

Metformin Alleviates Epirubicin-Induced Endothelial Impairment by Restoring Mitochondrial Homeostasis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
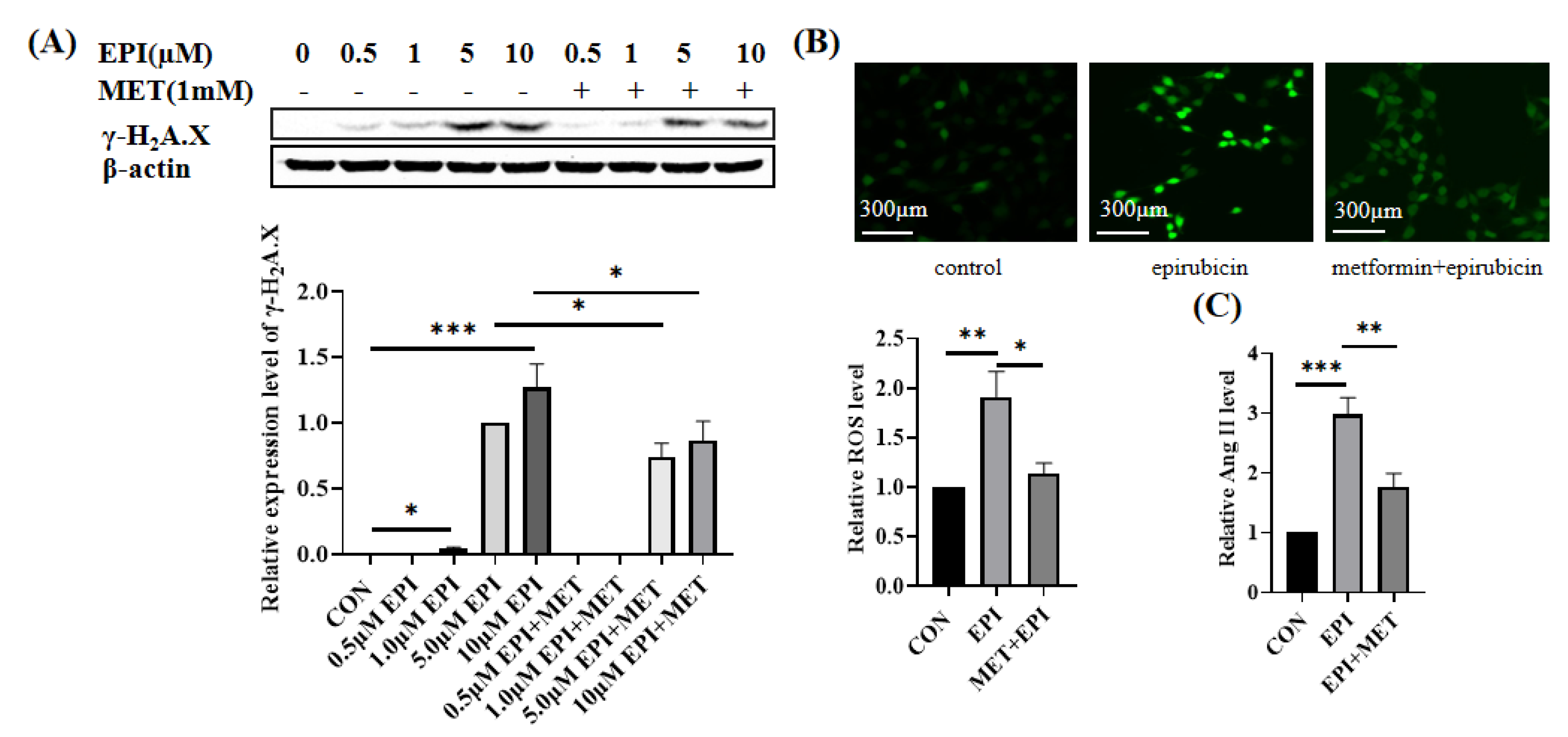
2.1. Metformin Alleviates Epirubicin-Induced HUVEC Cells Dysfunction
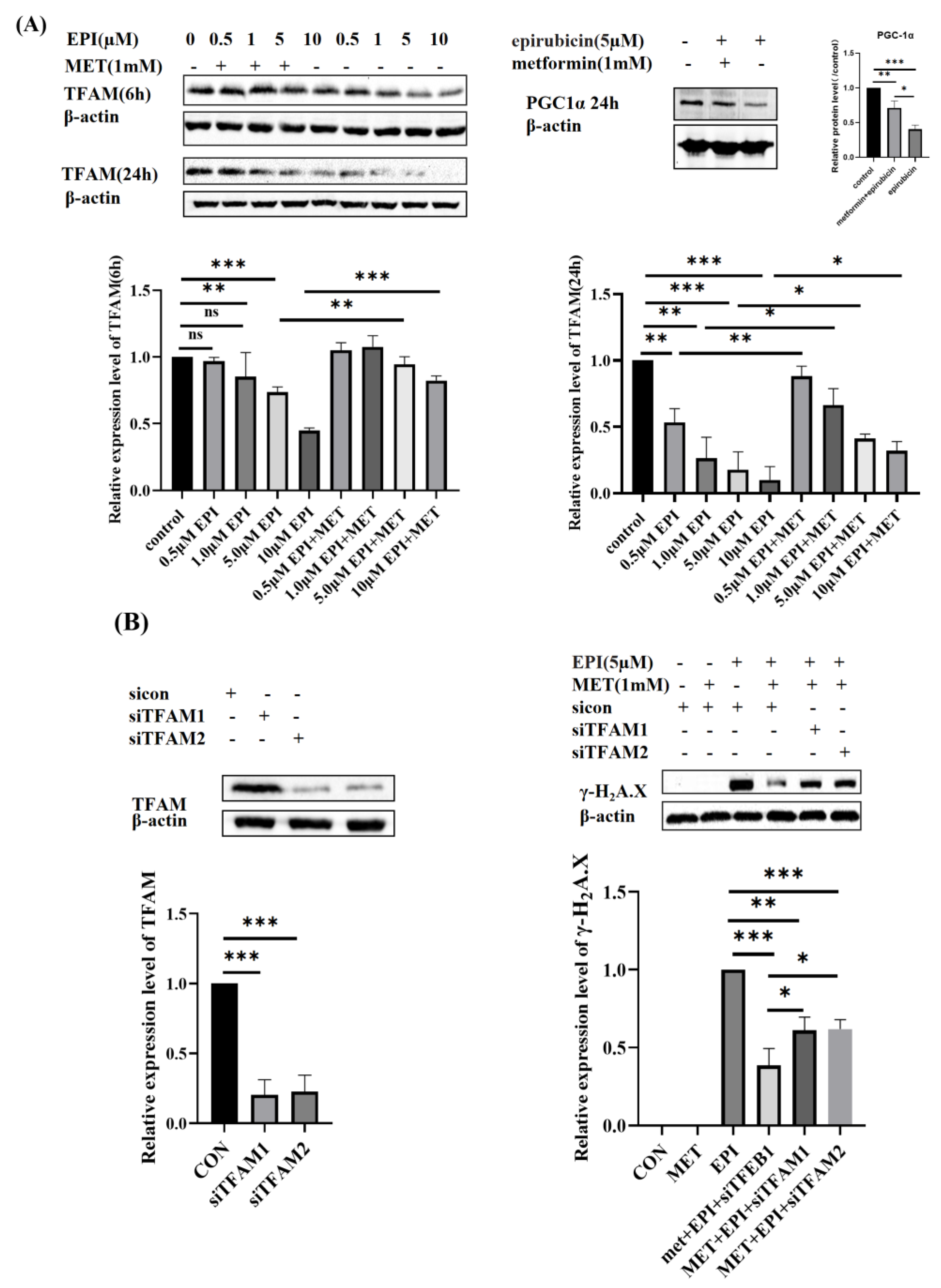
2.2. Metformin Restores Mitochondrial Biogenesis
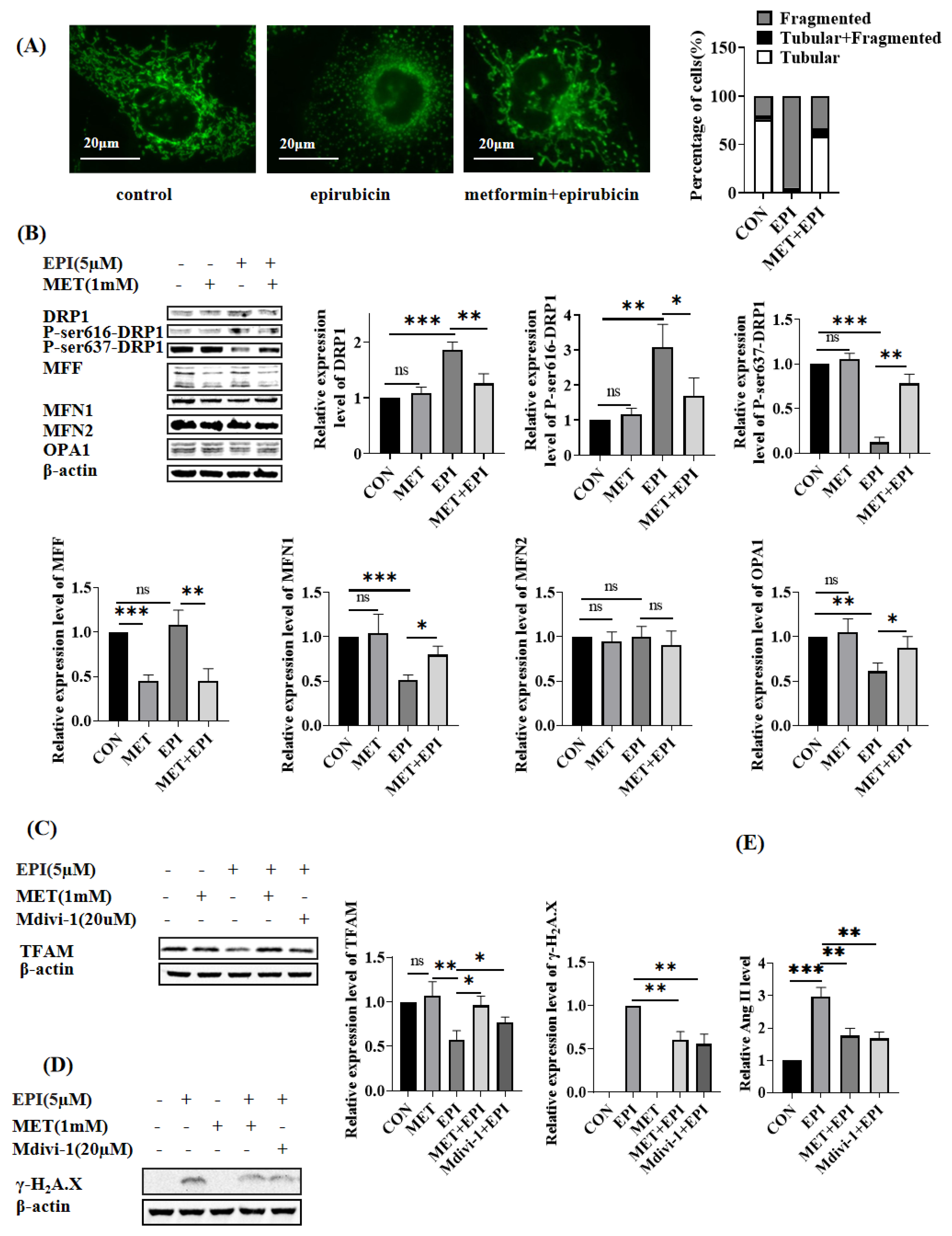
2.3. Metformin Restores Mitochondrial Dynamics
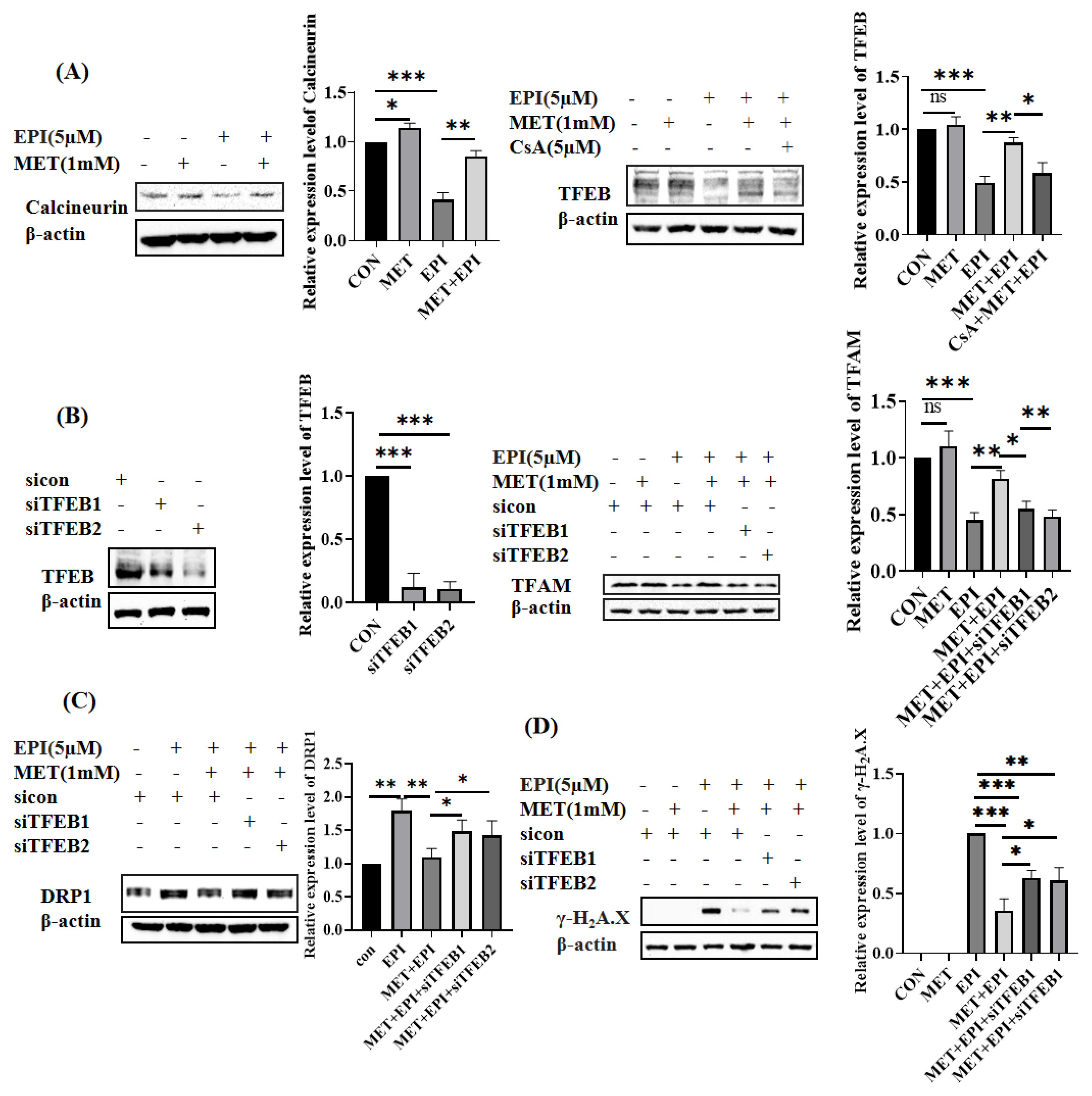
2.4. Metformin Restores Mitochondrial Dynamics via Calcineurin/TFEB Pathway
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Enzyme-Linked Immunosorbent Assays (ELISA)
4.4. Measurement of Cellular Reactive Oxygen Species
4.5. Western Blot
4.6. SiRNA Transfection
4.7. Mitochondrial Morphology Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin—A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Bosch, X.; Rovira, M.; Sitges, M.; Domenech, A.; Ortiz-Perez, J.T.; de Caralt, T.M.; Morales-Ruiz, M.; Perea, R.J.; Monzo, M.; Esteve, J. Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: The OVERCOME trial (preventiOn of left Ventricular dysfunction with Enalapril and caRvedilol in patients submitted to intensive ChemOtherapy for the treatment of Malignant hEmopathies). J. Am. Coll. Cardiol. 2013, 61, 2355–2362. [Google Scholar] [PubMed]
- Heck, S.L.; Mecinaj, A.; Ree, A.H.; Hoffmann, P.; Schulz-Menger, J.; Fagerland, M.W.; Gravdehaug, B.; Røsjø, H.; Steine, K.; Geisler, J.; et al. Prevention of Cardiac Dysfunction During Adjuvant Breast Cancer Therapy (PRADA): Extended Follow-Up of a 2 × 2 Factorial, Randomized, Placebo-Controlled, Double-Blind Clinical Trial of Candesartan and Metoprolol. Circulation 2021, 143, 2431–2440. [Google Scholar] [CrossRef] [PubMed]
- Gujral, D.M.; Lloyd, G.; Bhattacharyya, S. Effect of prophylactic betablocker or ACE inhibitor on cardiac dysfunction & heart failure during anthracycline chemotherapy ± trastuzumab. Breast 2018, 37, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Vejpongsa, P.; Yeh, E.T. Prevention of anthracycline-induced cardiotoxicity: Challenges and opportunities. J. Am. Coll. Cardiol. 2014, 64, 938–945. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, X.; Zhang, N.; Wei, W.Y.; Li, L.L.; Ma, Z.G.; Tang, Q.Z. Osteocrin attenuates inflammation, oxidative stress, apoptosis, and cardiac dysfunction in doxorubicin-induced cardiotoxicity. Clin. Transl. Med. 2020, 10, e124. [Google Scholar] [CrossRef]
- Luu, A.Z.; Chowdhury, B.; Al-Omran, M.; Teoh, H.; Hess, D.A.; Verma, S. Role of Endothelium in Doxorubicin-Induced Cardiomyopathy. JACC Basic Transl. Sci. 2018, 3, 861–870. [Google Scholar] [CrossRef]
- Sawyer, D.B. Anthracycline-Induced Vascular Dysfunction: Is MitoQ the Answer? JACC CardioOncol. 2020, 2, 489–490. [Google Scholar] [CrossRef]
- Tao, R.H.; Kobayashi, M.; Yang, Y.; Kleinerman, E.S. Exercise Inhibits Doxorubicin-Induced Damage to Cardiac Vessels and Activation of Hippo/YAP-Mediated Apoptosis. Cancers 2021, 13, 2740. [Google Scholar] [CrossRef]
- Bar-Joseph, H.; Ben-Aharon, I.; Tzabari, M.; Tsarfaty, G.; Stemmer, S.M.; Shalgi, R. In vivo bioimaging as a novel strategy to detect doxorubicin-induced damage to gonadal blood vessels. PLoS ONE 2011, 6, e23492. [Google Scholar] [CrossRef]
- Khanna, A.; Pequeno, P.; Gupta, S.; Thavendiranathan, P.; Lee, D.S.; Abdel-Qadir, H.; Nathan, P.C. Increased Risk of All Cardiovascular Disease Subtypes Among Childhood Cancer Survivors: Population-Based Matched Cohort Study. Circulation 2019, 140, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Rasanen, M.; Degerman, J.; Nissinen, T.A.; Miinalainen, I.; Kerkela, R.; Siltanen, A.; Backman, J.T.; Mervaala, E.; Hulmi, J.J.; Kivela, R.; et al. VEGF-B gene therapy inhibits doxorubicin-induced cardiotoxicity by endothelial protection. Proc. Natl. Acad. Sci. USA 2016, 113, 13144–13149. [Google Scholar] [CrossRef] [PubMed]
- Todorova, V.K.; Hsu, P.C.; Wei, J.Y.; Lopez-Candales, A.; Chen, J.Z.N.; Su, L.J.; Makhoul, I. Biomarkers of inflammation, hypercoagulability and endothelial injury predict early asymptomatic doxorubicin-induced cardiotoxicity in breast cancer patients. Am. J. Cancer Res. 2020, 10, 2933–2945. [Google Scholar] [PubMed]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Munoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef] [PubMed]
- Stohr, W.; Paulides, M.; Brecht, I.; Kremers, A.; Treuner, J.; Langer, T.; Beck, J.D. Comparison of epirubicin and doxorubicin cardiotoxicity in children and adolescents treated within the German Cooperative Soft Tissue Sarcoma Study (CWS). J. Cancer Res. Clin. 2006, 132, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Egashira, N.; Bando, A.; Nishime, Y.; Tonogai, Y.; Imuta, M.; Yano, T.; Oishi, R. Activation of p38 MAPK by oxidative stress underlying epirubicin-induced vascular endothelial cell injury. Free Radic Biol. Med. 2012, 52, 1285–1293. [Google Scholar] [CrossRef]
- Sarvazyan, N. Visualization of doxorubicin-induced oxidative stress in isolated cardiac myocytes. Am. J. Physiol. Heart C 1996, 271, H2079–H2085. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, X.; Lee, C.P.; Chua, B.H.; Ho, Y.S. Attenuation of doxorubicin-induced contractile and mitochondrial dysfunction in mouse heart by cellular glutathione peroxidase. Free Radic Biol. Med. 2006, 41, 46–55. [Google Scholar] [CrossRef]
- Yen, H.C.; Oberley, T.D.; Gairola, C.G.; Szweda, L.I.; St Clair, D.K. Manganese superoxide dismutase protects mitochondrial complex I against adriamycin-induced cardiomyopathy in transgenic mice. Arch. Biochem. Biophys. 1999, 362, 59–66. [Google Scholar] [CrossRef]
- Kelleni, M.T.; Amin, E.F.; Abdelrahman, A.M. Effect of Metformin and Sitagliptin on Doxorubicin-Induced Cardiotoxicity in Rats: Impact of Oxidative Stress, Inflammation, and Apoptosis. J. Toxicol. 2015, 2015, 424813. [Google Scholar] [CrossRef]
- Asensio-Lopez, M.C.; Lax, A.; Pascual-Figal, D.A.; Valdes, M.; Sanchez-Mas, J. Metformin protects against doxorubicin-induced cardiotoxicity: Involvement of the adiponectin cardiac system. Free Radic Biol. Med. 2011, 51, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Arinno, A.; Maneechote, C.; Khuanjing, T.; Ongnok, B.; Prathumsap, N.; Chunchai, T.; Arunsak, B.; Kerdphoo, S.; Shinlapawittayatorn, K.; Chattipakorn, S.C.; et al. Cardioprotective effects of melatonin and metformin against doxorubicin-induced cardiotoxicity in rats are through preserving mitochondrial function and dynamics. Biochem. Pharmacol. 2021, 192, 114743. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yuan, J.; Wang, Q.; Lei, T.; Shen, X.; Cui, B.; Zhang, F.; Ding, W.; Lu, Z. Metformin protects against PM2.5-induced lung injury and cardiac dysfunction independent of AMP-activated protein kinase alpha2. Redox Biol. 2020, 28, 101345. [Google Scholar] [CrossRef] [PubMed]
- Soberanes, S.; Misharin, A.V.; Jairaman, A.; Morales-Nebreda, L.; McQuattie-Pimentel, A.C.; Cho, T.; Hamanaka, R.B.; Meliton, A.Y.; Reyfman, P.A.; Walter, J.M.; et al. Metformin Targets Mitochondrial Electron Transport to Reduce Air-Pollution-Induced Thrombosis. Cell Metab. 2019, 29, 335–347.e5. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Maruthur, N.M.; Tseng, E.; Hutfless, S.; Wilson, L.M.; Suarez-Cuervo, C.; Berger, Z.; Chu, Y.; Iyoha, E.; Segal, J.B.; Bolen, S. Diabetes Medications as Monotherapy or Metformin-Based Combination Therapy for Type 2 Diabetes: A Systematic Review and Meta-analysis. Ann. Intern. Med. 2016, 164, 740–751. [Google Scholar] [CrossRef]
- Wallace, K.B. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc. Toxicol. 2007, 7, 101–107. [Google Scholar] [CrossRef]
- Chaiswing, L.; Cole, M.P.; Ittarat, W.; Szweda, L.I.; St Clair, D.K.; Oberley, T.D. Manganese superoxide dismutase and inducible nitric oxide synthase modify early oxidative events in acute Adriamycin-induced mitochondrial toxicity. Mol. Cancer Ther. 2005, 4, 1056–1064. [Google Scholar] [CrossRef]
- Zhou, S.Y.; Starkov, A.; Froberg, M.K.; Leino, R.L.; Wallace, K.B. Cumulative and irreversible cardiac mitochondrial dysfunction induced by doxorubicin. Cancer Res. 2001, 61, 771–777. [Google Scholar]
- Clayton, Z.S.; Brunt, V.E.; Hutton, D.A.; Van Dongen, N.S.; D’Alessandro, A.; Reisz, J.A.; Ziemba, B.P.; Seals, D.R. Doxorubicin-Induced Oxidative Stress and Endothelial Dysfunction in Conduit Arteries Is Prevented by Mitochondrial-Specific Antioxidant Treatment. JACC CardioOncol. 2020, 2, 475–488. [Google Scholar] [CrossRef]
- Rabbani, N.; Chittari, M.V.; Bodmer, C.W.; Zehnder, D.; Ceriello, A.; Thornalley, P.J. Increased glycation and oxidative damage to apolipoprotein B100 of LDL cholesterol in patients with type 2 diabetes and effect of metformin. Diabetes 2010, 59, 1038–1045. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mohan, M.; Al-Talabany, S.; McKinnie, A.; Mordi, I.R.; Singh, J.S.S.; Gandy, S.J.; Baig, F.; Hussain, M.S.; Bhalraam, U.; Khan, F.; et al. A randomized controlled trial of metformin on left ventricular hypertrophy in patients with coronary artery disease without diabetes: The MET-REMODEL trial. Eur. Heart J. 2019, 40, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.; Mauro, M.; Williams, Z. Direct toxicity of insulin on the human placenta and protection by metformin. Fertil. Steril. 2019, 111, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Kudabayeva, K.; Kosmuratova, R.; Bazargaliyev, Y.; Sartayeva, A.; Kereyeva, N. Effects of metformin on lymphocyte DNA damage in obese individuals among Kazakh population. Diabetes Metab. Syndr. 2022, 16, 102569. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Bai, N.; Zheng, M.; Wang, Y.; Wang, Y.; Zhang, L.; Li, J.; Li, G.; Zhao, H.; Liu, G.; et al. Tranilast prevents doxorubicin-induced myocardial hypertrophy and angiotensin II synthesis in rats. Life Sci. 2021, 267, 118984. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Kong, J.; Wang, Y.L.; Li, J.L.; Hei, N.H.; Cao, X.R.; Yang, J.J.; Yan, W.J.; Liang, W.J.; Dai, H.Y.; et al. Angiotensin-converting enzyme 2 overexpression protects against doxorubicin-induced cardiomyopathy by multiple mechanisms in rats. Oncotarget 2017, 8, 24548–24563. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Y.; Heims-Waldron, D.; Bezzerides, V.; Guatimosim, S.; Guo, Y.; Gu, F.; Zhou, P.; Lin, Z.; Ma, Q.; et al. Mitochondrial Cardiomyopathy Caused by Elevated Reactive Oxygen Species and Impaired Cardiomyocyte Proliferation. Circ. Res. 2018, 122, 74–87. [Google Scholar] [CrossRef]
- Koh, J.H.; Johnson, M.L.; Dasari, S.; LeBrasseur, N.K.; Vuckovic, I.; Henderson, G.C.; Cooper, S.A.; Manjunatha, S.; Ruegsegger, G.N.; Shulman, G.I.; et al. TFAM Enhances Fat Oxidation and Attenuates High-Fat Diet-Induced Insulin Resistance in Skeletal Muscle. Diabetes 2019, 68, 1552–1564. [Google Scholar] [CrossRef]
- Yin, J.; Guo, J.; Zhang, Q.; Cui, L.; Zhang, L.; Zhang, T.; Zhao, J.; Li, J.; Middleton, A.; Carmichael, P.L.; et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol. In Vitro 2018, 51, 1–10. [Google Scholar] [CrossRef]
- Liu, D.; Ma, Z.; Di, S.; Yang, Y.; Yang, J.; Xu, L.; Reiter, R.J.; Qiao, S.; Yuan, J. AMPK/PGC1alpha activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic Biol. Med. 2018, 129, 59–72. [Google Scholar] [CrossRef]
- Machado, I.F.; Teodoro, J.S.; Castela, A.C.; Palmeira, C.M.; Rolo, A.P. miR-378a-3p Participates in Metformin’s Mechanism of Action on C2C12 Cells under Hyperglycemia. Int. J. Mol. Sci. 2021, 22, 541. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Teague, A.M.; Tryggestad, J.B.; Jensen, M.E.; Chernausek, S.D. Role of metformin in epigenetic regulation of placental mitochondrial biogenesis in maternal diabetes. Sci. Rep. 2020, 10, 8314. [Google Scholar] [CrossRef] [PubMed]
- Lugus, J.J.; Ngoh, G.A.; Bachschmid, M.M.; Walsh, K. Mitofusins are required for angiogenic function and modulate different signaling pathways in cultured endothelial cells. J. Mol. Cell Cardiol. 2011, 51, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.Q.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.L.; Zhang, M.; Torres, G.; Wu, S.N.; Ouyang, C.H.; Xie, Z.L.; Zou, M.H. Metformin Suppresses Diabetes-Accelerated Atherosclerosis via the Inhibition of Drp1-Mediated Mitochondrial Fission. Diabetes 2017, 66, 193–205. [Google Scholar] [CrossRef]
- Wang, W.J.; Wang, Y.; Long, J.Y.; Wang, J.R.; Haudek, S.B.; Overbeek, P.; Chang, B.H.J.; Schumacker, P.T.; Danesh, F.R. Mitochondrial Fission Triggered by Hyperglycemia Is Mediated by ROCK1 Activation in Podocytes and Endothelial Cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef]
- Robert, P.; Nguyen, P.M.C.; Richard, A.; Grenier, C.; Chevrollier, A.; Munier, M.; Grimaud, L.; Proux, C.; Champin, T.; Lelievre, E.; et al. Protective role of the mitochondrial fusion protein OPA1 in hypertension. FASEB J. 2021, 35, e21678. [Google Scholar] [CrossRef]
- Li, L.; Li, J.H.; Wang, Q.L.; Zhao, X.; Yang, D.L.; Niu, L.; Yang, Y.Z.; Zheng, X.X.; Hu, L.M.; Li, Y.H. Shenmai Injection Protects Against Doxorubicin-Induced Cardiotoxicity via Maintaining Mitochondrial Homeostasis. Front. Pharmacol. 2020, 11, 815. [Google Scholar] [CrossRef]
- Catanzaro, M.P.; Weiner, A.; Kaminaris, A.; Li, C.R.; Cai, F.; Zhao, F.Y.; Kobayashi, S.; Kobayashi, T.; Huang, Y.; Sesaki, H.; et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J. 2019, 33, 11096–11108. [Google Scholar] [CrossRef]
- Maneechote, C.; Khuanjing, T.; Ongnok, B.; Arinno, A.; Prathumsap, N.; Chunchai, T.; Arunsak, B.; Nawara, W.; Chattipakorn, S.C.; Chattipakorn, N. Promoting mitochondrial fusion in doxorubicin-induced cardiotoxicity: A novel therapeutic target for cardioprotection. Clin. Sci. (Lond.) 2022, 136, 841–860. [Google Scholar] [CrossRef]
- Breitzig, M.T.; Alleyn, M.D.; Lockey, R.F.; Kolliputi, N. A mitochondrial delicacy: Dynamin-related protein 1 and mitochondrial dynamics. Am. J. Physiol. Cell Physiol. 2018, 315, C80–C90. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, X.; Jiang, A.; Li, X.; Chang, W.; Chen, J.; Ye, F. Metformin alleviates lead-induced mitochondrial fragmentation via AMPK/Nrf2 activation in SH-SY5Y cells. Redox Biol. 2020, 36, 101626. [Google Scholar] [CrossRef] [PubMed]
- Li, A.Y.; Zhang, S.H.; Li, J.; Liu, K.; Huang, F.; Liu, B.L. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef] [PubMed]
- de Maranon, A.M.; Canet, F.; Abad-Jimenez, Z.; Jover, A.; Morillas, C.; Rocha, M.; Victor, V.M. Does Metformin Modulate Mitochondrial Dynamics and Function in Type 2 Diabetic Patients? Antioxid Redox Sign. 2021, 35, 377–385. [Google Scholar] [CrossRef]
- Martina, J.A.; Diab, H.I.; Brady, O.A.; Puertollano, R. TFEB and TFE3 are novel components of the integrated stress response. Embo J. 2016, 35, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Pastore, N.; Brady, O.A.; Diab, H.I.; Martina, J.A.; Sun, L.; Huynh, T.; Lim, J.A.; Zare, H.; Raben, N.; Ballabio, A.; et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 2016, 12, 1240–1258. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.C.; Liu, H.Y.; Murphy, J.T.; Foyil, S.R.; Godar, R.J.; Abuirqeba, H.; Weinheimer, C.J.; Barger, P.M.; Diwan, A. Regulation of the Transcription Factor EB-PGC1 alpha Axis by Beclin-1 Controls Mitochondrial Quality and Cardiomyocyte Death under Stress. Mol. Cell Biol. 2015, 35, 956–976. [Google Scholar] [CrossRef]
- Wang, S.J.; Chen, Y.S.; Li, X.Y.; Zhang, W.H.; Liu, Z.J.; Wu, M.; Pan, Q.J.; Liu, H.F. Emerging role of transcription factor EB in mitochondrial quality control. Biomed. Pharmacother. 2020, 128, 110271. [Google Scholar] [CrossRef]
- Lu, H.C.; Fan, Y.B.; Qiao, C.Z.; Liang, W.Y.; Hu, W.T.; Zhu, T.Q.; Zhang, J.F.; Chen, Y.E. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci. Signal. 2017, 10, eaah4214. [Google Scholar] [CrossRef]
- Song, W.; Zhang, C.L.; Gou, L.; He, L.; Gong, Y.Y.; Qu, D.; Zhao, L.; Jin, N.; Chan, T.F.; Wang, L.; et al. Endothelial TFEB (Transcription Factor EB) Restrains IKK (IkappaB Kinase)-p65 Pathway to Attenuate Vascular Inflammation in Diabetic db/db Mice. Arterioscler Thromb. Vasc. Biol. 2019, 39, 719–730. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, J.N.; Zhou, M.; Zhang, Y.; Liu, Y.; Hou, P.F.; Zeng, X.L.; Yi, L.; Mi, M.T. Resveratrol attenuates endothelial oxidative injury by inducing autophagy via the activation of transcription factor EB. Nutr. Metab. 2019, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Doronzo, G.; Astanina, E.; Cora, D.; Chiabotto, G.; Comunanza, V.; Noghero, A.; Neri, F.; Puliafito, A.; Primo, L.; Spampanato, C.; et al. TFEB controls vascular development by regulating the proliferation of endothelial cells. EMBO J. 2019, 38, e98250. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Lu, H.; Liang, W.; Garcia-Barrio, M.T.; Guo, Y.; Zhang, J.; Zhu, T.; Hao, Y.; Zhang, J.; Chen, Y.E. Endothelial TFEB (Transcription Factor EB) Positively Regulates Postischemic Angiogenesis. Circ. Res. 2018, 122, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.J.; Trivedi, P.C.; Yeung, P.; Kienesberger, P.C.; Pulinilkunnil, T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem. J. 2016, 473, 3769–3789. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Q.; Li, W.; Zhang, Q.; Jiang, Y.; Guo, D.; Sun, X.; Lu, W.; Li, C.; Wang, Y. TFEB-NF-kappaB inflammatory signaling axis: A novel therapeutic pathway of Dihydrotanshinone I in doxorubicin-induced cardiotoxicity. J. Exp. Clin. Cancer Res. 2020, 39, 93. [Google Scholar] [CrossRef] [PubMed]
- Mansueto, G.; Armani, A.; Viscomi, C.; D’Orsi, L.; De Cegli, R.; Polishchuk, E.V.; Lamperti, C.; Di Meo, I.; Romanello, V.; Marchet, S.; et al. Transcription Factor EB Controls Metabolic Flexibility during Exercise. Cell Metab. 2017, 25, 182–196. [Google Scholar] [CrossRef]
- Kim, H.J.; Joe, Y.; Rah, S.Y.; Kim, S.K.; Park, S.U.; Park, J.; Kim, J.; Ryu, J.; Cho, G.J.; Surh, Y.J.; et al. Carbon monoxide-induced TFEB nuclear translocation enhances mitophagy/mitochondrial biogenesis in hepatocytes and ameliorates inflammatory liver injury. Cell Death Dis. 2018, 9, 1060. [Google Scholar] [CrossRef]
- Santin, Y.; Sicard, P.; Vigneron, F.; Guilbeau-Frugier, C.; Dutaur, M.; Lairez, O.; Couderc, B.; Manni, D.; Korolchuk, V.I.; Lezoualc’h, F.; et al. Oxidative Stress by Monoamine Oxidase-A Impairs Transcription Factor EB Activation and Autophagosome Clearance, Leading to Cardiomyocyte Necrosis and Heart Failure. Antioxid Redox Sign. 2016, 25, 10–27. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Pan, B.; Li, J.; Parajuli, N.; Tian, Z.W.; Wu, P.L.; Lewno, M.T.; Zou, J.Q.; Wang, W.J.; Bedford, L.; Mayer, R.J.; et al. The Calcineurin-TFEB-p62 Pathway Mediates the Activation of Cardiac Macroautophagy by Proteasomal Malfunction. Circ. Res. 2020, 127, 502–518. [Google Scholar] [CrossRef]
- Chowdhury, A.R.; Zielonka, J.; Kalyanaraman, B.; Hartley, R.C.; Murphy, M.P.; Avadhani, N.G. Mitochondria-targeted paraquat and metformin mediate ROS production to induce multiple pathways of retrograde signaling: A dose-dependent phenomenon. Redox Biol. 2020, 36, 101606. [Google Scholar] [CrossRef] [PubMed]
- Eraky, S.M.; Ramadan, N.M. Effects of omega-3 fatty acids and metformin combination on diabetic cardiomyopathy in rats through autophagic pathway. J. Nutr. Biochem. 2021, 97, 108789. [Google Scholar] [CrossRef] [PubMed]
- Kheirandish, M.; Mahboobi, H.; Yazdanparast, M.; Kamal, W.; Kamal, M.A. Anti-cancer Effects of Metformin: Recent Evidences for its Role in Prevention and Treatment of Cancer. Curr. Drug Metab. 2018, 19, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Tang, N.; Jiang, S.; Bai, Y.; Guan, C.; Zhang, W.; Fan, S.; Huang, Y.; Lin, H.; Ying, Y. The Chemosensitizing Role of Metformin in Anti-Cancer Therapy. Anticancer Agents Med. Chem. 2021, 21, 949–962. [Google Scholar] [CrossRef]
- Coronel-Hernandez, J.; Salgado-Garcia, R.; Cantu-De Leon, D.; Jacobo-Herrera, N.; Millan-Catalan, O.; Delgado-Waldo, I.; Campos-Parra, A.D.; Rodriguez-Morales, M.; Delgado-Buenrostro, N.L.; Perez-Plasencia, C. Combination of Metformin, Sodium Oxamate and Doxorubicin Induces Apoptosis and Autophagy in Colorectal Cancer Cells via Downregulation HIF-1 alpha. Front. Oncol. 2021, 11, 594200. [Google Scholar] [CrossRef]
- Pateliya, B.; Burade, V.; Goswami, S. Enhanced antitumor activity of doxorubicin by naringenin and metformin in breast carcinoma: An experimental study. Naunyn Schmiedebergs Arch. Pharmacol. 2021, 394, 1949–1961. [Google Scholar] [CrossRef]
- Li, Y.; Wang, M.; Zhi, P.; You, J.; Gao, J.Q. Metformin synergistically suppress tumor growth with doxorubicin and reverse drug resistance by inhibiting the expression and function of P-glycoprotein in MCF7/ADR cells and xenograft models. Oncotarget 2018, 9, 2158–2174. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Q.; Jia, H.; Cheng, S.; Wang, Y.; Wang, J. Metformin Alleviates Epirubicin-Induced Endothelial Impairment by Restoring Mitochondrial Homeostasis. Int. J. Mol. Sci. 2023, 24, 343. https://doi.org/10.3390/ijms24010343
Sun Q, Jia H, Cheng S, Wang Y, Wang J. Metformin Alleviates Epirubicin-Induced Endothelial Impairment by Restoring Mitochondrial Homeostasis. International Journal of Molecular Sciences. 2023; 24(1):343. https://doi.org/10.3390/ijms24010343
Chicago/Turabian StyleSun, Qi, Huiling Jia, Shuo Cheng, Yujuan Wang, and Jun Wang. 2023. "Metformin Alleviates Epirubicin-Induced Endothelial Impairment by Restoring Mitochondrial Homeostasis" International Journal of Molecular Sciences 24, no. 1: 343. https://doi.org/10.3390/ijms24010343
APA StyleSun, Q., Jia, H., Cheng, S., Wang, Y., & Wang, J. (2023). Metformin Alleviates Epirubicin-Induced Endothelial Impairment by Restoring Mitochondrial Homeostasis. International Journal of Molecular Sciences, 24(1), 343. https://doi.org/10.3390/ijms24010343