
Moving Average-Based Multitasking In Silico Classification Modeling: Where Do We Stand and What Is Next?



Abstract
:1. Introduction
2. Multitasking QSAR Modeling: Rationale and Existing Challenges
3. Multitasking In Silico Modeling Methodologies
3.1. Moving Average Approach
3.2. Descriptor Calculation
3.3. Data Pooling, Databases, and Inclusion/Exclusion Criteria
3.4. Dataset Division
3.5. Set-Up of the MA-Mtk Model
3.6. Statistical Analysis and Validation
4. Applications of Mtk-QSAR Modeling
4.1. MA-Mtk Modeling of the Activity against Cells/Organisms/Species
4.2. MA-Mtk Modeling of the Activity against Bio-Macromolecular Targets
5. Software Developed for Multitasking Modeling
5.1. QSAR-Co
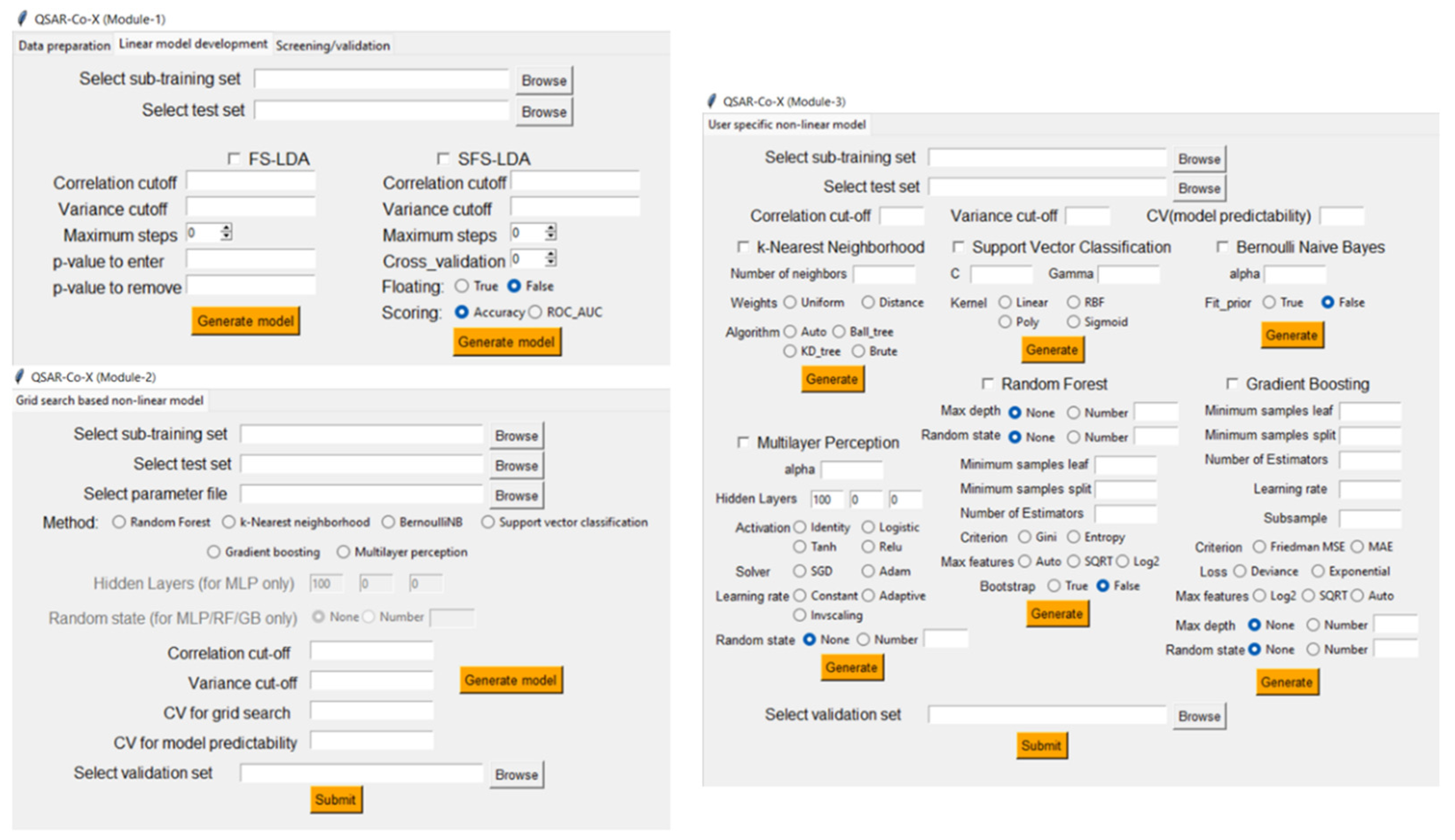
5.2. QSAR-Co-X
5.3. FRAMA
6. Future Scope
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hansch, C.; Maloney, P.P.; Fujita, T.; Muir, R.M. Correlation of Biological Activity of Phenoxyacetic Acids with Hammett Substituent Constants and Partition Coefficients. Nature 1962, 194, 178–180. [Google Scholar] [CrossRef]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR Without Borders. Chem. Soc. Rev. 2020, 49, 3525–3564. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; MacKerell, A.D. Computer-Aided Drug Design Methods. In Antibiotics. Methods in Molecular Biology; Sass, P., Ed.; Humana Press: New York, NY, USA, 2017; Volume 1520, pp. 85–106. ISBN 978-1-4939-6634-9. [Google Scholar]
- Abdolmaleki, A.; Ghasemi, J.; Ghasemi, F. Computer Aided Drug Design for Multi-Target Drug Design: SAR/QSAR, Molecular Docking and Pharmacophore Methods. Curr. Drug Targets 2017, 18, 556–575. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Halder, A.K.; Moura, A.S.; Cordeiro, M.N.D.S. QSAR modelling: A therapeutic patent review 2010-present. Expert Opin. Ther. Pat. 2018, 28, 467–476. [Google Scholar] [CrossRef]
- Speck-Planche, A. Recent advances in fragment-based computational drug design: Tackling simultaneous targets/biological effects. Future Med. Chem. 2018, 10, 2021–2024. [Google Scholar] [CrossRef] [Green Version]
- Searls, D.B. Data integration: Challenges for drug discovery. Nat. Rev. Drug Discov. 2005, 4, 45–58. [Google Scholar] [CrossRef]
- Ortega-Tenezaca, B.; Quevedo-Tumailli, V.; Bediaga, H.; Collados, J.; Arrasate, S.; Madariaga, G.; Munteanu, C.R.; Cordeiro, M.N.D.S.; Gonzalez-Díaz, H. PTML Multi-Label Algorithms: Models, Software, and Applications. Curr. Top. Med. Chem. 2020, 20, 2326–2337. [Google Scholar] [CrossRef]
- Halder, A.K.; Cordeiro, M.N.D.S. Development of Multi-Target Chemometric Models for the Inhibition of Class I PI3K Enzyme Isoforms: A Case Study Using QSAR-Co Tool. Int. J. Mol. Sci. 2019, 20, 4191. [Google Scholar] [CrossRef] [Green Version]
- Halder, A.K.; Cordeiro, M.N.D.S. QSAR-Co-X: An open source toolkit for multitarget QSAR modelling. J. Cheminform. 2021, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Multitasking models for quantitative structure–biological effect relationships: Current status and future perspectives to speed up drug discovery. Expert Opin. Drug Discov. 2015, 10, 245–256. [Google Scholar] [CrossRef]
- Lambrinidis, G.; Tsantili-Kakoulidou, A. Multi-objective optimization methods in novel drug design. Expert Opin. Drug Discov. 2020, 16, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A. Multi-Scale Modeling in Drug Discovery against Infectious Diseases. Mini Rev. Med. Chem. 2019, 19, 1560–1563. [Google Scholar] [CrossRef] [PubMed]
- Toropov, A.A.; Toropova, A.P. QSPR/QSAR: State-of-Art, Weirdness, the Future. Molecules 2020, 25, 1292. [Google Scholar] [CrossRef] [Green Version]
- Roy, K.; Kar, S.; Das, R.N. Chemical Information and Descriptors. In Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Academic Press: Amsterdam, The Netherlands, 2015; pp. 47–80. ISBN 9780128016336. [Google Scholar]
- Halder, A.K.; Delgado, A.H.S.; Cordeiro, M.N.D.S. First multi-target QSAR model for predicting the cytotoxicity of acrylic acid-based dental monomers. Dental Mater. 2022, 38, 333–346. [Google Scholar] [CrossRef]
- Prado-Prado, F.J.; Gonzalez-Díaz, H.; de la Vega, O.M.; Ubeira, F.M.; Chou, K.-C. Unified QSAR approach to antimicrobials. Part 3: First multi-tasking QSAR model for input-coded prediction, structural back-projection, and complex networks clustering of antiprotozoal compounds. Bioorg. Med. Chem. 2008, 16, 5871–5880. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Speck-Planche, A. The urgent need for pan-antiviral agents: From multitarget discovery to multiscale design. Future Med. Chem. 2021, 13, 5–8. [Google Scholar] [CrossRef]
- Halder, A.K.; Cordeiro, M.N.D.S. Multi-Target in Silico Prediction of Inhibitors for Mitogen-Activated Protein Kinase-Interacting Kinases. Biomolecules 2021, 11, 1670. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Scotti, M.T.; Speck-Planche, A. Computational Drug Repurposing for Antituberculosis Therapy: Discovery of Multi-Strain Inhibitors. Antibiotics 2021, 10, 1005. [Google Scholar] [CrossRef]
- Cumming, J.G.; Davis, A.M.; Muresan, S.; Haeberlein, M.; Chen, H. Chemical predictive modelling to improve compound quality. Nat. Rev. Drug Discov. 2013, 12, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Scotti, L.; Junior, F.J.B.M.; Muratov, E.; Scotti, M.T.; Speck-Planche, A. QSAR Modeling for Multi-Target Drug Discovery: Designing Simultaneous Inhibitors of Proteins in Diverse Pathogenic Parasites. Front. Chem. 2021, 9, 634663. [Google Scholar] [CrossRef] [PubMed]
- Halder, A.K.; Cordeiro, M.N.D.S. AKT Inhibitors: The Road Ahead to Computational Modeling-Guided Discovery. Int. J. Mol. Sci. 2021, 22, 3944. [Google Scholar] [CrossRef] [PubMed]
- Box, G.E.P.; Jenkins, G.M.; Reinsel, G.C.; Ljung, G.M. Time Series Analysis: Forecasting and Control; Holden-Day: San Francisco, CA, USA, 1970. [Google Scholar]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Fragment-based QSAR model toward the selection of versatile anti-sarcoma leads. Eur. J. Med. Chem. 2011, 46, 5910–5916. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Scotti, M.T. Fragment-based approach for the in silico discovery of multi-target insecticides. Chemom. Intell. Lab. Syst. 2012, 111, 39–45. [Google Scholar] [CrossRef]
- Halder, A.K.; Haghbakhsh, R.; Voroshylova, I.V.; Duarte, A.R.C.; Cordeiro, M.N.D.S. Density of Deep Eutectic Solvents: The Path Forward Cheminformatics-Driven Reliable Predictions for Mixtures. Molecules 2021, 26, 5779. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. De novo computational design of compounds virtually displaying potent antibacterial activity and desirable in vitro ADMET profiles. Med. Chem. Res. 2017, 26, 2345–2356. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Speeding up Early Drug Discovery in Antiviral Research: A Fragment-Based in Silico Approach for the Design of Virtual Anti-Hepatitis C Leads. ACS Comb. Sci. 2017, 19, 501–512. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Scotti, M.T.; Scotti, L.; Nayarisseri, A.; Speck-Planche, A. Cell-based multi-target QSAR model for design of virtual versatile inhibitors of liver cancer cell lines. SAR QSAR Environ. Res. 2020, 31, 815–836. [Google Scholar] [CrossRef]
- Speck-Planche, A. Multi-Scale QSAR Approach for Simultaneous Modeling of Ecotoxic Effects of Pesticides. In Ecotoxicological QSARs. Methods in Pharmacology and Toxicology; Roy, K., Ed.; Springer: New York, NY, USA, 2020; pp. 639–660. ISBN 978-1-0716-0150-1. [Google Scholar]
- Kleandrova, V.V.; Scotti, M.T.; Scotti, L.; Speck-Planche, A. Multi-target Drug Discovery via PTML Modeling: Applications to the Design of Virtual Dual Inhibitors of CDK4 and HER2. Curr. Top. Med. Chem. 2021, 21, 661–675. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Scotti, M.T. In Silico Drug Repurposing for Anti-Inflammatory Therapy: Virtual Search for Dual Inhibitors of Caspase-1 and TNF-Alpha. Biomolecules 2021, 11, 1832. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Martiní, J.R.; Marrero-Ponce, Y.; García-Jacas, C.R.; Martinez-Mayorga, K.; Barigye, S.J.; Vaz d‘Almeida, Y.S.; Pham-The, H.; Pérez-Giménez, F.; Morell, C.A. QuBiLS-MAS, open source multi-platform software for atom- and bond-based topological (2D) and chiral (2.5D) algebraic molecular descriptors computations. J. Cheminform. 2017, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- García, I.; Fall, Y.; Gómez, G.; Gonzalez-Díaz, H. First computational chemistry multi-target model for anti-Alzheimer, anti-parasitic, anti-fungi, and anti-bacterial activity of GSK-3 inhibitors in vitro, in vivo, and in different cellular lines. Mol. Divers. 2010, 15, 561–567. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Scotti, M.T. BET bromodomain inhibitors: Fragment-based in silico design using multi-target QSAR models. Mol. Divers. 2018, 23, 555–572. [Google Scholar] [CrossRef]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. DRAGON software: An easy approach to molecular descriptor calculations. MATCH Commun. Math. Comput. Chem. 2006, 56, 237–248. [Google Scholar]
- Kleandrova, V.V.; Speck-Planche, A. PTML Modeling for Pancreatic Cancer Research: In Silico Design of Simultaneous Multi-Protein and Multi-Cell Inhibitors. Biomedicines 2022, 10, 491. [Google Scholar] [CrossRef]
- Speck-Planche, A. Combining Ensemble Learning with a Fragment-Based Topological Approach to Generate New Molecular Diversity in Drug Discovery: In Silico Design of Hsp90 Inhibitors. ACS Omega 2018, 3, 14704–14716. [Google Scholar] [CrossRef]
- Speck-Planche, A. Multicellular Target QSAR Model for Simultaneous Prediction and Design of Anti-Pancreatic Cancer Agents. ACS Omega 2019, 4, 3122–3132. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Speck-Planche, A. Multitasking Model for Computer-Aided Design and Virtual Screening of Compounds with High Anti-HIV Activity and Desirable ADMET Properties. In Multi-Scale Approaches in Drug Discovery; Speck-Planche, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 55–81. ISBN 97800-81011294. [Google Scholar]
- Ambure, P.; Halder, A.K.; Gonzalez Diaz, H.; Cordeiro, M.N.D.S. QSAR-Co: An Open Source Software for Developing Robust Multitasking or Multitarget Classification-Based QSAR Models. J. Chem. Inf. Model. 2019, 59, 2538–2544. [Google Scholar] [CrossRef]
- Urias, R.W.P.; Barigye, S.J.; Marrero-Ponce, Y.; García-Jacas, C.R.; Valdes-Martiní, J.R.; Perez-Gimenez, F. IMMAN: Free software for information theory-based chemometric analysis. Mol. Divers. 2015, 19, 305–319. [Google Scholar] [CrossRef]
- Halder, A.K.; Giri, A.K.; Cordeiro, M.N.D.S. Multi-Target Chemometric Modelling, Fragment Analysis and Virtual Screening with ERK Inhibitors as Potential Anticancer Agents. Molecules 2019, 24, 3909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, K.; Kar, S.; Das, R.N. Validation of QSAR Models. In Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Academic Press: Amsterdam, The Netherlands, 2015; pp. 231–289. ISBN 97801-28016336. [Google Scholar]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Ambure, P.; Bhat, J.; Puzyn, T.; Roy, K. Identifying natural compounds as multi-target-directed ligands against Alzheimer’s disease: An in silico approach. J. Biomol. Struct. Dyn. 2018, 37, 1282–1306. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Ruso, J.M.; Speck-Planche, A.; Cordeiro, M.N.D.S. Enabling the Discovery and Virtual Screening of Potent and Safe Antimicrobial Peptides. Simultaneous Prediction of Antibacterial Activity and Cytotoxicity. ACS Comb. Sci. 2016, 18, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Simultaneous Modeling of Antimycobacterial Activities and ADMET Profiles: A Chemoinformatic Approach to Medicinal Chemistry. Curr. Top. Med. Chem. 2013, 13, 1656–1665. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Simultaneous Virtual Prediction of Anti-Escherichia coli Activities and ADMET Profiles: A Chemoinformatic Complementary Approach for High-Throughput Screening. ACS Comb. Sci. 2014, 16, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Enabling Virtual Screening of Potent and Safer Antimicrobial Agents against Noma: Mtk-QSBER Model for Simultaneous Prediction of Antibacterial Activities and ADMET Properties. Mini Rev. Med. Chem. 2015, 15, 194–202. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Multi-target drug discovery in anti-cancer therapy: Fragment-based approach toward the design of potent and versatile anti-prostate cancer agents. Bioorg. Med. Chem. 2011, 19, 6239–6244. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Chemoinformatics in anti-cancer chemotherapy: Multi-target QSAR model for the in silico discovery of anti-breast cancer agents. Eur. J. Pharm. Sci. 2012, 47, 273–279. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Chemoinformatics in Multi-target Drug Discovery for Anti-cancer Therapy: In Silico Design of Potent and Versatile Anti-brain Tumor Agents. Anti-Cancer Agents Med. Chem. 2012, 12, 678–685. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Rational drug design for anti-cancer chemotherapy: Multi-target QSAR models for the in silico discovery of anti-colorectal cancer agents. Bioorg. Med. Chem. 2012, 20, 4848–4855. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Unified Multi-target Approach for the Rational in silico Design of Anti-bladder Cancer Agents. Anti Cancer Agents Med. Chem. 2013, 13, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Chemoinformatics in Drug Design. Artificial Neural Networks for Simultaneous Prediction of Anti-Enterococci Activities and Toxicological Profiles. In Proceedings of the 5th International Joint Conference on Computational Intelligence, Algarve, Portugal, 20–22 September 2013; pp. 458–465. [Google Scholar]
- Speck-Planche, A.; Kleandrova, V.V.; Cordeiro, M.N.D.S. Chemoinformatics for rational discovery of safe antibacterial drugs: Simultaneous predictions of biological activity against streptococci and toxicological profiles in laboratory animals. Bioorg. Med. Chem. 2013, 21, 2727–2732. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Chemoinformatics for medicinal chemistry: In silico model to enable the discovery of potent and safer anti-cocci agents. Future Med. Chem. 2014, 6, 2013–2028. [Google Scholar] [CrossRef]
- Gonzalez-Díaz, H.; Herrera-Ibatá, D.M.; Duardo-Sánchez, A.; Munteanu, C.R.; Orbegozo-Medina, R.A.; Pazos, A. ANN Multiscale Model of Anti-HIV Drugs Activity vs. AIDS Prevalence in the US at County Level Based on Information Indices of Molecular Graphs and Social Networks. J. Chem. Inf. Mod. 2014, 54, 744–755. [Google Scholar] [CrossRef] [Green Version]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Computer-Aided Discovery in Antimicrobial Research: In Silico Model for Virtual Screening of Potent and Safe Anti-Pseudomonas Agents. Comb. Chem. High Throughput Screen 2015, 18, 305–314. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Ruso, J.M.; Cordeiro, M.N.D.S. First Multitarget Chemo-Bioinformatic Model to Enable the Discovery of Antibacterial Peptides against Multiple Gram-Positive Pathogens. J. Chem. Inf. Model. 2016, 56, 588–598. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V. Demystifying Artificial Neural Networks as Generators of New Chemical Knowledge: Antimalarial Drug Discovery as a Case Study. In Machine Learning in Chemistry; Cartwright, H.M., Ed.; Royal Society of Chemistry: Cambridge, UK, 2020; pp. 398–423. ISBN 978-1-78801-789-3. [Google Scholar]
- Kleandrova, V.V.; Luan, F.; Gonzalez-Díaz, H.; Ruso, J.M.; Melo, A.; Speck-Planche, A.; Cordeiro, M.N.D.S. Computational ecotoxicology: Simultaneous prediction of ecotoxic effects of nanoparticles under different experimental conditions. Environ. Int. 2014, 73, 288–294. [Google Scholar] [CrossRef]
- Halder, A.K.; Cordeiro, M.N.D.S. Probing the Environmental Toxicity of Deep Eutectic Solvents and Their Components: An In Silico Modeling Approach. ACS Sustain. Chem. Eng. 2019, 7, 10649–10660. [Google Scholar] [CrossRef]
- Marzaro, G.; Chilin, A.; Guiotto, A.; Uriarte, E.; Brun, P.; Castagliuolo, I.; Tonus, F.; Gonzalez-Díaz, H. Using the TOPS-MODE approach to fit multi-target QSAR models for tyrosine kinases inhibitors. Eur. J. Med. Chem. 2011, 46, 2185–2192. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Luan, F.; Cordeiro, M.N.D.S. Abelson Tyrosine-Protein Kinase 1 as Principal Target for Drug Discovery Against Leukemias. Role of the Current Computer-Aided Drug Design Methodologies. Curr. Top. Med. Chem. 2013, 12, 2745–2762. [Google Scholar] [CrossRef] [PubMed]
- Casañola-Martin, G.M.; Le-Thi-Thu, H.; Pérez-Giménez, F.; Marrero-Ponce, Y.; Merino-Sanjuán, M.; Abad, C.; Gonzalez-Díaz, H. Multi-output model with Box–Jenkins’s operators of linear indices to predict multi-target inhibitors of ubiquitin–proteasome pathway. Mol. Divers. 2015, 19, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Ouyang, S.S.; Yu, B.A.; Liu, Y.B.; Huang, K.; Gong, J.Y.; Zheng, S.Y.; Li, Z.H.; Li, H.L.; Jiang, H.L. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, W609–W614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pires, D.E.V.; Ascher, D.B. mycoCSM: Using Graph-Based Signatures to Identify Safe Potent Hits against Mycobacteria. J. Chem. Inf. Model. 2020, 60, 3450–3456. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Multi-Target QSAR Approaches for Modeling Protein Inhibitors. Simultaneous Prediction of Activities against Biomacromolecules Present in Gram-Negative Bacteria. Curr. Top. Med. Chem. 2015, 15, 1801–1813. [Google Scholar] [CrossRef] [PubMed]
- Kleandrova, V.V.; Luan, F.; Speck-Planche, A.; Cordeiro, M.N.D.S. Review of Structures Containing Fullerene-C60 for Delivery of Antibacterial Agents. Multitasking model for Computational Assessment of Safety Profiles. Curr. Bioinform. 2015, 10, 565–578. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V. In silico design of multi-target inhibitors for C–C chemokine receptors using substructural descriptors. Mol. Divers. 2011, 16, 183–191. [Google Scholar] [CrossRef]
- Kleandrova, V.V.; Rojas-Vargas, J.A.; Scotti, M.T.; Speck-Planche, A. PTML modeling for peptide discovery: In silico design of non-hemolytic peptides with antihypertensive activity. Mol. Divers. 2021, 1–12. [Google Scholar]
- Speck-Planche, A.; Luan, F.; Cordeiro, M.N.D.S. Role of Ligand-Based Drug Design Methodologies toward the Discovery of New Anti-Alzheimer Agents: Futures Perspectives in Fragment-Based Ligand Design. Curr. Med. Chem. 2012, 19, 1635–1645. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; Ortega-Tenezaca, B.; Quevedo-Tumailli, V. FRAMA 1.0: Framework for Moving Average Operators Calculation in Data Analysis. In Proceedings of the MOL2NET 2017, International Conference on Multidisciplinary Sciences, 3rd Ed, Basel, Switzerland, 15 January–15 December 2017. [Google Scholar]
- Le, N.Q.K.; Kha, Q.H.; Nguyen, V.H.; Chen, Y.-C.; Cheng, S.-J.; Chen, C.-Y. Machine Learning-Based Radiomics Signatures for EGFR and KRAS Mutations Prediction in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 9254. [Google Scholar] [CrossRef]
- Hung, T.N.K.; Le, N.Q.K.; Le, N.H.; Tuan, L.V.; Nguyen, T.P.; Thi, C.; Kang, J.H. An AI-based Prediction Model for Drug-drug Interactions in Osteoporosis and Paget’s Diseases from SMILES. Mol. Inform. 2022, e2100264. [Google Scholar] [CrossRef]
- Imrie, F.; Hadfield, T.E.; Bradley, A.R.; Deane, C.M. Deep generative design with 3D pharmacophoric constraints. Chem. Sci. 2021, 12, 14577–14589. [Google Scholar] [CrossRef]
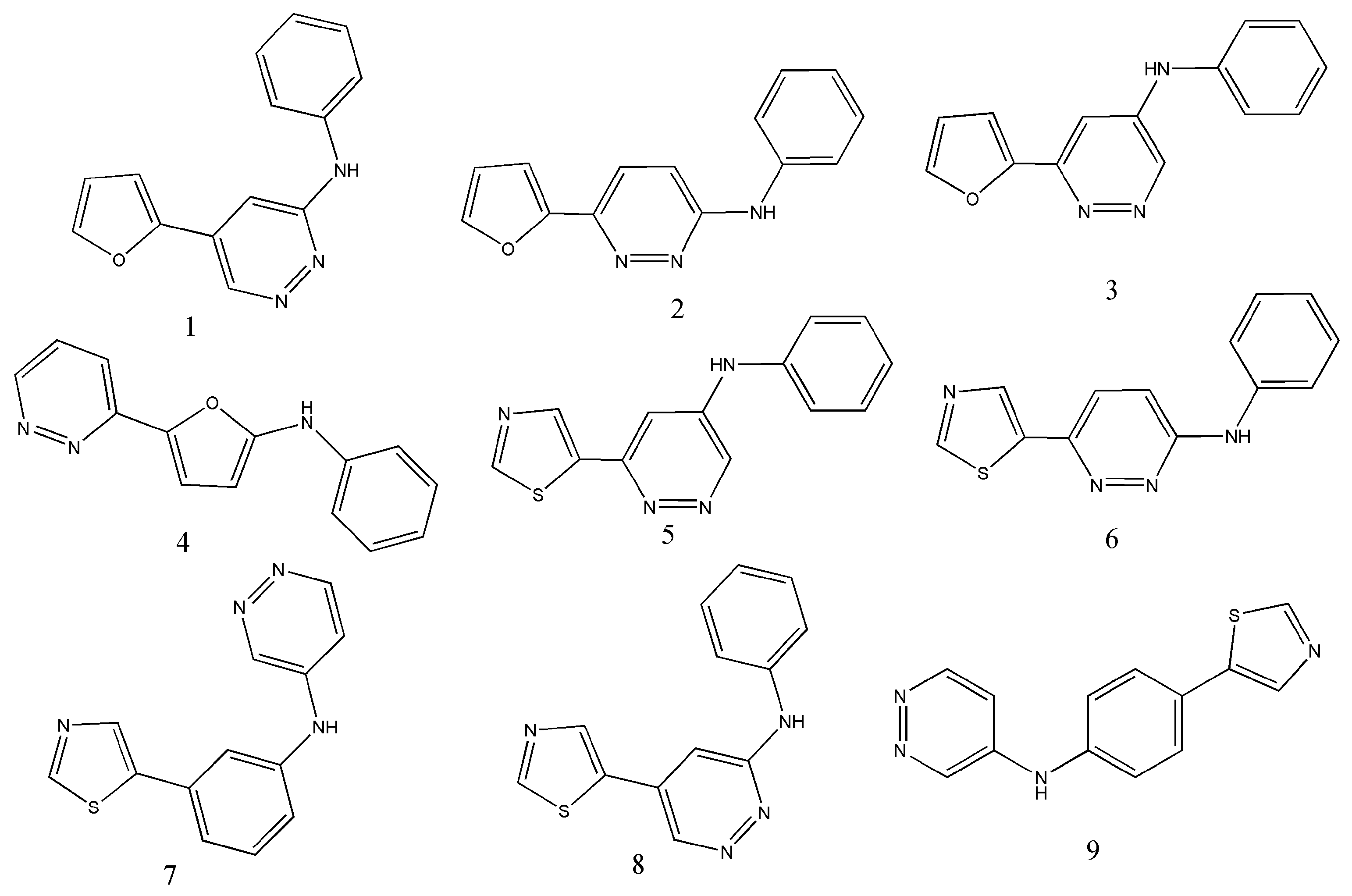
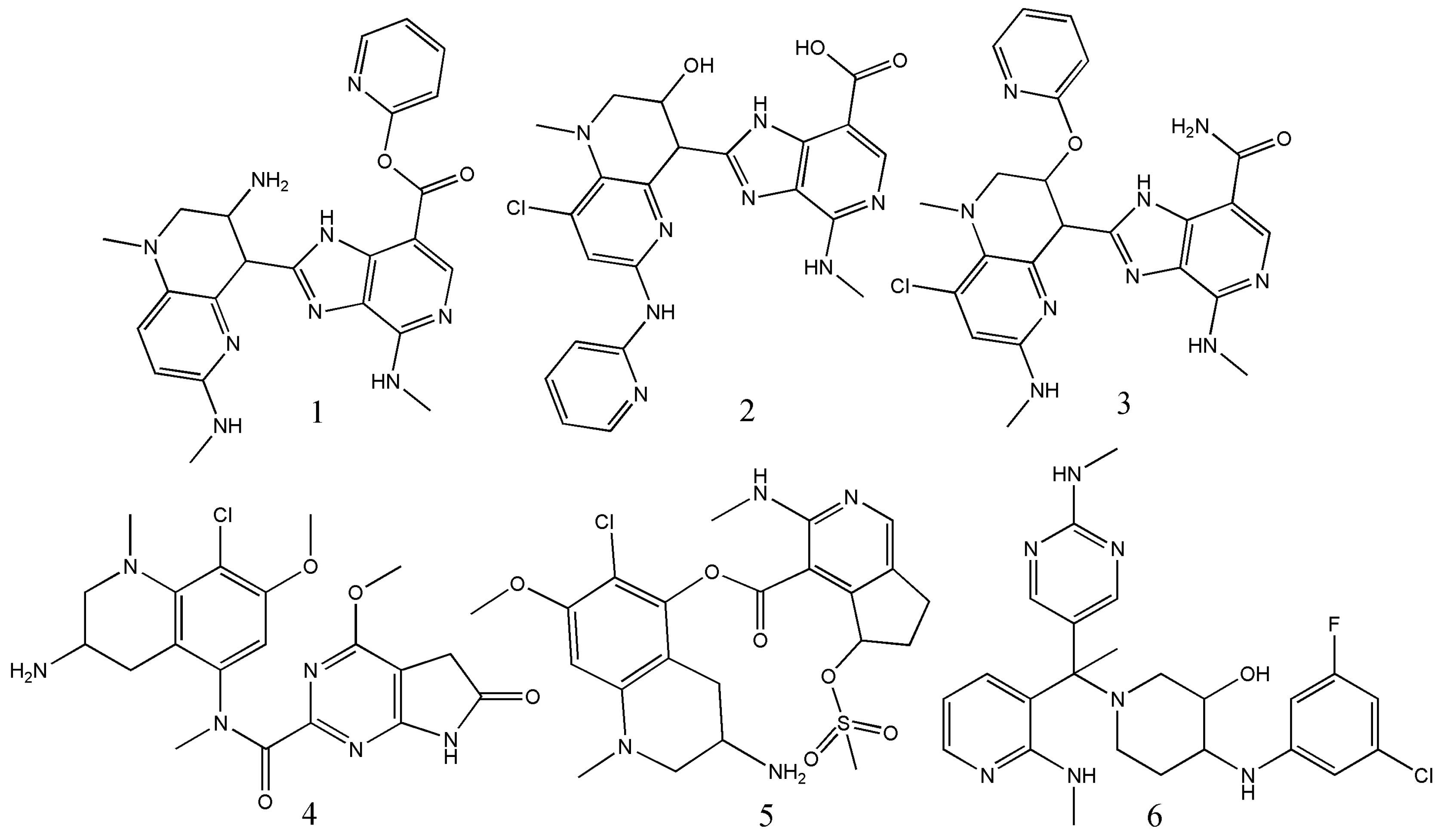
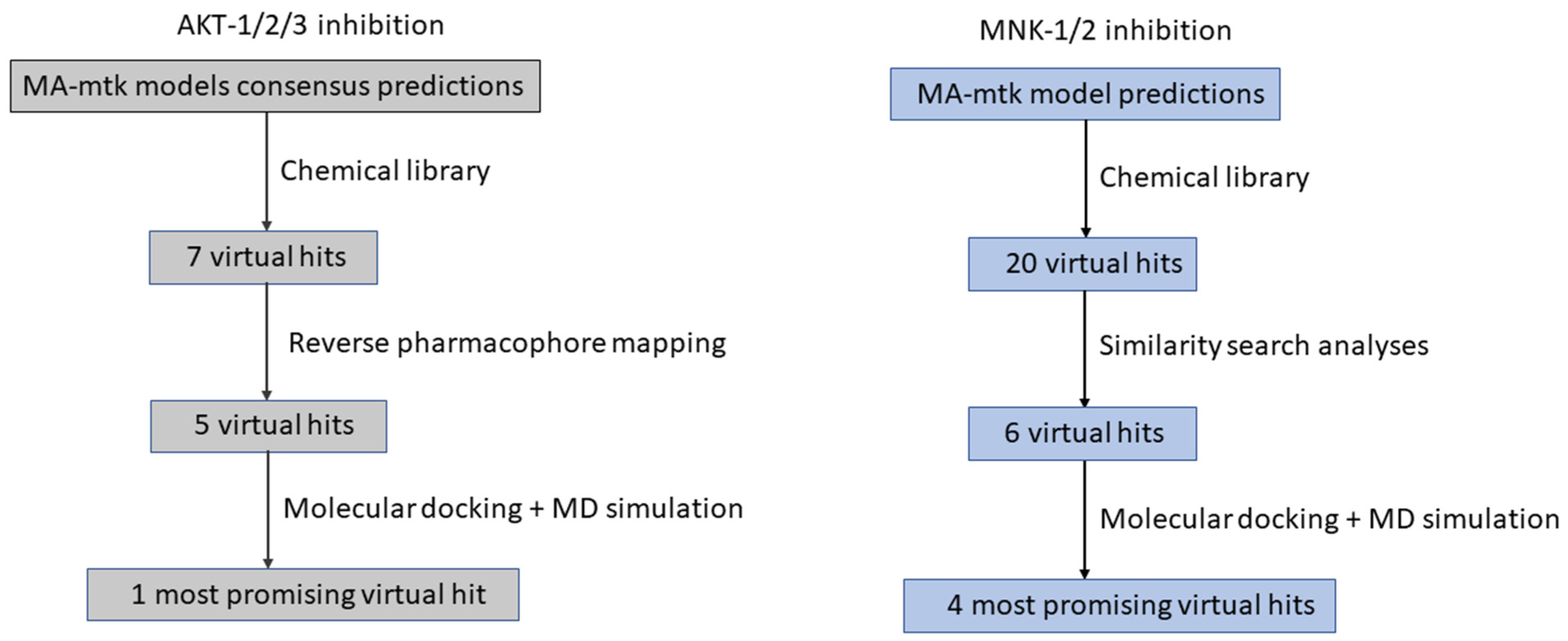


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Operators | Remarks |
|---|---|
| ∆(Di)cj = Di − avg(Di)cj | |
| ∆(Di)cj = pc·[Di − avg(Di)cj] | pc: A probabilistic term |
| ∆(Di)cj = [Di − avg(Di)cj]/(Dimax − Dimin) | Dimax: Maximum value of Di Dimin:Minimum value of Di |
| ∆(Di)cj = [Di − avg(Di)cj]/[(Dimax − Dimin) p(cj)c] | p(cj)c = n(cj)/N (N: Total number of data points in the modeling set) |
| ∆(Di)cj = [Di − avg(Di)cj]/[(Dimax − Dimin) √p(cj)c] | p(cj)c = n(cj)/N |
| ∆(Di)cj = [(Di − avg(Di)cj]/[SD(Di) √p(cj)c] | SD(Di): Standard deviation of Di |
| Feature Selection Tools—Linear Models (LDA) | Machine Learning Tools—Non-Linear Models |
|---|---|
| Fast stepwise (FS) selection | Decision trees (DT) |
| Sequential forward selection (SFS) | Random forests (RF) |
| Genetic algorithm (GA) selection | Gradient boosting (GB) |
| Post-selection similarity search modification (PS3M) | Support vector machines (SVM) |
| k-nearest neighborhood (kNN) | |
| Bernoulli naïve Bayes (NB) | |
| Artificial neural networks (ANN) | |
| Deep neural networks (DNN) |
| Year | Methodology a | No. of Chemicals (Ndp) b | Endpoint Responses c | Bio-Targets d | Acc (%) e | Ref. |
|---|---|---|---|---|---|---|
| 2013 | RBF-ANN | 8560 (10,918) | Anti-Enterococci activities and toxicological profiles | Enterococci strains; Mus musculus; Rattus norvegicus; human lymphocytes | 92.30 | [59] |
| 2013 | RBF-ANN | 6974 (11,576) | Anti-Streptococci activities and toxicological profiles | Streptococci strains; Mus musculus; Rattus norvegicus | 98.08 | [60] |
| 2013 | FS-LDA | 20,863 (34,629) | Anti-Mycobacterial activity and ADMET properties | Mycobacterium spp. strains; proteins; Mus musculus; Rattus norvegicus; Homo sapiens | 94.80 | [51] |
| 2014 | FS-LDA | 23,705 (37,834) | Anti-Escherichia coli activities and ADMET properties | Escherichia coli strains; proteins; laboratory animals (mice and rats); Homo sapiens | 95.85 | [52] |
| 2014 | FS-LDA | 26,945 (48,874) | Anti-cocci activities and ADMET properties | Gram-positive cocci strains; proteins; cell lines; laboratory animals; humans | 92.89 | [61] |
| 2014 | LNN-LDA | 21,582 (43,249) | Anti-HIV-1 activity and epidemiological profile | Viral or human proteins/enzymes (e.g., CC-CKR-5, HIV-1 RT, and HIV-1 PR); laboratory animals; humans | 76.76 | [62] |
| 2015 | FS-LDA | 30,738 (54,682) | Anti-Pseudomonas activities and ADMET properties | Pseudomonas spp. strains; proteins/enzymes; Mus musculus; Rattus norvegicus; Homo sapiens | 90.62 | [63] |
| 2015 | FS-LDA | 22,009 (30,181) | Anti-NOMA activity and ADMET profiles | Bacteria linked to NOMA infections (e.g., Fusobacterium spp., Prevotella spp., Bacillus, etc.); cell lines; laboratory animals; humans | 92.12 | [53] |
| 2016 | FS-LDA | 2123 (3592) | Anti-microbial peptides (AMP) activity and cytotoxicity | Gram-negative bacterial strains; mammalian cell types | 97.40 | [50] |
| 2016 | FS-LDA | 1581 (2488) | AMP activity | Gram-positive bacterial strains | 94.57 | [64] |
| 2017 | FS-LDA | 20,562 (29,682) | Anti-HIV activity and ADMET properties | HIV; proteins/enzymes; cell lines; laboratory animals; humans | 96.26 | [43] |
| 2017 | FS-LDA | 29,863 (40,158) | Anti-Hepatitis C activity and ADMET properties | Hepatitis C; proteins/enzymes; mammalian cells | 95.35 | [31] |
| 2020 | MLP-ANN | 18,798 (21,369) | Anti-malarial activity, cytotoxicity, and pharmacokinetic properties | Plasmodium falciparum strains; proteins; mammalian cells; plasma and liver microsomes | 90.49 | [65] |
| Method | Software/Webserver |
|---|---|
| Pharmacophore mapping | PharmMapper [25,71] |
| Molecular docking | AutoDock [21,25,72], AutoDock Vina [25,73], Molegro Virtual Docker [24,74] |
| Similarity search | SIMSEARCH [21] |
| Molecular dynamics simulations | Amber [21,75], Gromacs [46,76] |
| Homology modeling | SwissModel [25,77] |
| Drug-likeness | SwissADME [21,78] |
| Synthetic accessibility | SwissADME [21,78] |
| Graph-based signature | MycoCSM [22,79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halder, A.K.; Moura, A.S.; Cordeiro, M.N.D.S. Moving Average-Based Multitasking In Silico Classification Modeling: Where Do We Stand and What Is Next? Int. J. Mol. Sci. 2022, 23, 4937. https://doi.org/10.3390/ijms23094937
Halder AK, Moura AS, Cordeiro MNDS. Moving Average-Based Multitasking In Silico Classification Modeling: Where Do We Stand and What Is Next? International Journal of Molecular Sciences. 2022; 23(9):4937. https://doi.org/10.3390/ijms23094937
Chicago/Turabian StyleHalder, Amit Kumar, Ana S. Moura, and Maria Natália D. S. Cordeiro. 2022. "Moving Average-Based Multitasking In Silico Classification Modeling: Where Do We Stand and What Is Next?" International Journal of Molecular Sciences 23, no. 9: 4937. https://doi.org/10.3390/ijms23094937
APA StyleHalder, A. K., Moura, A. S., & Cordeiro, M. N. D. S. (2022). Moving Average-Based Multitasking In Silico Classification Modeling: Where Do We Stand and What Is Next? International Journal of Molecular Sciences, 23(9), 4937. https://doi.org/10.3390/ijms23094937