
Integrated Degradome and Srna Sequencing Revealed miRNA-mRNA Regulatory Networks between the Phloem and Developing Xylem of Poplar


Abstract
:1. Introduction
2. Results
2.1. Construction of Small RNA Libraries and Sequencing
2.2. Identification of MiRNAs Involved in the Developing Xylem and Phloem
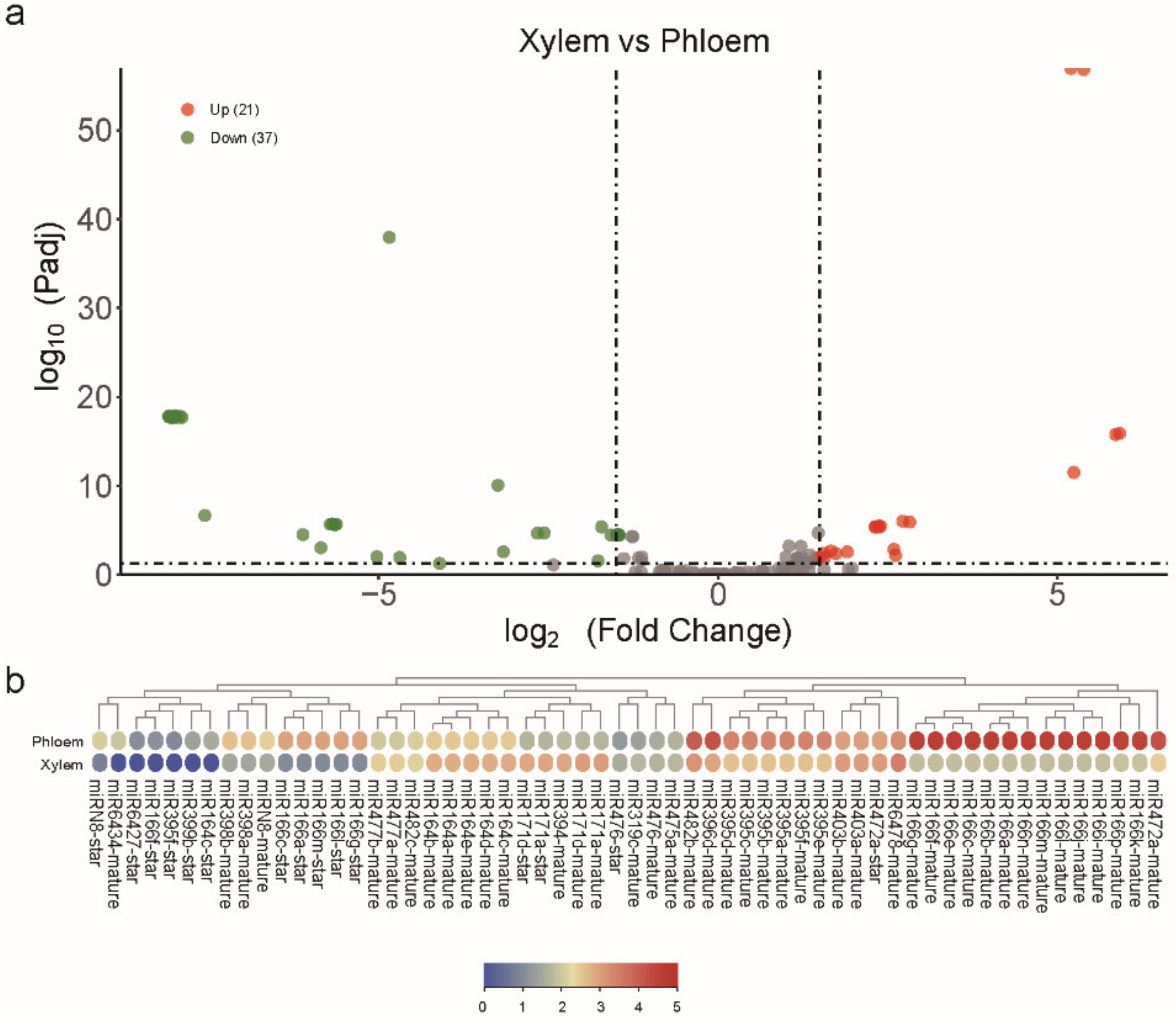
2.3. Identification of Differentially Expressed miRNAs
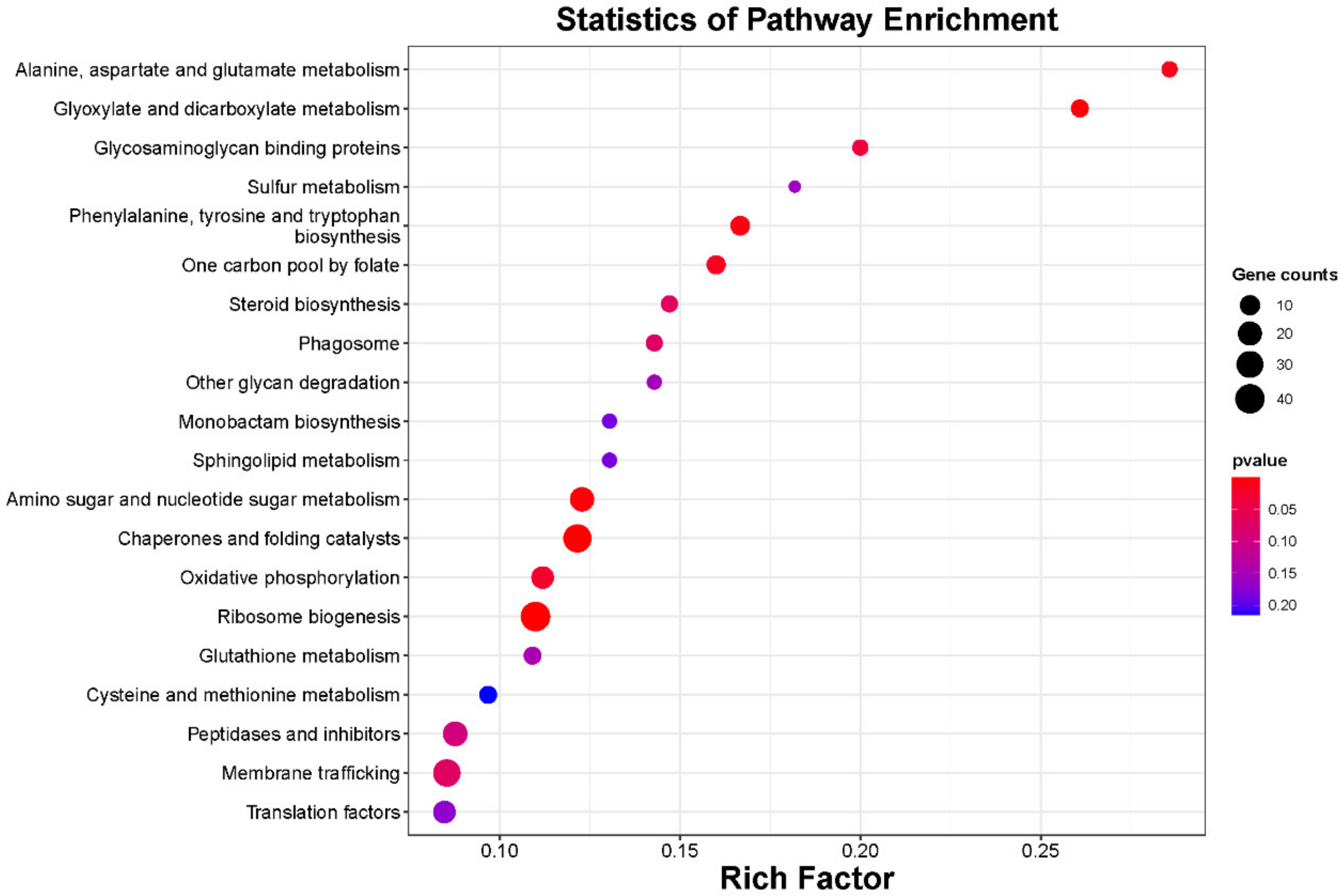
2.4. Identification of MiRNA Target Genes via Degradome Sequencing
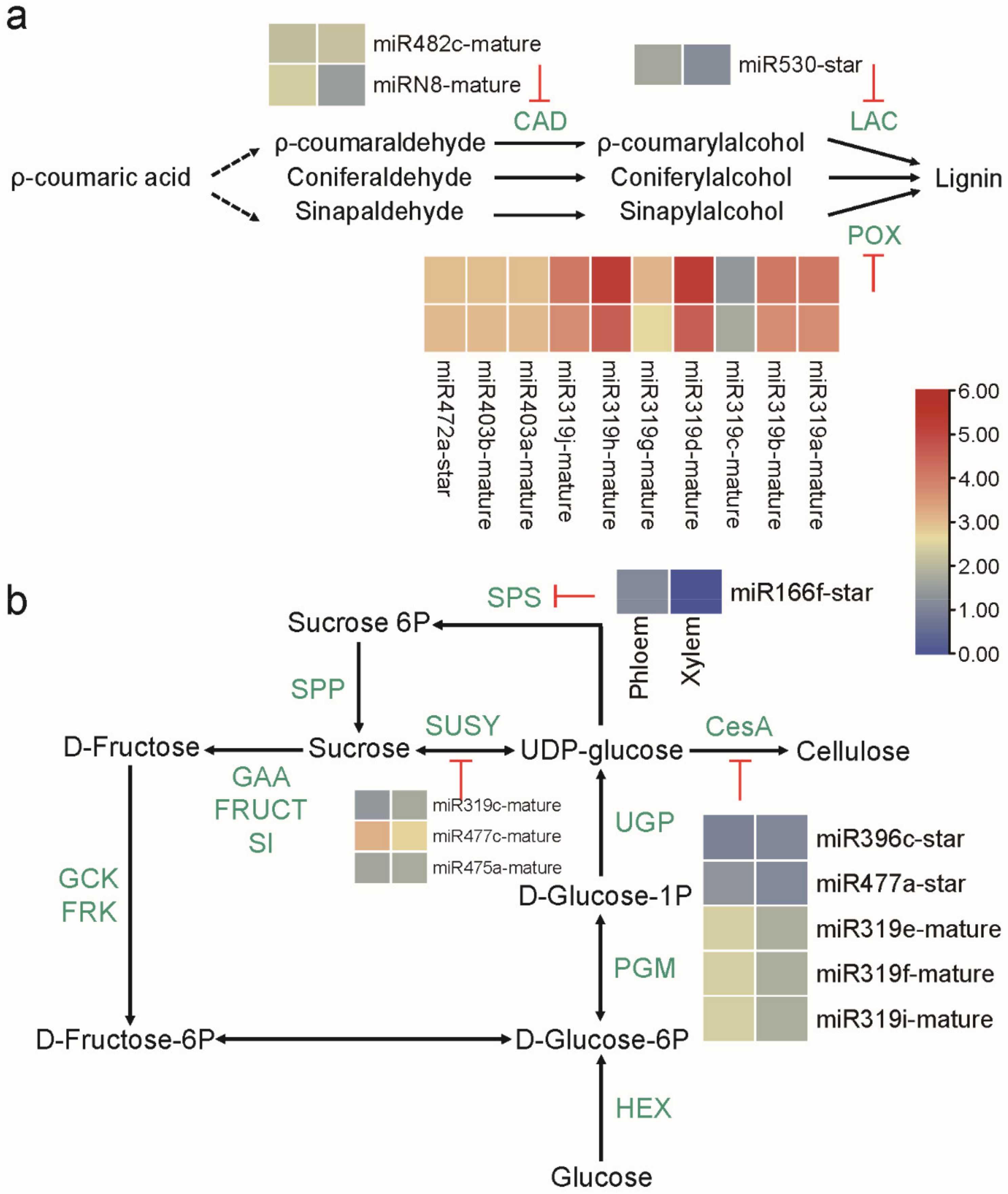
2.5. MicroRNAs Involved in the Regulation of Lignin and Cellulose Biosynthesis
3. Discussion
3.1. Annotation of MiRNAs Involved in the Developing Xylem and Phloem of P. deltoides
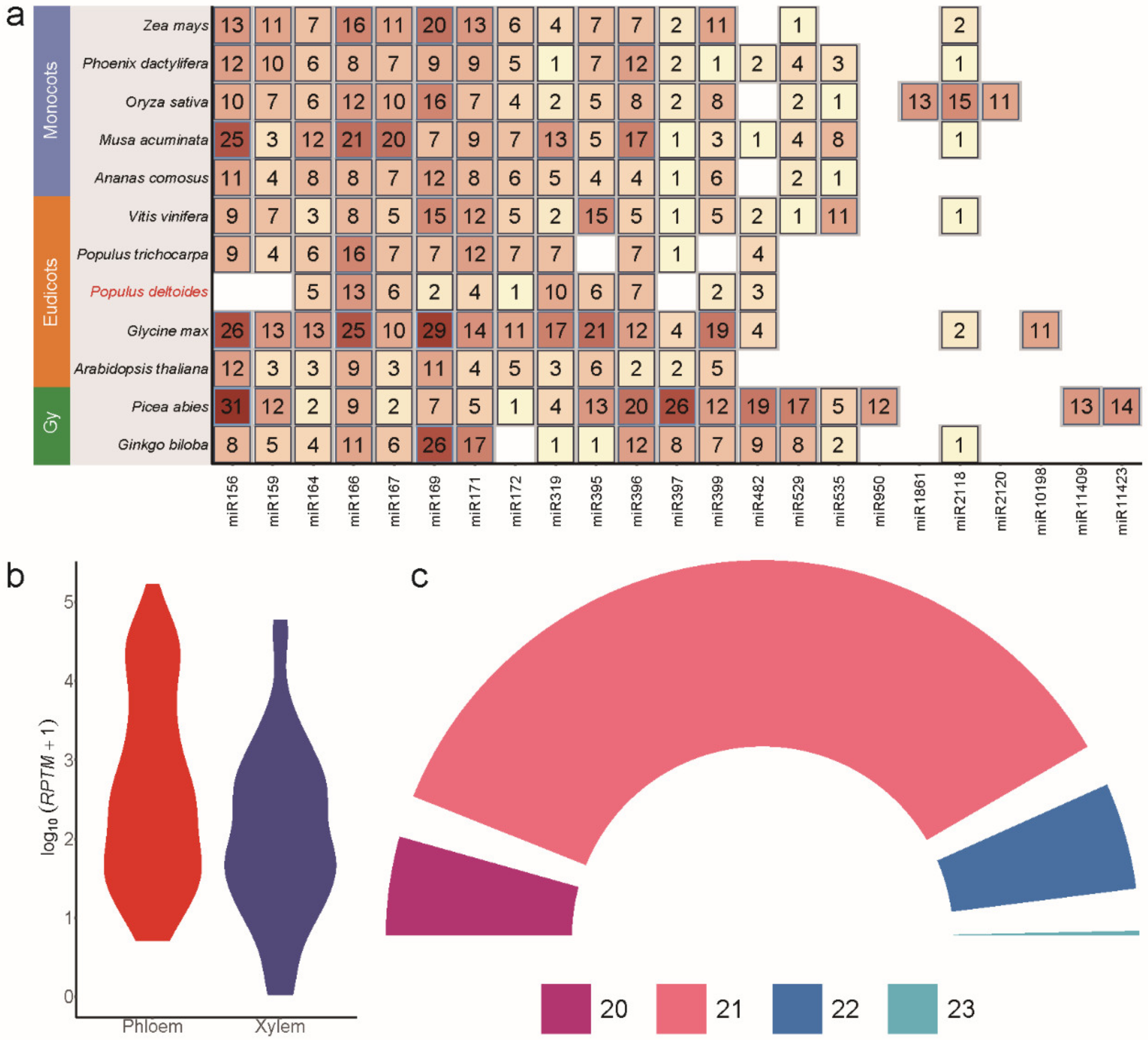
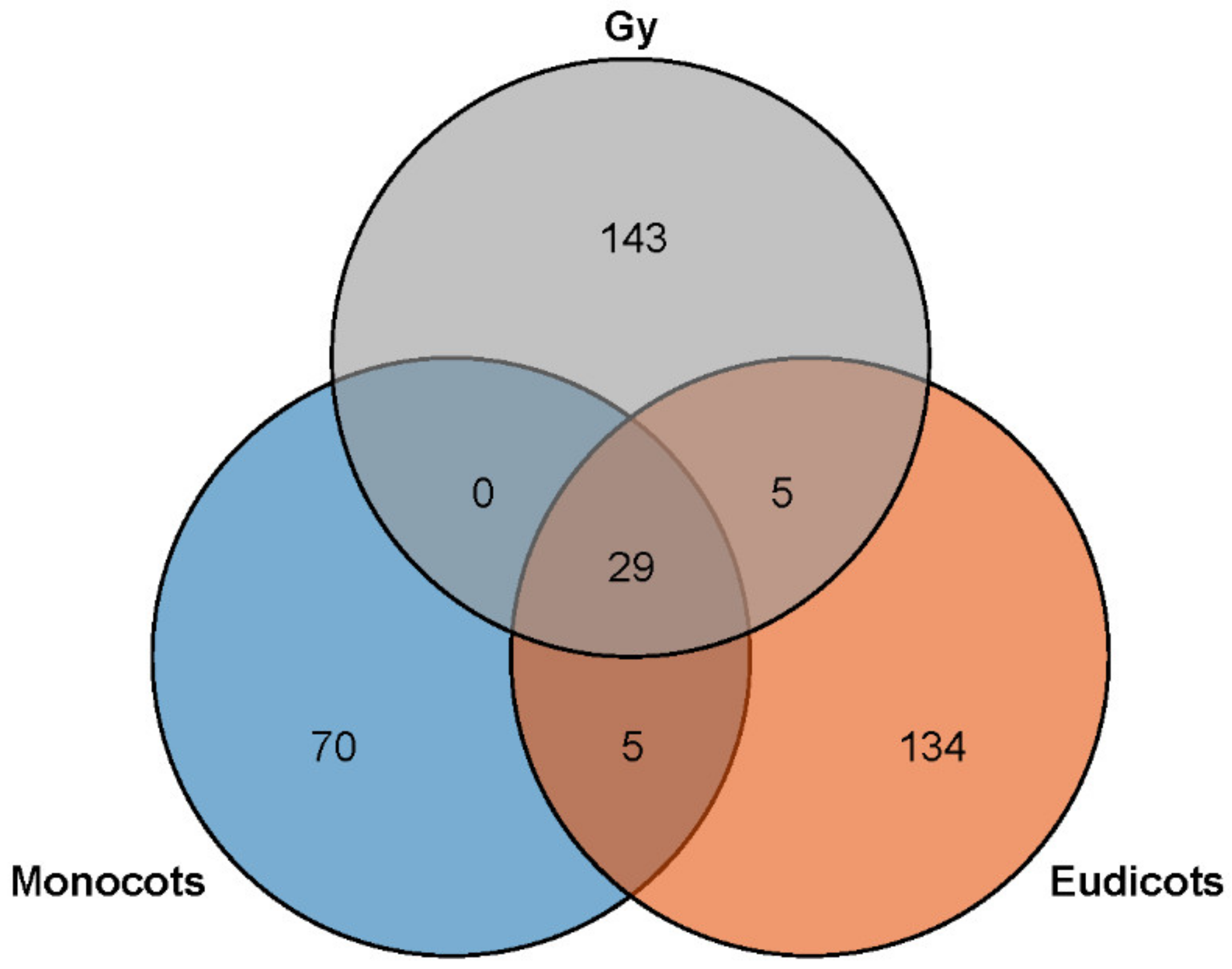
3.2. MiRNAs Families in Monocots, Eudicots, and Gymnosperms
3.3. Most Target Genes Are Transcription Factors
3.4. Conserved MicroRNAs-mRNAs Modules Involved in the Biosynthesis of Lignin and Cellulose
4. Materials and Methods
4.1. Plant Materials and Small RNA Sequencing
4.2. MiRNA Annotation Pipeline
4.3. Identification of MiRNA Target Genes
4.4. qRT-PCR Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fukuda, H.; Ohashi-Ito, K. Chapter Six—Vascular tissue development in plants. In Current Topics in Developmental Biology; Grossniklaus, U., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 131, pp. 141–160. [Google Scholar]
- De Rybel, B.; Mähönen, A.P.; Helariutta, Y.; Weijers, D. Plant vascular development: From early specification to differentiation. Nat. Rev. Mol. Cell Biol. 2016, 17, 30–40. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, H.; Döring, A.; Persson, S. The Cell Biology of Cellulose Synthesis. Annu. Rev. Plant Biol. 2014, 65, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D. Loosening of plant cell walls by expansins. Nature 2000, 407, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Scheller, H.V.; Ulvskov, P. Hemicelluloses. Annu. Rev. Plant Biol. 2010, 61, 263–289. [Google Scholar] [CrossRef]
- Vaahtera, L.; Schulz, J.; Hamann, T. Cell wall integrity maintenance during plant development and interaction with the environment. Nat. Plants 2019, 5, 924–932. [Google Scholar] [CrossRef]
- Liu, Q.; Luo, L.; Zheng, L. Lignins: Biosynthesis and Biological Functions in Plants. Int. J. Mol. Sci. 2018, 19, 335. [Google Scholar] [CrossRef] [Green Version]
- Amin, N.; McGrath, A.; Chen, Y.-P.P. Evaluation of deep learning in non-coding RNA classification. Nat. Mach. Intell. 2019, 1, 246–256. [Google Scholar] [CrossRef]
- Axtell, M. Classification and comparison of SMALL RNAs from plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Li, Y.; Cao, X.; Qi, Y. MicroRNAs and Their Regulatory Roles in Plant–Environment Interactions. Annu. Rev. Plant Biol. 2019, 70, 489–525. [Google Scholar] [CrossRef]
- Yu, Y.; Jia, T.; Chen, X. The ‘how’ and ‘where’ of plant microRNAs. New Phytol. 2017, 216, 1002–1017. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Zheng, B.; Yu, Y.; Won, S.; Mo, B.; Chen, X. The role of Mediator in small and long noncoding RNA production in Arabidopsis thaliana. EMBO J. 2011, 30, 814–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Allen, E.; Fahlgren, N.; Calamar, A.; Givan, S.; Carrington, J. Expression of Arabidopsis miRNA Genes. Plant Physiol. 2005, 138, 2145–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchert, G.; Lanier, W.; Davidson, B. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2007, 13, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Johansen, L.; Gustafson, A.; Kasschau, K.; Lellis, A.; Zilberman, D.; Jacobsen, S.; Carrington, J. Genetic and Functional Diversification of Small RNA Pathways in Plants. PLoS Biol. 2004, 2, E104. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Spector, D. Identification of Nuclear Dicing Bodies Containing Proteins for MicroRNA Biogenesis in Living Arabidopsis Plants. Curr. Biol. CB 2007, 17, 818–823. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yang, Z.; Yu, B.; Liu, J.; Chen, X. Methylation Protects miRNAs and siRNAs from a 3′-End Uridylation Activity in Arabidopsis. Curr. Biol. CB 2005, 15, 1501–1507. [Google Scholar] [CrossRef] [Green Version]
- Park, W.; Li, J.; Song, R.; Messing, J.; Chen, X. CARPEL FACTORY, a dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr. Biol. CB 2002, 12, 1484–1495. [Google Scholar] [CrossRef] [Green Version]
- Bollman, K.; Aukerman, M.; Park, M.Y.; Hunter, C.; Berardini, T.; Poethig, R. HASTY, the Arabidopsis ortholog of exportin 5/MSN5, regulates phase change and morphogenesis. Development 2003, 130, 1493–1504. [Google Scholar] [CrossRef] [Green Version]
- Bajczyk, M.; Lange, H.; Bielewicz, D.; Szewc, L.; Bhat, S.; Dolata, J.; Kuhn, L.; Gagliardi, D.; Jarmolowski, A. SERRATE interacts with the nuclear exosome targeting (NEXT) complex to degrade primary miRNA precursors in Arabidopsis. Nucleic Acids Res. 2020, 48, 6839–6854. [Google Scholar] [CrossRef]
- Xie, D.; Chen, M.; Niu, J.; Wang, L.; Li, Y.; Fang, X.; Li, P.; Qi, Y. Phase separation of SERRATE drives dicing body assembly and promotes miRNA processing in Arabidopsis. Nat. Cell Biol. 2021, 23, 1–8. [Google Scholar] [CrossRef]
- Lee, S.; Martienssen, R. Phase separation in plant miRNA processing. Nat. Cell Biol. 2020, 23, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Wang, J.; Jiang, N.; Zhang, S.; Wang, Y.; Zhang, J.; Li, N.; Fang, Y.; Yang, L.; Chen, S.; et al. Hyponastic Leaves 1 protects pri-miRNAs from nuclear exosome attack. Proc. Natl. Acad. Sci. USA 2020, 117, 17429–17437. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ma, Z.; Castillo-Gonzalez, C.; Sun, D.; Li, Y.; Yu, B.; Zhao, B.; Li, P.; Zhang, X. SWI2/SNF2 ATPase CHR2 remodels pri-miRNAs via Serrate to impede miRNA production. Nature 2018, 557, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; You, C.; Zhang, Y.; Zeng, L.; Hu, J.; Zhao, M.; Chen, X. Linking key steps of microRNA biogenesis by TREX-2 and the nuclear pore complex in Arabidopsis. Nat. Plants 2020, 6, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Cambiagno, D.; Giudicatti, A.; Arce, A.; Gagliardi, D.; Lei, L.; Yuan, W.; Lundberg, D.; Weigel, D.; Pablo, M. HASTY modulates miRNA biogenesis by linking pri-miRNA transcription and processing. Mol. Plant 2020, 14, 426–439. [Google Scholar] [CrossRef]
- Li, S.; Li, M.; Liu, K.; Zhang, H.; Zhang, S.; Zhang, C.; Yu, B. MAC5, an RNA-binding protein, protects pri-miRNAs from SERRATE-dependent exoribonuclease activities. Proc. Natl. Acad. Sci. USA 2020, 117, 23982–23990. [Google Scholar] [CrossRef]
- Li, S.; Xu, R.; Li, A.; Liu, K.; Gu, L.; Li, M.; Zhang, H.; Zhang, Y.; Zhuang, S.; Wang, Q.; et al. SMA1, a Homolog of the Splicing Factor Prp28, Has a Multifaceted Role in miRNA Biogenesis in Arabidopsis. Nucleic Acids Res. 2018, 46, 9148–9159. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Quan, L.; Li, S.; You, C.; Zhang, Y.; Gao, l.; Zeng, L.; Liu, L.; Qi, Y.; Mo, B.; et al. The Protein Phosphatase 4 Complex Promotes Transcription and Processing of Primary microRNAs in Arabidopsis. Plant Cell 2019, 31, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Khvorova, A.; Reynolds, A.; Jayasena, S. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P. RISC—Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Gyorgy, H.; Simard, M.J. Argonaute proteins: Key players in RNA silencing. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar]
- Liu, S.; Xu, M. Identification and characterization of long non-coding RNAs involved in the formation and development of poplar adventitious roots. Ind. Crops Prod. 2018, 118, 334–346. [Google Scholar] [CrossRef]
- Tang, F.; Wei, H.; Zhao, S.; Wang, L.; Zheng, H.; Lu, M. Identification of microRNAs Involved in Regeneration of the Secondary Vascular System in Populus tomentosa Carr. Front. Plant Sci. 2016, 7, 724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puzey, J.R.; Karger, A.; Axtell, M.; Kramer, E.M. Deep Annotation of Populus trichocarpa microRNAs from Diverse Tissue Sets. PLoS ONE 2012, 7, e33034. [Google Scholar] [CrossRef]
- Lu, S.; Li, Q.; Wei, H.; Chang, M.-J.; Tunlaya-Anukit, S.; Kim, H.; Liu, J.; Song, J.; Sun, Y.-H.; Yuan, L.; et al. Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Acad. Sci. USA 2013, 110, 10848–10853. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-Y.; Zhang, S.; Yu, Y.; Luo, Y.-C.; Liu, Q.; Ju, C.; Zhang, Y.-C.; Qu, L.-H.; Lucas, W.J.; Wang, X.; et al. MiR397b regulates both lignin content and seed number in Arabidopsis via modulating a laccase involved in lignin biosynthesis. Plant Biotechnol. J. 2014, 12, 1132–1142. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Liu, X.; Yang, J.; Liu, W.; Du, Q.; Wang, H.; Fu, C.; Li, W.-X. MicroRNA528 Affects Lodging Resistance of Maize by Regulating Lignin Biosynthesis under Nitrogen-Luxury Conditions. Mol. Plant 2018, 11, 806–814. [Google Scholar] [CrossRef] [Green Version]
- Shen, T.; Xu, M.; Qi, H.; Feng, Y.; Yang, Z.; Xu, M. Uncovering miRNA-mRNA Regulatory Modules in Developing Xylem of Pinus massoniana via Small RNA and Degradome Sequencing. Int. J. Mol. Sci. 2021, 22, 10154. [Google Scholar] [CrossRef]
- Reyes, J.L.; Chua, N.-H. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007, 49, 592–606. [Google Scholar] [CrossRef]
- Baldoni, E.; Genga, A.; Cominelli, E. Plant MYB Transcription Factors: Their Role in Drought Response Mechanisms. Int. J. Mol. Sci. 2015, 16, 15811–15851. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Ding, D.; Shi, C.; Xue, Y.; Zhang, Z.; Tang, G.; Tang, J. microRNA-dependent gene regulatory networks in maize leaf senescence. BMC Plant Biol. 2016, 16, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Wang, Q.; Sun, R.; Zhang, B. Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J. Exp. Bot. 2015, 66, 789–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Duan, X.; Zhang, Q.; Li, X.; Wang, B.; Huang, L.; Wang, X.; Kang, Z. The target gene of tae-miR164, a novel NAC transcription factor from the NAM subfamily, negatively regulates resistance of wheat to stripe rust. Mol. Plant Pathol. 2014, 15, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, J.; Watanabe, Y. miR165/166 and the development of land plants. Dev. Growth Differ. 2012, 54, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.; Meyers, B. Revisiting criteria for plant miRNA annotation in the era of big data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.; Tarver, J.; Hiscock, S.; Donoghue, P. Evolutionary history of plant microRNAs. Trends Plant Sci. 2014, 19, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Jangra, S.; Chaudhary, V.; Yadav, N. Transcription Factors and MicroRNA Interplay: A New Strategy for Crop Improvement; Books on Demand: Norderstedt, Germany, 2018; pp. 103–123. [Google Scholar]
- Mullany, L.; Herrick, J.; Wolff, R.; Stevens, J.; Samowitz, W.; Slattery, M. MicroRNA-Transcription Factor Interactions and Their Combined Effect on Target Gene Expression in Colon Cancer Cases. Genes Chromosom. Cancer 2017, 57, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Park, M.Y.; Conway, S.; Wang, J.-W.; Weigel, D.; Poethig, R. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Hossain, M.S.; Wang, J.; Valdés-López, O.; Liang, Y.; Libault, M.; Qiu, L.; Stacey, G. miR172 Regulates Soybean Nodulation. Mol. Plant-Microbe Interact. MPMI 2013, 26, 1371–1377. [Google Scholar] [CrossRef] [Green Version]
- Nova-Franco, B.; Iñiguez, L.; Valdés-López, O.; Alvarado-Affantranger, X.; Leija Salas, A.; Fuentes, S.; Ramirez, M.; Paul, S.; Reyes, J.; Girard, L.; et al. The miR172c-AP2-1 Node as a Key Regulator of the Common Bean—Rhizobia Nitrogen Fixation Symbiosis. Plant Physiol. 2015, 168, 273–291. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-W.; Wang, L.-J.; Mao, Y.-B.; Cai, W.-J.; Xue, H.-W.; Chen, X.-Y. Control of Root Cap Formation by MicroRNA-Targeted Auxin Response Factors in Arabidopsis. Plant Cell 2005, 17, 2204–2216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S. Function of MYB domain transcription factors in abiotic stress and epigenetic control of stress response in plant genome. Plant Signal. Behav. 2015, 11, e1117723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efroni, I.; Blum, E.; Goldshmidt, A.; Eshed, Y. A Protracted and Dynamic Maturation Schedule Underlies Arabidopsis Leaf Development. Plant Cell 2008, 20, 2293–2306. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Ali, A.; Saifi, M.; Saxena, P.; Ahlawat, S.; Abdin, M. Identification and the potential involvement of miRNAs in the regulation of artemisinin biosynthesis in A. annua. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Zhang, Q.; Cui, R.; Xu, X.; Zhu, F.-Y.; Cheng, Q.; Li, X. High-Throughput Sequencing Reveals the Regulatory Networks of Transcriptome and Small RNAs During the Defense Against Marssonina brunnea in Poplar. Front. Plant Sci. 2021, 12, 719549. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, J.; Blervacq, A.-S.; Creach, A.; Huss, B.; Hawkins, S.; Neutelings, G. Spatial regulation of monolignol biosynthesis and laccase genes control developmental and stress-related lignin in flax. BMC Plant Biol. 2017, 17, 1–20. [Google Scholar] [CrossRef]
- Xue, C.; Yao, J.L.; Qin, M.-F.; Zhang, M.; Allan, A.; Wang, D.-F.; Wu, J. PbrmiR397a regulates lignification during stone cell development in pear fruit. Plant Biotechnol. J. 2018, 17, 103–117. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Gulyas, Z.; Kalapos, B.; Boldizsar, A.; Liu, X.; Pal, M.; Yao, Y.; Galiba, G.; Kocsy, G. Identification of a redox-dependent regulatory network of miRNAs and their targets in wheat. J. Exp. Bot. 2019, 70, 85–99. [Google Scholar] [CrossRef]
- Stein, O.; Granot, D. An Overview of Sucrose Synthases in Plants. Front. Plant Sci. 2019, 10, 95. [Google Scholar] [CrossRef] [Green Version]
- Doblin, M.; Kurek, I.; Jacob-Wilk, D.; Delmer, D. Cellulose biosynthesis in plants: From genes to rosettes. Plant Cell Physiol. 2003, 43, 1407–1420. [Google Scholar] [CrossRef]
- Turner, S.; Kumar, M. Cellulose synthase complex organization and cellulose microfibril structure. Philos. Trans. A Math. Phys. Eng. Sci. 2018, 376, 20170048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, B.; Habermann, K.; Arif, M.A.; Top, O. Identification of Small RNAs During High Light Acclimation in Arabidopsis thaliana. Front. Plant Sci. 2021, 12, 1179. [Google Scholar] [CrossRef] [PubMed]
- Langenkamper, G.; Fung, R.W.M.; Newcomb, R.; Atkinson, R.; Gardner, R.C.; Macrae, E. Sucrose phosphate synthase genes in plants belong to three different families. J. Mol. Evol. 2002, 54, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Chua, T.; Bujnicki, J.; Tan, T.-C.; Huynh, F.; Patel, B.; Sivaraman, J. The Structure of Sucrose Phosphate Synthase from Halothermothrix orenii Reveals Its Mechanism of Action and Binding Mode. Plant Cell 2008, 20, 1059–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyawali, B.; Barozai, M.Y.; Aziz, A. Comparative expression analysis of microRNAs and their targets in emerging bio-fuel crop sweet sorghum (Sorghum bicolor L.). Plant Gene 2021, 26, 100274. [Google Scholar] [CrossRef]
- Ohashi-Ito, K.; Kubo, M.; Demura, T.; Fukuda, H. Class III Homeodomain Leucine-Zipper Proteins Regulate Xylem Cell Differentiation. Plant Cell Physiol. 2005, 46, 1646–1656. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Reng, M.; Tian, S.; Liu, C.; Cheng, H.; Liu, Y.; Zhang, H.; Wei, H.; Wei, Z. Transcriptome-wide identification and characterization of microRNAs in diverse phases of wood formation in Populus trichocarpa. G3-Genes Genomes Genet. 2021, 11, jkab195. [Google Scholar] [CrossRef]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 January 2020).
- Martin, M. CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Tsuji, J.; Deng, Z. DNApi: A De Novo Adapter Prediction Algorithm for Small RNA Sequencing Data. PLoS ONE 2016, 11, e0164228. [Google Scholar] [CrossRef]
- Veneziano, D.; Di Bella, S.; Giovanni, N.; Laganà, A.; Ferro, A.; Croce, C. Non-coding RNA: Current Deep Sequencing Data Analysis Approaches and Challenges. Hum. Mutat. 2016, 37, 1283–1298. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Li, J.; Feng, J.; Bo, L.; Feng, L.; Yu, X.; Li, G.; Zhai, J.; Meyers, B.; Xia, R. sRNAanno—a database repository of uniformly annotated small RNAs in plants. Hortic. Res. 2021, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Xia, R.; Liu, Y. Comprehensive Characterization of miRNA and PHAS Loci in the Diploid Strawberry (Fragaria vesca) Genome. Hortic. Plant J. 2019, 5, 255–267. [Google Scholar] [CrossRef]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodstein, D.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2011, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Q30 (%) | GC (%) | Mapped Rate (%) |
|---|---|---|---|---|---|
| P1 | 11,419,418 | 11,161,174 | 95.96 | 50.74 | 90.07 |
| P2 | 11,768,425 | 11,432,450 | 95.47 | 51.42 | 95.10 |
| P3 | 11,997,478 | 11,760,792 | 95.83 | 50.77 | 91.37 |
| X1 | 10,320,359 | 10,100,623 | 95.85 | 50.13 | 97.40 |
| X2 | 10,796,007 | 10,499,531 | 95.17 | 50.58 | 97.22 |
| X3 | 11,069,777 | 10,824,465 | 95.52 | 50.27 | 97.39 |
| miRNA | Transcript Annotation | Degradome Category | Degradome p Value |
|---|---|---|---|
| miR171a-mature | GRAS | 0 | 0.000271151 |
| miR160f-mature | ARF17 | 0 | 0.000271151 |
| miR167d-mature | ARF8 | 0 | 0.000271151 |
| miR396b-mature | GRF4 | 0 | 0.000271151 |
| miR472a-star | AGO3 | 0 | 0.000271151 |
| miR319a-mature | MYB65 | 0 | 0.000542229 |
| miR164d-mature | NAC80 | 0 | 0.000542229 |
| miR172c-mature | AP2 | 0 | 0.000813233 |
| miR160d-mature | ARF10 | 0 | 0.002708204 |
| miR160e-mature | ARF16 | 0 | 0.002708204 |
| miR396a-mature | GRF9 | 0 | 0.002708204 |
| miR530-mature | bHLH | 0 | 0.017206273 |
| miR396d-mature | SCL13 | 0 | 0.019336135 |
| miR171d-star | WRKY | 1 | 0.027125399 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, C.; Shen, T.; Ran, N.; Zhang, H.; Pan, H.; Su, X.; Xu, M. Integrated Degradome and Srna Sequencing Revealed miRNA-mRNA Regulatory Networks between the Phloem and Developing Xylem of Poplar. Int. J. Mol. Sci. 2022, 23, 4537. https://doi.org/10.3390/ijms23094537
Ding C, Shen T, Ran N, Zhang H, Pan H, Su X, Xu M. Integrated Degradome and Srna Sequencing Revealed miRNA-mRNA Regulatory Networks between the Phloem and Developing Xylem of Poplar. International Journal of Molecular Sciences. 2022; 23(9):4537. https://doi.org/10.3390/ijms23094537
Chicago/Turabian StyleDing, Changjun, Tengfei Shen, Na Ran, Heng Zhang, Huixin Pan, Xiaohua Su, and Meng Xu. 2022. "Integrated Degradome and Srna Sequencing Revealed miRNA-mRNA Regulatory Networks between the Phloem and Developing Xylem of Poplar" International Journal of Molecular Sciences 23, no. 9: 4537. https://doi.org/10.3390/ijms23094537