
The Combination of Conventional QTL Analysis, Bulked-Segregant Analysis, and RNA-Sequencing Provide New Genetic Insights into Maize Mesocotyl Elongation under Multiple Deep-Seeding Environments


Abstract
:1. Introduction
2. Results
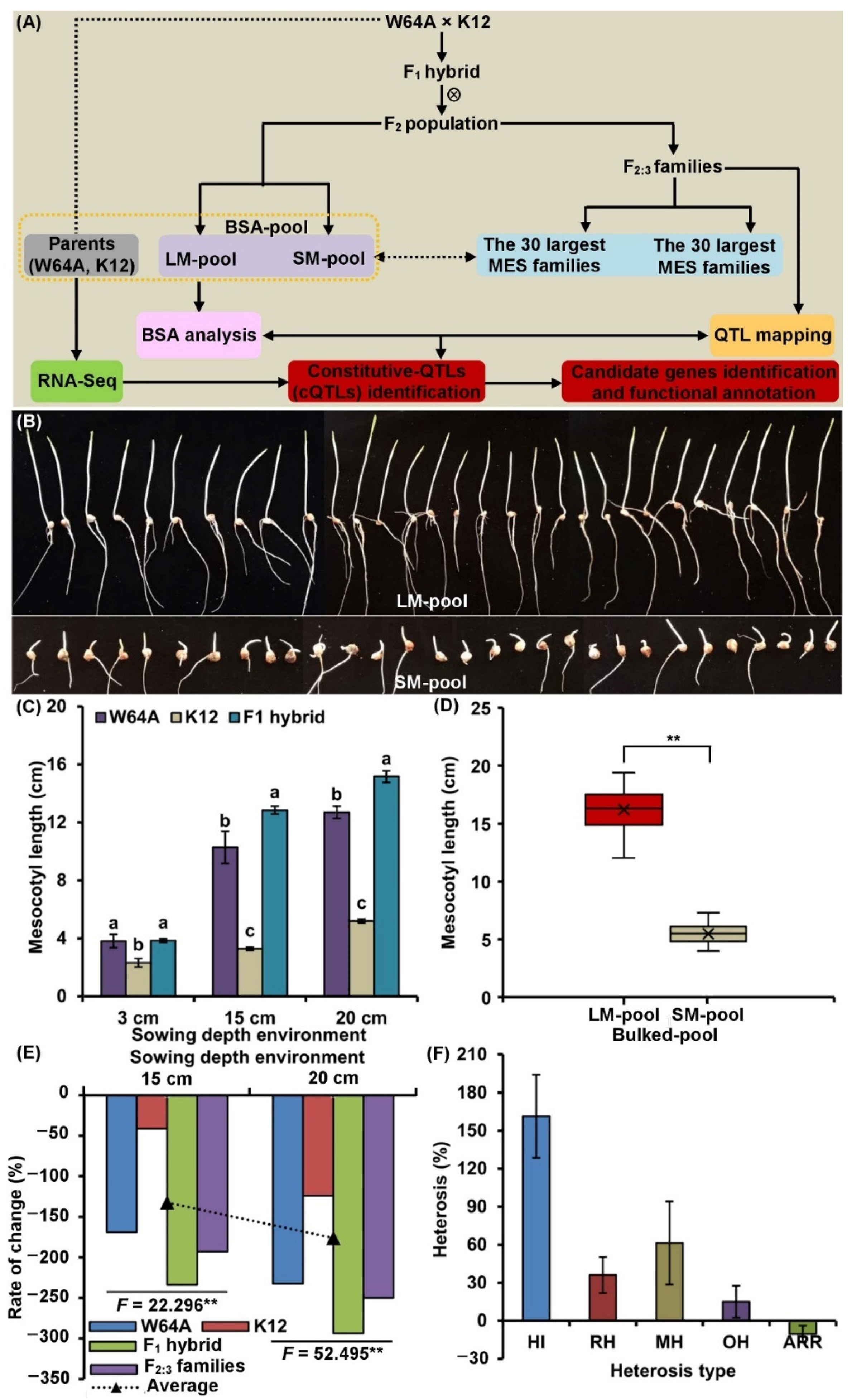
2.1. MES Variation and Genetic Analysis
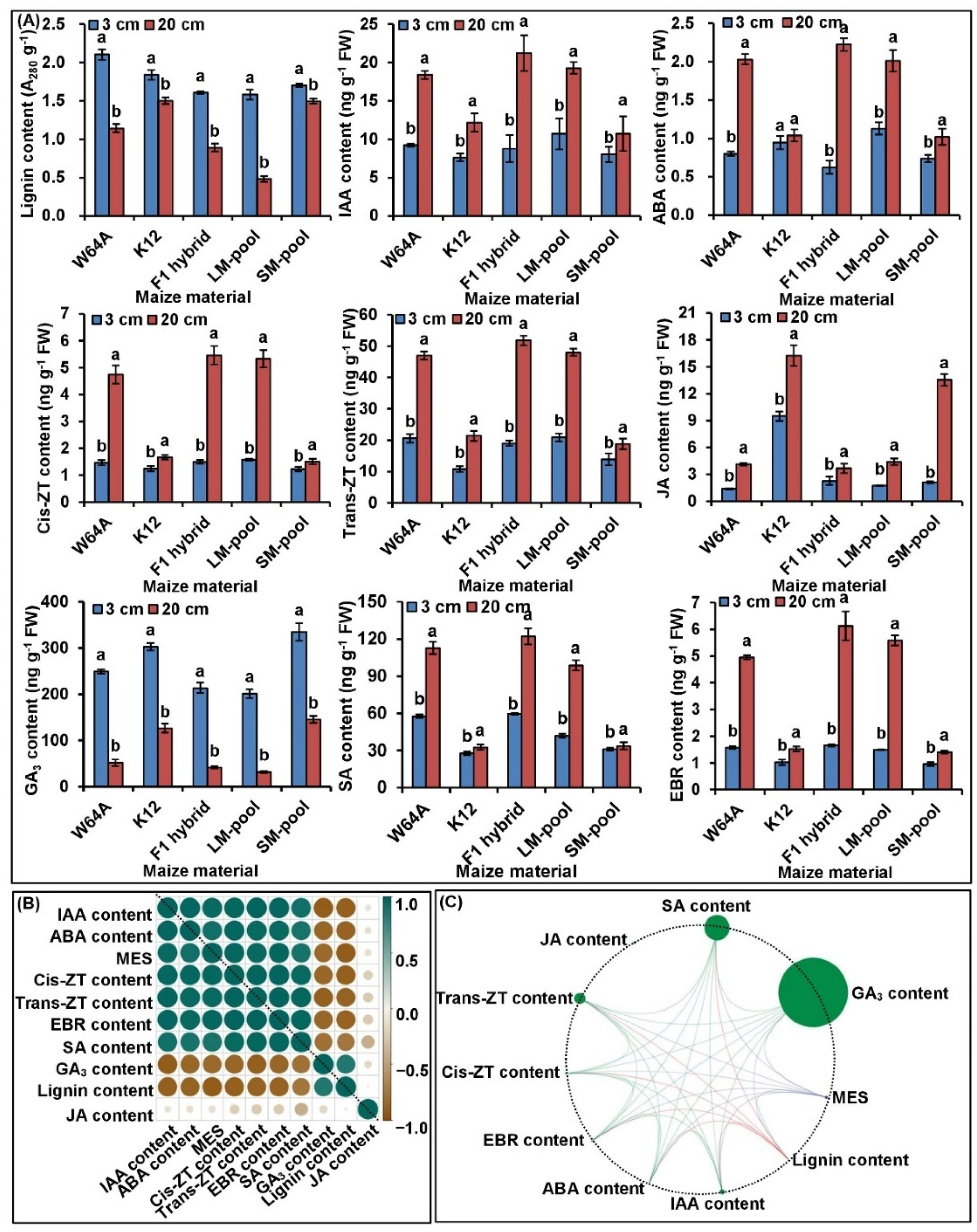
2.2. Framework of Phenotype and Physiological Metabolisms of MES
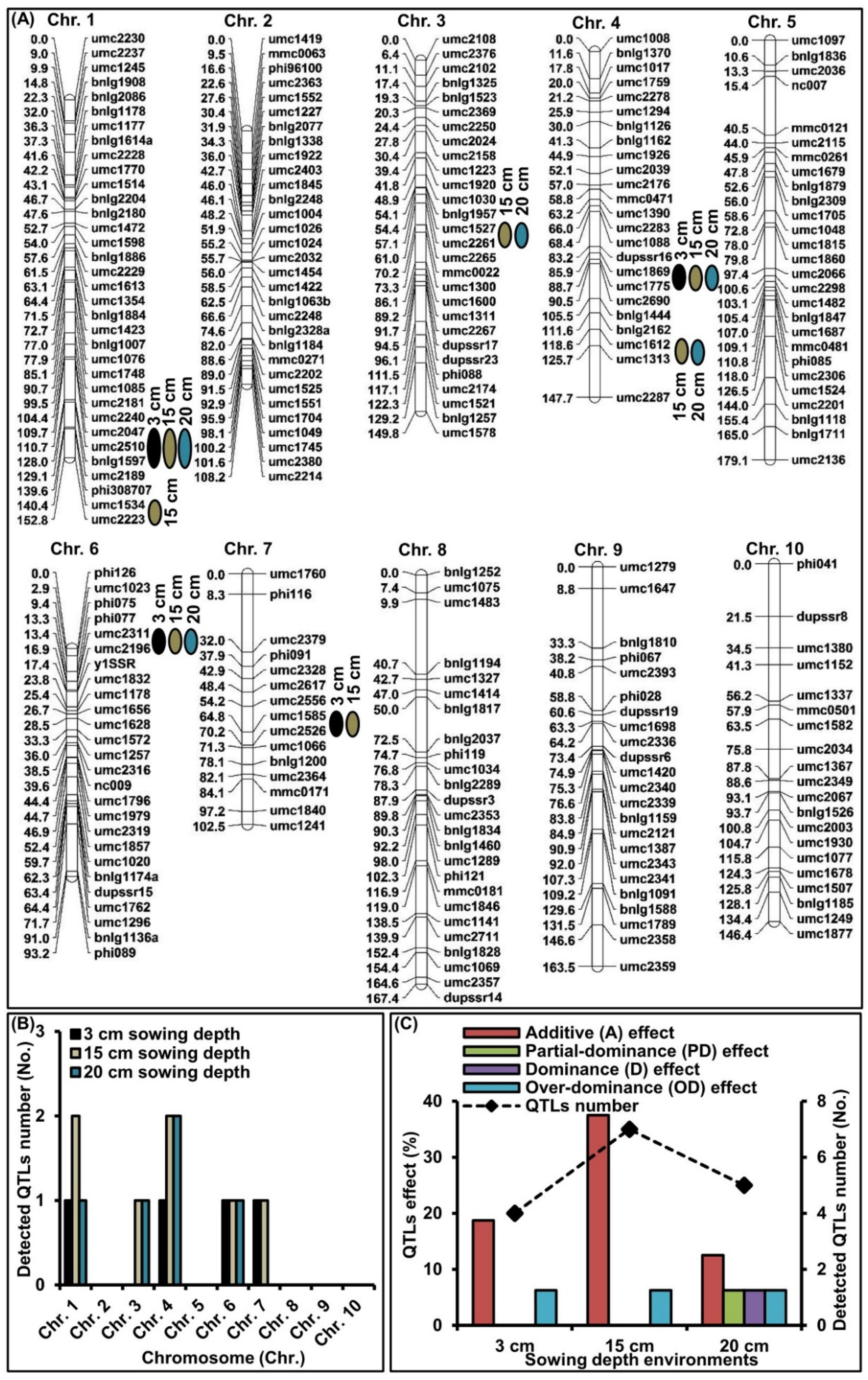
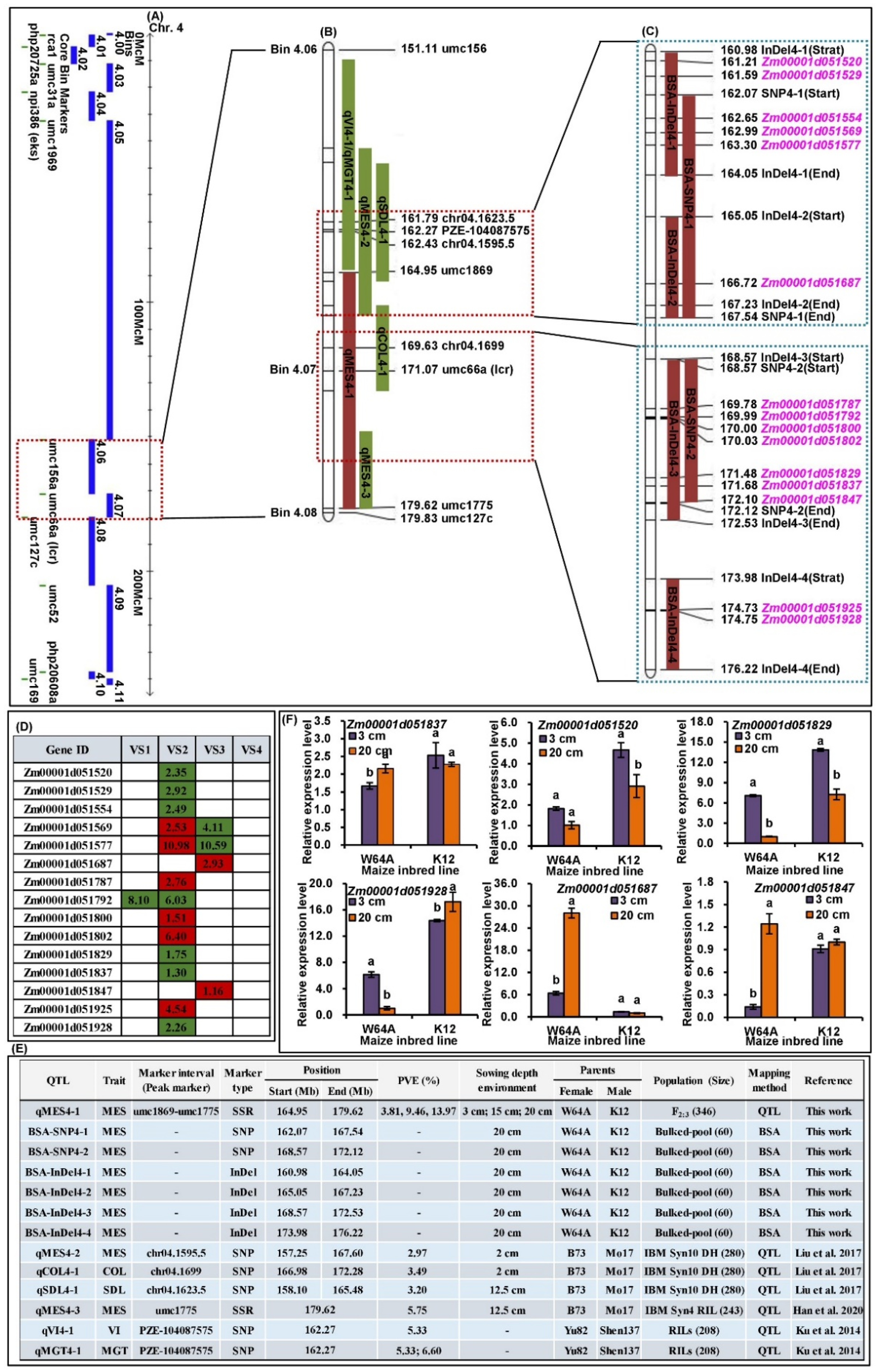
2.3. QTLs Analysis of MES across Three Sowing Conditions
2.4. BSA of MES under 20 cm Sowing Conditions
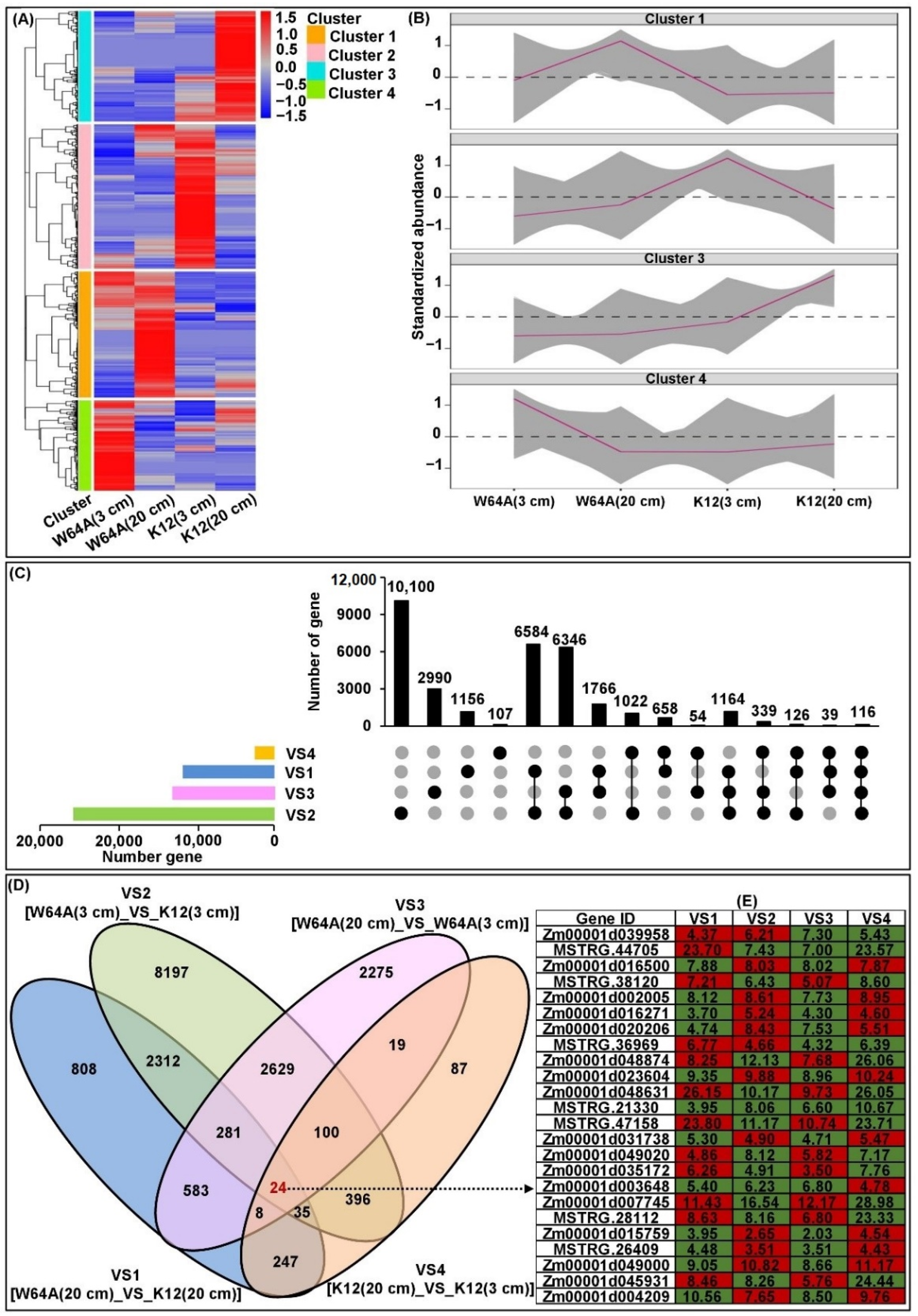
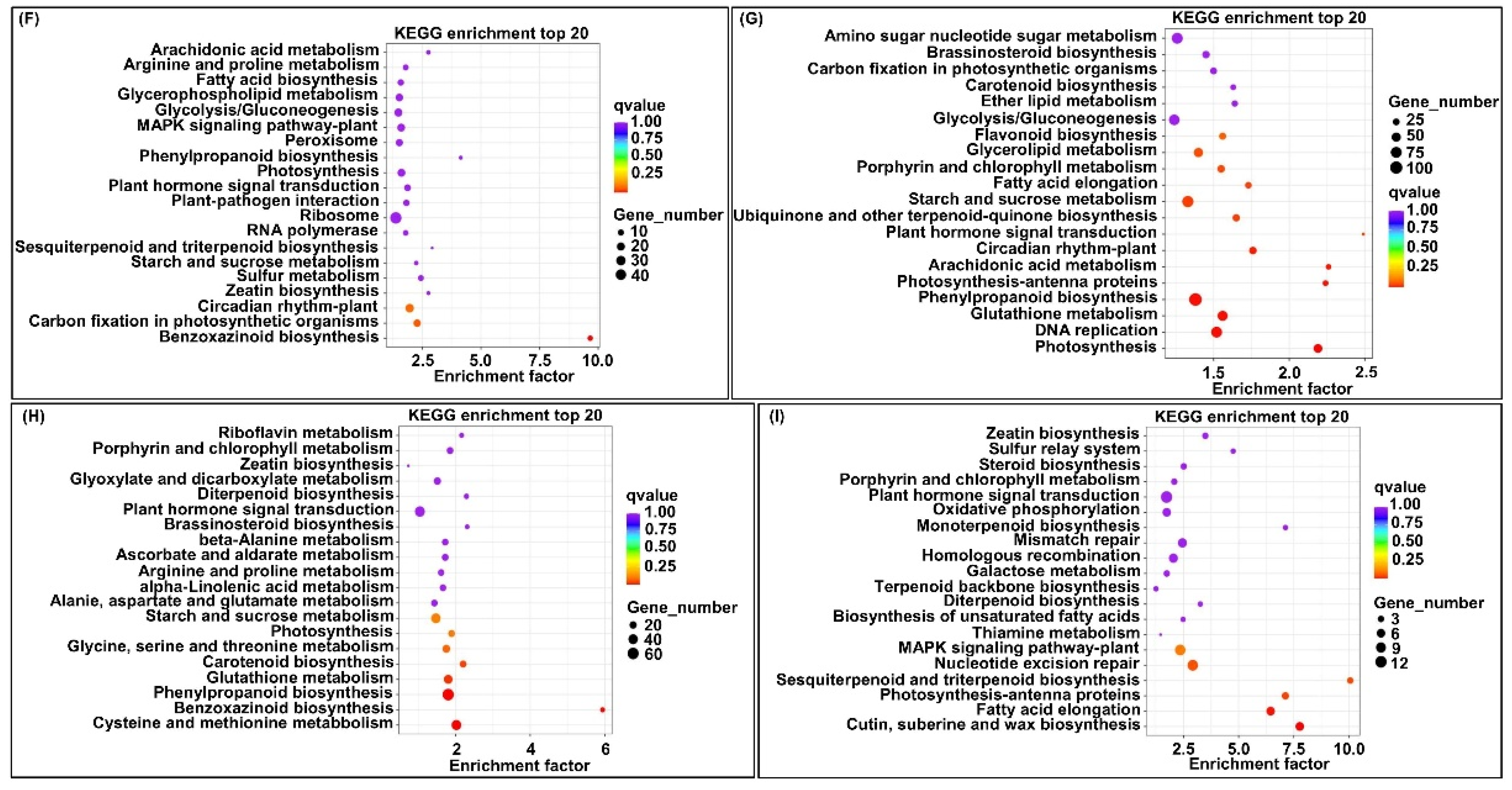
2.5. Transcriptome Analysis of Two Parent Mesocotyls
2.6. cQMES4 Identification and Candidate Genes Analysis in cQMES4
2.7. qRT-PCR Analysis of Six Candidate Genes in cQMES4
3. Discussion and Conclusions
3.1. The Relationship of Mesocotyls, Abiotic Stress, and Agronomic Traits
3.2. Genetic Dissection of MES in Maize and the Identification of cQTLs via Different Methods
3.3. Validation of Candidate Genes in the cQMES4 Region and Their Functions Analysis
4. Materials and Methods
4.1. Development of Maize Populations
4.2. Phenotypic and Physiological Evaluation of Mesocotyls
4.3. SSRs Analysis, Genetic Linkage Map Contruction, and QTL Analysis
4.4. BSA-Sequencing and Sequence Alignment
4.5. RNA-Seq and DEGs Analysis
4.6. cQTL Detection and the Dissection of Candidate Genes
4.7. qRT-PCR Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| BSA | bulked-segregant analysis |
| CIM | composite interval mapping |
| DEGs | Differentially expressed genes |
| InDel | insertion-deletion |
| MES | mesocotyl length |
| MAS | marker-assisted selection |
| qRT-PCR | Quantitative real-time PCR |
| QTL | quantitative trait locus |
| RNA-Seq | RNA-Sequencing |
| SNP | single-nucleotide polymorphism |
| SSRs | simple sequence repeats |
References
- Rodríguez, M.N.S.; Cassab, G.I. Primary Root and Mesocotyl Elongation in Maize Seedlings: Two organs with antagonistic growth below the soil surface. Plants 2021, 10, 1274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Q.; Zhong, Y.; Zhou, W.Q. Molecular mechanism of mesocotyl elongation induced by brassinosteroid in maize under deep-seeding stress by RNA-sequencing, microstructure observation, and physiological metabolism. Genomics 2021, 113, 3565–3581. [Google Scholar] [CrossRef]
- Zhao, X.Q.; Zhang, J.W.; Fang, P.; Peng, Y.L. Comparative QTL analysis for yield components and morphological traits in maize (Zea mays L.) under water-stressed and well-watered conditions. Breed. Sci. 2019, 69, 621–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.Q.; Zhong, Y. 24-Epibrassinolide mediated interaction among antioxidant defense, lignin metabolism, and phytohormones signaling promoted better cooperative elongation of maize mesocotyl and coleoptile under deep-seeding stress. Russ. J. Plant Physiol. 2021, 68, 1194–1207. [Google Scholar] [CrossRef]
- Liu, H.J.; Zhang, L.; Wang, J.C.; Li, C.S.; Zeng, X.; Xie, S.P.; Zhang, Y.Z.; Liu, S.S.; Hu, S.L.; Wang, J.H.; et al. Quantitative trait locus analysis for deep-sowing germination ability in the maize IBM Syn10 DH population. Front. Plant Sci. 2017, 8, 813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.W.; Ma, P.; Zhao, Z.N.; Zhao, G.W.; Tian, B.H.; Wang, J.H.; Wang, G.Y. Mapping QTLcontrolling maize deep-seeding tolerance-related traits and confirmation of a major QTL for mesocotyl length. Theor. Appl. Genet. 2012, 124, 223–232. [Google Scholar] [CrossRef]
- Troyer, A.F. The location of genes governing long first internode of corn. Genetics 1997, 145, 1149–1154. [Google Scholar] [CrossRef]
- Blackburn, A.; Sidhu, G.; Schillinger, W.F.; Skinner, D.; Gill, K. QTL mapping using GBS and SSR genotyping reveals genomic regions controlling wheat coleoptile length and seedling emergence. Euphytica 2021, 217, 45. [Google Scholar] [CrossRef]
- Singh, K.; Shukla, S.; Kadam, S.; Semwal, V.K.; Singh, N.K.; Khanna-Chopra, R. Genomic regions and underlying candidate genes associated with coleoptile length under deep-sowing conditions in a wheat RIL population. J. Plant Biochem. Biotechnol. 2015, 24, 324–330. [Google Scholar] [CrossRef]
- Paynter, B.H.; Clarke, G.P.Y. Coleoptile length of barely (Hordeum vulgare L.) cultivars. Genet. Resour. Crop Evol. 2010, 57, 395–403. [Google Scholar] [CrossRef]
- Takahashi, H.; Sato, K.; Takeda, K. Mapping genes for deep-seeding tolerance in barely. Euphytica 2001, 122, 37–43. [Google Scholar] [CrossRef]
- Zhan, J.H.; Lu, X.; Liu, H.Y.; Zhao, Q.Z.; Ye, G.Y. Mesocotyl elongation, an essential trait for dry-seeded rice (Oryza sativa L.): A review of physiological and genetic basis. Planta 2020, 251, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.S.; Sasaki, K.; Kang, J.W.; Sato, T.; Song, W.Y.; Ahn, S.N. Mesocotyl elongation is essential for seedling emergence under deep-seeding condition in rice. Rice 2017, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Q.; Zhong, Y.; Shi, J.; Zhou, W.Q. 24-Epibrassinolide confers tolerance against deep-seeding stress in Zea mays L. coleoptile development by phytohormones signaling transduction and their interaction network. Plant Signal. Behav. 2021, 16, 1963583. [Google Scholar] [CrossRef] [PubMed]
- Dubois, P.G.; Olsefski, G.T.; Flint-Garcia, S. Physiological and genetic characterization of end-of-day far-red light response in maize seedlings. Plant Physisol. 2010, 154, 173–186. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.Y.; Zhan, J.H.; Li, J.L.; Lu, X.; Liu, J.D.; Wang, Y.M.; Zhao, Q.Z.; Ye, G.Y. Genome-wide association study (GWAS) for mesocotyl elongation in rice (Oryza sativa L.) under multiple culture conditions. Genes 2019, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.H.; Shen, Y.; Lv, L.; Lee, M.; Lübberstedt, T.; Zhao, G.W. QTL analysis of deep-sowing tolerance during seed germination in the maize IBM Syn4 RIL population. Plant Breed. 2020, 139, 1125–1134. [Google Scholar] [CrossRef]
- Zhao, G.W.; Wang, J.H. Effect of gibberellin and uniconazole on mesocotyl elongation of dark-grown maize under different seeding depths. Plant Prod. Sci. 2008, 11, 423–429. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, J.Y.; Li, L.; Ma, Y.W.; Weng, Z.P.; Duan, Y.P.; Hao, Z.F. Genome-wide association analysis of deep-planting related traits in maize. Crops 2014, 2, 43–47. [Google Scholar]
- Li, C.L.; Ling, F.L.; Su, G.H.; Sun, W.F.; Liu, H.H.; Su, Y.C.; Qi, X. Location and mapping of the NCLB resistance genes in maize by bulked segregant analysis (BSA) using whole genome re-sequencing. Mol. Breed. 2020, 40, 92–103. [Google Scholar] [CrossRef]
- Kamara, N.; Jiao, Y.M.; Lu, Z.J.; Aloryi, K.D.; Wu, J.W.; Liu, X.D.; Shahid, M.Q. Cytological observations and bulked-segregant analysis coupled global genome sequencing reveal two genes associated with pollen fertility in tetraploid rice. Int. J. Mol. Sci. 2020, 22, 841. [Google Scholar] [CrossRef] [PubMed]
- Xin, F.; Zhu, T.; Wei, S.W.; Han, Y.C.; Zhao, Y.; Zhang, D.Z.; Ma, L.J.; Ding, Q. QTL mapping of kernel traits and validation of a major QTL for kernel length-width ratio using SNP and bulked segregant analysis in wheat. Sci. Rep. 2020, 10, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.J.; Hao, R.Q.; Wu, X.L.; Wang, W. Maize mesocotyl: Role in response to stress and deep-sowing tolerance. Plant Breed. 2020, 139, 466–473. [Google Scholar] [CrossRef]
- Yang, Z.J.; Yan, L.; Song, M.F.; Su, L.; Meng, F.H.; Li, H.D.; Bai, J.R.; Guo, L.; Yang, J.P. Transcription characteristics of ZmPHYA1 and ZmPHYAA under different light treatments in maize. Acta Agron. Sin. 2016, 42, 1462–1470. [Google Scholar] [CrossRef]
- Schopfer, P.; Lapierre, C.; Nolte, T. Light-controlled growth of the maize seedling mesocotyl: Mechanical cell-wall changes in the elongation zone and related changes in lignification. Physiol. Plant 2001, 111, 83. [Google Scholar] [CrossRef]
- Gao, C.; Jin, H.; Sheng, Z.; Zheng, Y.; Knapp, A. Association of polyamines in governing the chilling sensitivity of maize genotypes. Plant Growth Regul. 2009, 57, 31–38. [Google Scholar] [CrossRef]
- Takahashi, N. Adaptive importance of mesocotyl and coleoptile growth in rice under different moisture regions. Aust. J. Plant Physiol. 1978, 5, 511–517. [Google Scholar]
- Mgonja, M.A.; Ladeinde, T.A.O.; Aken-Ova, M.E. Genetic analysis of mesocotyl length and its relationship with other agronomic characters in rice (Oryza sativa L.). Euphytica 1993, 72, 189–195. [Google Scholar] [CrossRef]
- Dilday, R.H.; Mgonja, M.A.; Amonsilpa, S.A.; Collins, F.C.; Wells, B.R. Plant height vs. mesocotyl and coleoptile elongation in rice: Linkage or pleiotropism? Crop Sci. 1990, 30, 815–818. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhao, X.Q.; Li, W.; Zhang, D.; Zhou, W.Q. Heterosis and genetic effects analysis of deep-seeding traits in maize under different sowing environments. J. Nuclear Agric. Sci. 2021, 35, 556–566. [Google Scholar]
- Ku, L.X.; Cui, X.J.; Cheng, F.F.; Guo, S.L.; Qi, J.S.; Tian, Z.Q.; Han, T.; Ren, Z.Z.; Zhang, L.K.; Su, H.H.; et al. Genetic dissection of seed vigor under artificial ageing conditions using two joined maize recombinant inbred line populations. Plant Breed. 2014, 133, 728–737. [Google Scholar] [CrossRef]
- Kircher, S.; Terecskei, K.; Wolf, I.; Sipos, M.; Adam, E. Phytochrome A-specific signaling in Arabidopsis thaliana. Plant Signal. Behav. 2011, 6, 1714–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoopes, G.M.; Hamilton, J.P.; Wood, J.C.; Esteban, E.; Pasha, A.; Vaillancourt, B.; Provart, N.J.; Buell, C.R. An updated gene atlas for maize reveals organ-specific and stress-induced genes. Plant J. 2019, 97, 1154–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zheng, L.; He, F.; Zhao, F.J.; Shen, Z.; Zheng, L. Transcriptional and physiological analyses identify a regulatory role for hydrogen peroxide in the lignin biosynthesis of copper-stressed rice roots. Plant Soil 2015, 387, 323–336. [Google Scholar] [CrossRef]
- Spartz, A.K.; Lee, S.H.; Wenger, J.P.; Gonzalez, N.; Itoh, H.; Inz, D.; Peer, W.A.; Murphy, A.S.; Overvoorde, P.J.; Gray, W.M. The SAUR19 subfamily of SMALL AUXIN UP RNA genes promote cell expansion. Plant J. 2012, 70, 978–990. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.G.; Qu, Z.S.; Zhang, L.J.; Zhao, S.J.; Bi, Z.H.; Ji, X.H.; Wang, X.W.; Wei, H.R. Overexpression of poplar xylem sucrose synthase in tobacco leads to thicked cell wall and increased height. PLoS ONE 2015, 10, e0120669. [Google Scholar]
- Du, L.G.; Jiang, H.Y.; Zhao, G.W.; Ren, J.Y. Gene cloning of ZmMYB59 transcription factor in maize and its expression during seed germination in response to deep-sowing and exogenous hormones. Plant Breed. 2017, 136, 834–844. [Google Scholar] [CrossRef]
- Zhao, X.Q.; Zhong, Y. Genetic dissection of the photosynthetic parameters of maize (Zea mays L.) in drought-stressed and well-watered environments. Russ. J. Plant Physiol. 2021, 68, 1125–1134. [Google Scholar] [CrossRef]
- Zhao, X.Q.; Ren, B.; Peng, Y.L.; Xu, M.X.; Fang, P.; Zhuang, Z.L.; Zhang, J.W.; Zeng, W.J.; Gao, Q.H.; Ding, Y.F.; et al. Epistatic and QTL ×environment interaction effects for ear related traits in two maize (Zea mays) populations under eight watering environments. Acta Agronomic Sinica 2019, 45, 856–871. [Google Scholar]
- Zhao, X.Q.; Peng, Y.L.; Zhang, J.W.; Fang, P.; Wu, B.Y. Identification of QTLs and meta-QTLs for seven agronomic traits in multiple maize populations under well-watered and water-stressed conditions. Crop Sci. 2018, 58, 507–520. [Google Scholar] [CrossRef]
- Stuber, C.W.; Edwards, M.D.; Wendel, J. Molecular marker-facilitated investigations of quantitative trait loci in maize. II. Factors influencing yield and its component traits. Crop Sci. 1987, 27, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Ren, R.; Xu, J.; Zhang, M.; Liu, G.; Yao, X.; Zhu, L.; Hou, Q. Identification and molecular mapping of a gummay stem blight resistance gene in wild watermelon (Citrullus amarus) germplasm PI 189225. Plant Dis. 2020, 104, 16–24. [Google Scholar] [CrossRef]
- Begali, H. A pipeline for markers selection using restriction site associated DNA sequencing. J. Appl. Bioinf. Comput. Biol. 2018, 6, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Bguyen, T.; Wang, L.; Land, S.J.; Lu, X.Y.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, snpEFF: SNPs in the genome of drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosnowski, O.; Chaarcosset, A.; Joets, J. BioMercator V3: An upgrade of genetic map compuilation and quantitative trait loci meta-analysis algorithms. Bioinformatics 2012, 28, 2082–2083. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QTL | Bin | Sowing Depth Environment | QTL Position | LOD | Additive (a) | Dominance (d) | Gene Action | PVE (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| cM | Marker Interval | |d/a| | Type | |||||||
| qMES1-1 | 1.09 | 3 cm | 110.9 | umc2047-bnlg1597 | 3.10 | 0.25 | 0.03 | 0.12 | A | 2.89 |
| 15 cm | 110.8 | umc2047-bnlg1597 | 8.84 | −0.75 | −0.11 | 0.10 | A | 8.34 | ||
| 20 cm | 110.8 | umc2047-bnlg1597 | 10.43 | −0.91 | −0.15 | 0.16 | A | 10.01 | ||
| qMES1-2 | 1.10 | 15 cm | 145.2 | umc1534-umc2223 | 13.96 | −1.17 | 0.10 | 0.09 | A | 13.12 |
| qMES3-1 | 3.04 | 15 cm | 55.3 | umc1527-umc2261 | 7.10 | −1.88 | −0.21 | 0.11 | A | 11.60 |
| 20 cm | 56.1 | umc1527-umc2261 | 4.77 | −1.03 | −1.20 | 1.17 | D | 4.94 | ||
| qMES4-1 | 4.06–4.07 | 3 cm | 88.1 | umc1869-umc1775 | 4.08 | −0.34 | −0.02 | 0.06 | A | 3.81 |
| 15 cm | 88.0 | umc1869-umc1775 | 10.02 | −0.83 | −0.09 | 0.11 | A | 9.46 | ||
| 20 cm | 88.2 | umc1869-umc1775 | 14.84 | −1.25 | −0.18 | 0.14 | A | 13.97 | ||
| qMES4-2 | 4.05–4.08 | 15 cm | 119.1 | umc1612-umc1313 | 4.44 | −1.10 | 0.08 | 0.07 | A | 6.98 |
| 20 cm | 119.4 | umc1612-umc1313 | 4.61 | 0.17 | 0.10 | 0.59 | PD | 8.42 | ||
| qMES6-1 | 6.01 | 3 cm | 14.3 | umc2311-umc2196 | 9.30 | −1.01 | −1.51 | 1.50 | OD | 8.11 |
| 15 cm | 14.3 | umc2311-umc2196 | 5.81 | −0.61 | 0.97 | 1.59 | OD | 5.06 | ||
| 20 cm | 14.3 | umc2311-umc2196 | 11.45 | −1.14 | −1.43 | 1.25 | OD | 10.03 | ||
| qMES7-1 | 7.02 | 3 cm | 67.0 | umc1585-umc2526 | 3.08 | −0.80 | −0.03 | 0.04 | A | 4.93 |
| 15 cm | 66.9 | umc1585-umc2526 | 2.29 | −0.61 | 0.08 | 0.13 | A | 3.67 | ||
| Linked SNP-/InDel-Region | Chromosome | Start (bp) | End (bp) | Region Length (Mb) | Genes Number | Unique Genes Number |
|---|---|---|---|---|---|---|
| BSA-SNP1-1 | 1 | 243,149,664 | 251,888,006 | 8.74 | 169 | 169 |
| BSA-SNP4-1 | 4 | 162,067,875 | 167,544,471 | 5.48 | 131 | 329 |
| BSA-SNP4-2 | 4 | 168,572,722 | 172,116,409 | 3.54 | 82 | |
| BSA-InDel4-1 | 4 | 160,984,132 | 164,049,553 | 3.07 | 64 | |
| BSA-InDel4-2 | 4 | 165,049,976 | 167,225,404 | 2.18 | 65 | |
| BSA-InDel4-3 | 4 | 168,572,674 | 172,533,611 | 3.96 | 96 | |
| BSA-InDel4-4 | 4 | 173,979,835 | 176,222,964 | 2.24 | 74 |
| Gene ID (Encoded Protein) | Primer Sequence (5′ to 3′) |
|---|---|
| Zm00001d051837 (Sucrose synthase 6) | F: ATGGACCACAGGTATCATTTCTCA R: TGGCATTGTGAACGCATAGTG |
| Zm00001d051520 (MYB-related protein) | F: GCTGGAGAATTTGGAGAAGGAG R: CTGGAAGTAGGTGGAGACTGGG |
| Zm00001d051829 (U-box domain-containing protein 44) | F: TCCTCAAGTGGTTCAGGGAGT R: CAGGTCAGCTATGGTGGGTATG |
| Zm00001d051928 (Glutathione S-transferase family protein) | F: TTCACGACGCTCATCCGA R: CCTCAAGAGCGACTCCCTATC |
| Zm00001d051687 (Rho GDP-dissociation inhibitor 1) | F: GAGATGCTCGGCACGTTCA R: AGGCGTAGTTGATCTCCAGGTA |
| Zm00001d051847 (Nana2-like1) | F: GATGATGACGGGCGTGTATG R: CTTGAACAGGCGATGCGG |
| ZmActin1 (Actin 1) | F: CGATTGAGCATGGCATTGTCA R: CCCACTAGCGTACAACGAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.; Niu, Y. The Combination of Conventional QTL Analysis, Bulked-Segregant Analysis, and RNA-Sequencing Provide New Genetic Insights into Maize Mesocotyl Elongation under Multiple Deep-Seeding Environments. Int. J. Mol. Sci. 2022, 23, 4223. https://doi.org/10.3390/ijms23084223
Zhao X, Niu Y. The Combination of Conventional QTL Analysis, Bulked-Segregant Analysis, and RNA-Sequencing Provide New Genetic Insights into Maize Mesocotyl Elongation under Multiple Deep-Seeding Environments. International Journal of Molecular Sciences. 2022; 23(8):4223. https://doi.org/10.3390/ijms23084223
Chicago/Turabian StyleZhao, Xiaoqiang, and Yining Niu. 2022. "The Combination of Conventional QTL Analysis, Bulked-Segregant Analysis, and RNA-Sequencing Provide New Genetic Insights into Maize Mesocotyl Elongation under Multiple Deep-Seeding Environments" International Journal of Molecular Sciences 23, no. 8: 4223. https://doi.org/10.3390/ijms23084223
APA StyleZhao, X., & Niu, Y. (2022). The Combination of Conventional QTL Analysis, Bulked-Segregant Analysis, and RNA-Sequencing Provide New Genetic Insights into Maize Mesocotyl Elongation under Multiple Deep-Seeding Environments. International Journal of Molecular Sciences, 23(8), 4223. https://doi.org/10.3390/ijms23084223