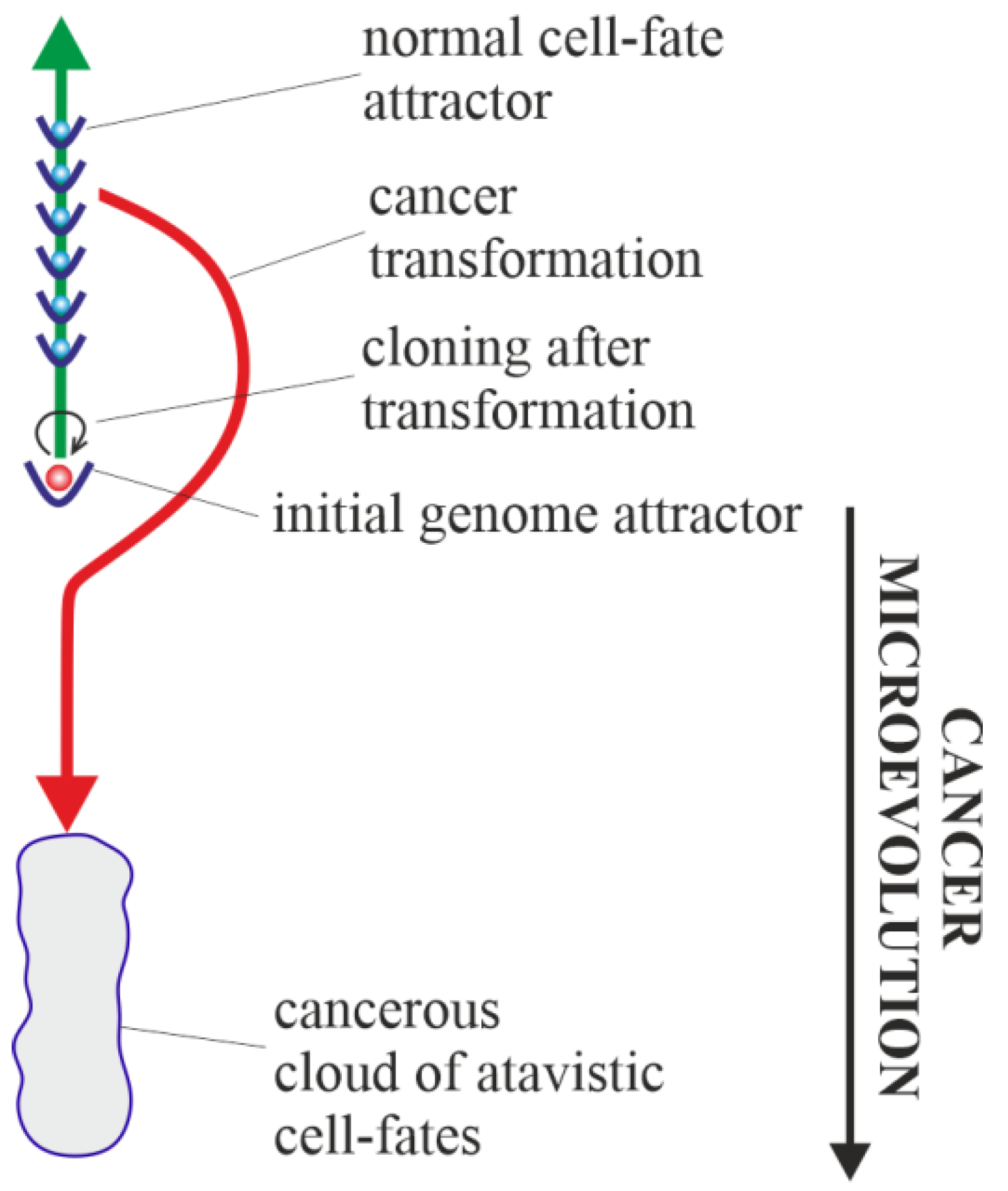
3.1. Cancer Transformation as a Loss of Control over Atavistic Functionalities
3.2. Vertical and Horizontal Cancer Development
3.3. Vertical Cancer Development
3.4. Horizontal Cancer Development
- (a)
- Cancer transformation is caused by a loss of control over atavistic functionalities;
- (b)
- Vertical cancer development is caused by recurring (repeated) losses of current cell-fate stability; at
- (c)
- Horizontal cancer development is optional and is caused by recurring (repeated) losses of genomic integrity.
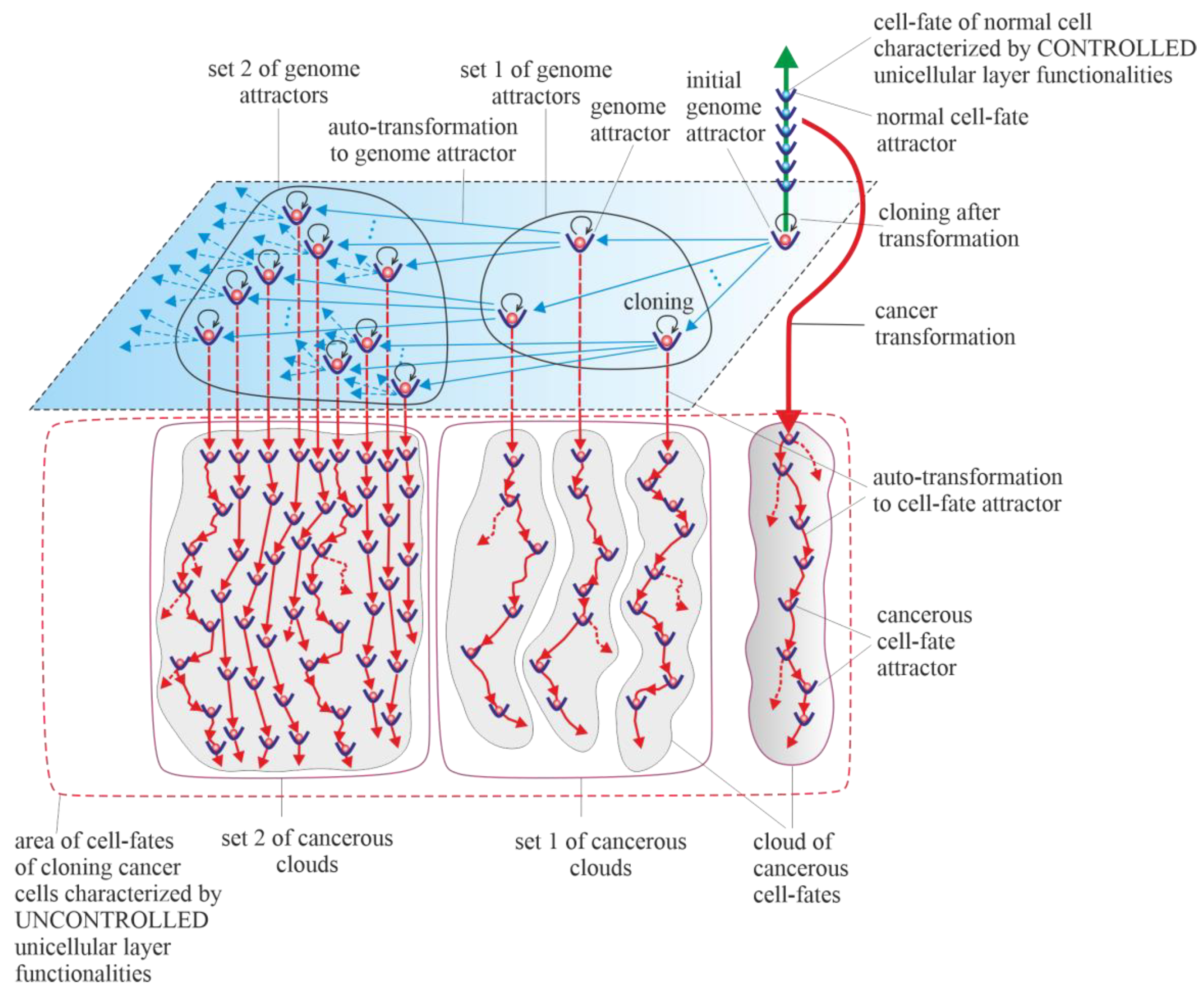
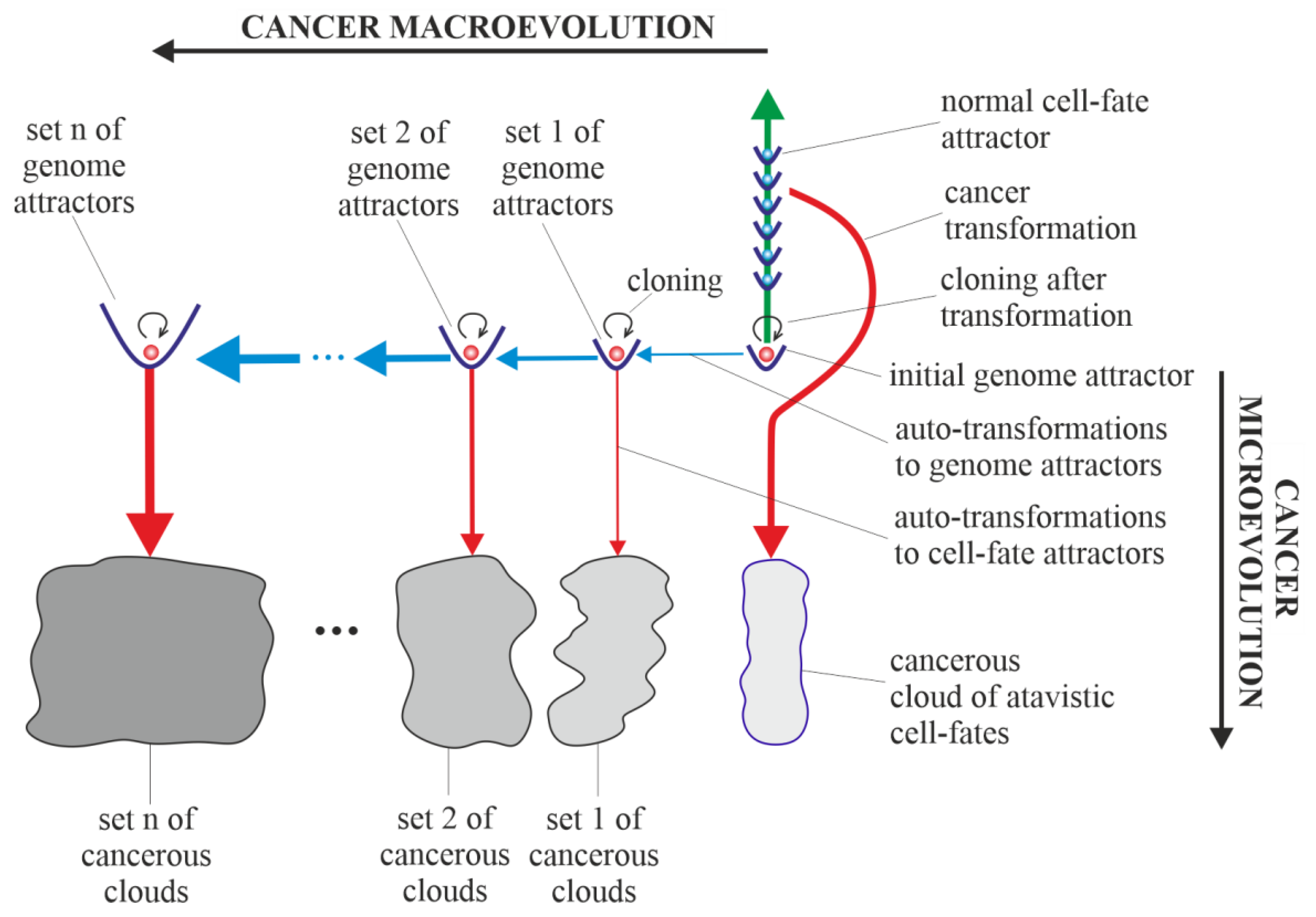
3.5. Cancerous Clouds of Atavistic Cell-Fates
3.6. Purely Vertical Cancer Development
3.7. Cancers without Mutation
3.8. Cancer Development as a Learning Process
4. Conclusions
- (a)
- It is better not to concentrate on destruction of unicellular layer functionalities (for example, aerobic glycolysis, which occurs during the Warburg effect); such functionalities are very conservative and secured by many redundant metabolic pathways;
- (b)
- In order to stop permanent changes of genome attractors, cancerous mitochondria should be discharged from high-energy molecules, among others, by increasing physical activity, intensive aeration and ensuring a proper diet with food limitation. This conclusion is in accordance with the mitochondrial correction method, which represents a new therapeutic paradigm for cancer [97]. The disadvantage of the discharging mitochondria method is that after discharge, driven by uncontrolled atavistic functionalities, cancerous cell fates are most likely still active;
- (c)
- In order to regain control over atavistic functionalities of the unicellular layer, functionalities of the multicellular layer should be reconstructed and reactivated. This idea is related to an old concept of tumor reversion [98]. Studies demonstrated that when tumor cells are placed within a “normal” morphogenetic milieu, they can be “reprogrammed” (thus acquiring a healthy phenotype de novo) and can then behave as native cells [99]. For example, several cancers undergo partial or complete reversion after exposure to embryonic environments [100]. This phenotypic reversion can be achieved despite the large number of genome alterations presented in cancerous cells and is related to inhibition of the migrating/invasive phenotype of cancer cells by some low-molecular-weight factors expressed by early embryonic microenvironments [101,102,103,104]. In this light, one possibilities for reconstruction and reactivation of functionalities of the multicellular layer is to expose cancer to embryo and/or egg extracts [105,106]. The disadvantage of this method is that after reconstruction and reactivation of functionalities of the multicellular layer, mitochondria are most likely still overenergized, which threatens repeated cancer transformation;
- (d)
- The best solution seems to be the discharge of cancerous mitochondria from high-energy molecules, along with the reconstruction and reactivation of functionalities of the multicellular layer.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heng, J.; Heng, H.H. Genome Chaos, Information Creation, and Cancer Emergence: Searching for New Frameworks on the 50th Anniversary of the “War on Cancer”. Genes 2022, 13, 101. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of cancer: Survival at the brink. Semin. Cancer Biol. 2020; in press. [Google Scholar] [CrossRef]
- Heng, H.H. Debating Cancer: The Paradox in Cancer Research; World Scientific Publishing Co.: Singapore, 2015; ISBN 978-981-4520-84-3. [Google Scholar]
- Ye, C.J.; Sharpe, Z.; Heng, H.H. Origins and Consequences of Chromosomal Instability: From Cellular Adaptation to Genome Chaos-Mediated System Survival. Genes 2020, 11, 1162. [Google Scholar] [CrossRef]
- Baker, S.G. The detached pericyte hypothesis: A novel explanation for many puzzling aspects of tumorigenesis. Org. J. Biol. Sci. 2018, 2, 25–41. [Google Scholar]
- Vainshelbaum, N.M.; Salmina, K.; Gerashchenko, B.I.; Lazovska, M.; Zayakin, P.; Cragg, M.S.; Pjanova, D.; Erenpreisa, J. Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance. Cells 2022, 11, 880. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. [Google Scholar] [CrossRef]
- Ivanov, A.; Cragg, M.S.; Erenpreisa, J.; Emzinsh, D.; Lukman, H.; Illidge, T.M. Endopolyploid cells produced after severe genotoxic damage have the potential to repair DNA double strand breaks. J. Cell. Sci. 2003, 116, 4095–4106. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.G. Paradoxes in carcinogenesis should spur new avenues of research: An historical perspective. Disrupt. Sci. Technol. 2012, 1, 100–107. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W., Jr. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Baker, S.G. The case for a cancer paradox initiative. Carcinogenesis 2021, 42, 1023–1025. [Google Scholar] [CrossRef]
- Arguello, F. Atavistic Metamorphosis: A New and Logical Explanation for the Origin and Biological Nature of Cancer: With a Discussion on a Novel Approach to Treat Cancer; CreateSpace: Scotts Valley, CA, USA, 2011; ISBN 13:978-1460968994. [Google Scholar]
- Davies, P. Exposing cancer’s deep evolutionary roots. Phys. World 2013, 26, 37–40. [Google Scholar] [CrossRef]
- Davies, P.C.W.; Lineweaver, C.H. Cancer tumors as Metazoa 1.0: Tapping genes of ancient ancestors. Phys. Biol. 2011, 8, 15001. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V.; Vazquez-Martin, A.; Salmina, K.; Cragg, M.S. Stress-induced polyploidy shifts somatic cells towards a pro-tumourogenic unicellular gene transcription network. Cancer Hypotheses 2018, 1, 1–20. [Google Scholar]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Life-cycle features of tumour cells. In Evolutionary Biology from Concept to Application; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 61–71. [Google Scholar]
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol. Int. 2005, 29, 1012–1018. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Wheatley, D. Endopolyploidy in development and cancer; “survival of the fattest?”. Cell Biol. Int. 2005, 29, 981–982. [Google Scholar] [CrossRef]
- Lineweaver, C.H.; Davies, P.C.W.; Vincent, M.D. Targeting cancer’s weaknesses (not its strengths): Therapeutic strategies suggested by the atavistic model. BioEssays 2014, 36, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. Developmental and non developmental polyploidy in xenic and axenic cultured stem cell lines of Entamoeba invadens and E. histolytica. Insights Stem. Cells 2016, 2, 1–9. [Google Scholar]
- Vincent, M.D. Cancer: A de-repression of a default survival program common to all cells? BioEssays 2012, 34, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.D. Cancer: Beyond speciation. Adv. Cancer Res. 2011, 112, 283–350. [Google Scholar]
- Lineweaver, C.H.; Bussey, K.J.; Blackburn, A.C.; Davies, P.C.W. Cancer progression as a sequence of atavistic reversions. BioEssays 2021, 43, 2000305. [Google Scholar] [CrossRef]
- Yett, J.R. Eukaryotes. In Salem Press Encyclopedia of Science; Grey House Publishing: Amenia, NY, USA, 2015. [Google Scholar]
- Khozouz, R. Atavistic cancermodel: A new theory of cancer? BioEssays 2021, 43, 2100206. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Wen, S.; Huang, P. Targeting cancer cell mitochondria as a therapeutic approach: Recent updates. Future Med. Chem. 2017, 9, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Kasperski, A.; Kasperska, R. Study on attractors during organism evolution. Sci. Rep. 2021, 11, 9637. [Google Scholar] [CrossRef] [PubMed]
- Cross, W.; Kovac, M.; Mustonen, V.; Temko, D.; Davis, H.; Baker, A.; Biswas, S.; Arnold, R.; Chegwidden, L.; Gatenbee, C.; et al. The evolutionary landscape of colorectal tumorigenesis. Nat. Ecol. Evol. 2018, 2, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Regoes, R.R. Population genetics meets cancer genomics. Proc. Natl. Acad. Sci. USA 2010, 107, 18241–18242. [Google Scholar] [CrossRef]
- Kasperski, A.; Kasperska, R. Bioenergetics of life, disease and death phenomena. Theory Biosci. 2018, 137, 155–168. [Google Scholar] [CrossRef]
- Kasperski, A. Modelling of cells bioenergetics. Acta Biotheor. 2008, 56, 233–247. [Google Scholar] [CrossRef]
- Noda, M.; Yamashita, S.; Takahashi, N.; Eto, K.; Shen, L.M.; Izumi, K.; Daniel, S.; Tsubamoto, Y.; Nemoto, T.; Iino, M.; et al. Switch to anaerobic glucose metabolism with NADH accumulation in the beta-cell model of mitochondrial diabetes. Characteristics of betaHC9 cells deficient in mitochondrial DNA transcription. J. Biol. Chem. 2002, 277, 41817–41826. [Google Scholar] [CrossRef]
- Kasperski, A. Genome Attractors as Places of Evolution and Oases of Life. Processes 2021, 9, 1646. [Google Scholar] [CrossRef]
- Kasperski, A.; Kasperska, R. Selected disease fundamentals based on the unified cell bioenergetics. J. Investig. Biochem. 2013, 2, 93–100. [Google Scholar] [CrossRef]
- Bakker, B.M.; Overkamp, K.M.; van Maris, A.J.; Kötter, P.; Luttik, M.A.; van Dijken, J.P.; Pronk, J.T. Stoichiometry and compartmentation of NADH metabolism in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2001, 25, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Overkamp, K.M.; Bakker, B.M.; Kötter, P.; van Tuijl, A.; de Vries, S.; van Dijken, J.P.; Pronk, J.T. In vivo analysis of the mechanisms for oxidation of cytosolic NADH by Saccharomyces cerevisiae mitochondria. J. Bacteriol. 2000, 182, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Aon, M.A.; Winslow, R.L.; O’Rourke, B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys. J. 2004, 87, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Xu, R.H.; Du, M.; Feng, L.; Sasaki, R.; Carew, J.S.; Hu, Y.; Ramdas, L.; Hu, L.; Keating, M.J.; et al. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanizm. J. Cell. Biol. 2006, 175, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Heikal, A.A. Two-photon autofluorescence dynamics imaging reveals sensitivity of intracellular NADH concentration and conformation to cell physiology at the single-cell level. J. Photochem. Photobio. B Biol. 2009, 95, 46–57. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Bose, C.K. Debating cancer: The paradox in cancer research. Indian J. Med. Res. 2017, 146, 435–436. [Google Scholar]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Crabtree, H.G. Observations on the carbohydrate metabolism of tumours. Biochem. J. 1929, 23, 536–545. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Shayoub, M.E.; Muddathir, A.K.; Elhassan, G.O.; Bashir, A.H. Evolution of Tumor Metabolism might Reflect Carcinogenesis as a Reverse Evolution process (Dismantling of Multicellularity). Cancers 2011, 3, 3002–3017. [Google Scholar] [CrossRef]
- Diaz-Ruiz, R.; Uribe-Carvajal, S.; Devin, A.; Rigoulet, M. Tumor cell energy metabolism and its common features with yeast metabolism. Biochim. Biophys. Acta 2009, 1796, 252–265. [Google Scholar] [CrossRef]
- Boese, A.C.; Kang, S. Mitochondrial metabolism-mediated redox regulation in cancer progression. Redox Biol. 2021, 42, 101870. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Ogasawara, M.A.; Huang, P. Models of reactive oxygen species in cancer. Drug Discov. Today Dis. Models 2007, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Chandel, N.S. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol. Life Sci. 2009, 66, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Zhu, D.; Huang, P. Targeting cancer cell mitochondria as a therapeutic approach. Future Med. Chem. 2013, 5, 53–67. [Google Scholar] [CrossRef]
- Yang, Y.; Karakhanova, S.; Werner, J.; Bazhin, A.V. Reactive oxygen species in cancer biology and anticancer therapy. Curr. Med. Chem. 2013, 20, 3677–3692. [Google Scholar] [CrossRef]
- Aktipis, C.A.; Nesse, R.M. Evolutionary foundations for cancer biology. Evol. Appl. 2013, 6, 144–159. [Google Scholar] [CrossRef]
- Thomas, F.; Ujvari, B.; Renaud, F.; Vincent, M. Cancer adaptations: Atavism, de novo selection, or something in between? BioEssays 2017, 39, 1700039. [Google Scholar] [CrossRef]
- Mazzocca, A. The Systemic–Evolutionary Theory of the Origin of Cancer (SETOC): A New Interpretative Model of Cancer as a Complex Biological System. Int. J. Mol. Sci. 2019, 20, 4885. [Google Scholar] [CrossRef]
- Kováč, L. Lamarck and Darwin revisited. EMBO Rep. 2019, 20, e47922. [Google Scholar] [CrossRef]
- Lineweaver, C.H.; Davies, P.C.W. Comparison of the Atavistic Model of Cancer to Somatic Mutation Theory: Phylostratigraphic Analyses Support the Atavistic Model. In The Physics of Cancer Research Advances; Gerstman, B.S., Ed.; World Scientific: Singapore, 2020; Volume 12, pp. 243–261. [Google Scholar]
- Cornish-Bowden, A. Lynn Margulis and the origin of the eukaryotes. J. Theor. Biol. 2017, 434, 1. [Google Scholar] [CrossRef]
- Grosberg, R.K.; Strathmann, R.R. The Evolution of Multicellularity: A Minor Major Transition? Annu. Rev. Ecol. Evol. Syst. 2007, 38, 621–654. [Google Scholar] [CrossRef]
- Nedelcu, A.M. The evolution of multicellularity and cancer: Views and paradigms. Biochem. Soc. Trans. 2020, 48, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. How the evolution of multicellularity set the stage for cancer. Br. J. Cancer 2018, 118, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim. Biophys. Acta 2017, 1867, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, UK, 2000. [Google Scholar]
- Huang, S.; Ernberg, I.; Kaufman, S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 2009, 20, 869–876. [Google Scholar] [CrossRef]
- Kaufman, S. Diferentiation of malignant to benign cells. J. Theor. Biol. 1971, 31, 429–451. [Google Scholar] [CrossRef]
- Vinogradov, A.E.; Anatskaya, O.V. Evolutionary framework of the human interactome: Unicellular and multicellular giant clusters. Biosystems 2019, 181, 82–87. [Google Scholar] [CrossRef]
- Giuliani, A. Models portability: Some considerations about transdisciplinary approaches, Studies of Nonlinear Phenomena in Life Science. In The Complex Matters of the Mind; World Scientific: Singapore, 1998; pp. 193–210. [Google Scholar] [CrossRef]
- Lewin, R. Complexity: Life at the Edge of Chaos; Maxwell Macmillan International: New York, NY, USA, 1993; ISBN 1857990285. [Google Scholar]
- Krigerts, J.; Salmina, K.; Freivalds, T.; Zayakin, P.; Rumnieks, F.; Inashkina, I.; Giuliani, A.; Hausmann, M.; Erenpreisa, J. Differentiating cancer cells reveal early large-scale genome regulation by pericentric domains. Biophys. J. 2021, 120, 711–724. [Google Scholar] [CrossRef]
- Zimatore, G.; Tsuchiya, M.; Hashimoto, M.; Kasperski, A.; Giuliani, A. Self-organization of whole-gene expression through coordinated chromatin structural transition. Biophys. Rev. 2021, 2, 031303. [Google Scholar] [CrossRef]
- Zimatore, G.; Tsuchiya, M.; Hashimoto, M.; Kasperski, A.; Giuliani, A. Self-organization of whole gene expression through coordinated chromatin structural transition: Validation of self-organized critical control of genome expression. bioRxiv 2019. bioRxiv: 852681. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Stevens, J.; Horne, S.; Abdallah, B.; Ye, K.; Bremer, S.; Ye, C.; Chen, D.J.; Heng, H. Genome chaos: Survival strategy during crisis. Cell Cycle 2014, 13, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Heng, H.H. Two-phased evolution: Genome chaos-mediated information creation and maintenance. Prog. Biophys. Mol. Biol. 2021, 165, 29–42. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2020; in press. [Google Scholar] [CrossRef]
- Heng, H.H. Genome Chaos: Rethinking Genetics, Evolution, and Molecular Medicine; Academic Press Elsevier: Cambridge, MA, USA, 2019; ISBN 978-012-8136-35-5. [Google Scholar]
- Ye, C.J.; Sharpe, Z.; Alemara, S.; Mackenzie, S.; Liu, G.; Abdallah, B.; Horne, S.; Regan, S.; Heng, H.H. Micronuclei and Genome Chaos: Changing the System Inheritance. Genes 2019, 10, 366. [Google Scholar] [CrossRef] [PubMed]
- Pavlopoulou, A.; Spandidos, D.A.; Michalopoulos, I. Human cancer databases (review). Oncol. Rep. 2015, 33, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome Doubling Shapes the Evolution and Prognosis of Advanced Cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef]
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with Purpose. Genes Dev. 2009, 23, 2461–2477. [Google Scholar] [CrossRef]
- Pienta, K.J.; Hammarlund, E.U.; Axelrod, R.; Brown, J.S.; Amend, S.R. Poly-Aneuploid Cancer Cells Promote Evolvability, Generating Lethal. Cancer Evol. Appl. 2020, 13, 1626–1634. [Google Scholar] [CrossRef]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The Evolutionary Significance of Polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Karyotype coding: The creation and maintenance of system information for complexity and biodiversity. Biosystems 2021, 208, 104476. [Google Scholar] [CrossRef] [PubMed]
- Rancati, G.; Pavelka, N.; Fleharty, B.; Noll, A.; Trimble, R.; Walton, K.; Perera, A.; Staehling-Hampton, K.; Seidel, C.W.; Li, R. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved. Cell 2008, 135, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.; Rasnick, D. Aneuploidy, the somatic mutation that makes cancer a species of its own. Cell Motil. Cytoskelet. 2000, 47, 81–107. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.H. The Metabolism of Tumours: Investigations from the Kaiser Wilhelm Institute for Biology; Arnold Constable: Berlin, Germany; Dahlem, Germany; London, UK, 1930. [Google Scholar]
- Larson, S.M. Positron emission tomography-based molecular imaging in human cancer: Exploring the link between hypoxia and accelerated glucose metabolism. Clin. Cancer Res. 2004, 10, 2203–2204. [Google Scholar] [CrossRef][Green Version]
- Szigeti, G.P.; Szasz, O.; Hegyi, G. Connections between Warburg’s and Szentgyorgyi’s Approach about the Causes of Cancer. J. Neoplasm 2017, 1, 1–13. [Google Scholar]
- Blagosklonny, M.V. Molecular theory of cancer. Cancer Biol. Ther. 2005, 4, 621–627. [Google Scholar] [CrossRef]
- Shih, I.M. Gestational trophoblastic neoplasia—Pathogenesis and potential therapeutic targets. Lancet Oncol. 2007, 8, 642–650. [Google Scholar] [CrossRef]
- Shomar, A.; Barak, O.; Brenner, N. Cancer progression as a learning process. Iscience 2022, 25, 103924. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Malaise, E.P.; Chavaudra, N.; Tubiana, M. The relationship between growth rate, labelling index and histological type of human solid tumours. Eur. J. Cancer 1973, 9, 305–312. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef]
- Gonzalez, M.J.; Seyfried, T.; Nicolson, G.L.; Barclay, B.J.; Matta, J.; Vasquez, A.; D’Agostino, D.; Olalde, J.; Duconge, J.; Hunninghake, R.; et al. Mitochondrial Correction: A New Therapeutic Paradigm for Cancer and Degenerative Diseases. J. Orthomol. Med. 2018, 33, 4. [Google Scholar]
- Seilern-Aspang, F.; Kratochwil, K. Induction and differentiation of an epithelial tumour in the newt (Triturus cristatus). J. Embryol. Exp. Morphol. 1962, 10, 337–356. [Google Scholar] [CrossRef]
- Bizzarri, M.; Giuliani, A.; Cucina, A. Minini, M. Redifferentiation therapeutic strategies in cancer. Drug Discov. Today 2020, 25, 731–738. [Google Scholar] [CrossRef]
- Proietti, S.; Cucina, A.; Pensotti, A.; Fuso, A.; Marchese, C.; Nicolini, A.; Bizzarri, M. Tumor reversion and embryo morphogenetic factors. Semin. Cancer Biol. 2020, 79, 83–90. [Google Scholar] [CrossRef]
- Li, L.; Connelly, M.C.; Wetmore, C.; Curran, T.; Morgan, J.I. Mouse embryos cloned from brain tumors. Cancer Res. 2003, 63, 2733–2736. [Google Scholar]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Kasemeier-Kulesa, J.; Kulesa, P.M.; Postovit, L.M. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat. Rev. Cancer 2007, 7, 246–255. [Google Scholar] [CrossRef]
- Joel, M.; Sandberg, C.J.; Boulland, J.L.; Vik-Mo, E.O.; Langmoen, I.A.; Glover, J.C. Inhibition of tumor formation and redirected differentiation of glioblastoma cells in a xenotypic embryonic environment. Dev. Dyn. 2013, 242, 1078–1093. [Google Scholar] [CrossRef] [PubMed]
- Proietti, S.; Cucina, A.; Catizone, A.; Ricci, G.; Pensotti, A.; Bizzarri, M. Zebrafish embryo extracts enhance 5-FU anti-cancer effects upon breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 3235–3245. [Google Scholar] [PubMed]
- Allegrucci, C.; Rushton, M.D.; Dixon, J.E.; Sottile, V.; Shah, M.; Kumari, R.; Watson, S.; Alberio, R.; Johnson, A.D. Epigenetic reprogramming of breast cancer cells with oocyte extracts. Mol. Cancer 2011, 10, 7. [Google Scholar] [CrossRef]
- Bizzarri, M.; Cucina, A.M.; Biava, P.; Proietti, S.; D’Anselmi, F.; Dinicola, S.; Pasqualato, A.; Lisi, E. Embryonic morphogenetic field induces phenotypic reversion in cancer cells. Curr. Pharm. Biotechnol. 2011, 12, 243–253. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasperski, A. Life Entrapped in a Network of Atavistic Attractors: How to Find a Rescue. Int. J. Mol. Sci. 2022, 23, 4017. https://doi.org/10.3390/ijms23074017
Kasperski A. Life Entrapped in a Network of Atavistic Attractors: How to Find a Rescue. International Journal of Molecular Sciences. 2022; 23(7):4017. https://doi.org/10.3390/ijms23074017
Chicago/Turabian StyleKasperski, Andrzej. 2022. "Life Entrapped in a Network of Atavistic Attractors: How to Find a Rescue" International Journal of Molecular Sciences 23, no. 7: 4017. https://doi.org/10.3390/ijms23074017
APA StyleKasperski, A. (2022). Life Entrapped in a Network of Atavistic Attractors: How to Find a Rescue. International Journal of Molecular Sciences, 23(7), 4017. https://doi.org/10.3390/ijms23074017