_Putnam.png)
HPLC Method Validation for the Estimation of Lignocaine HCl, Ketoprofen and Hydrocortisone: Greenness Analysis Using AGREE Score

 , , , and
, , , and 
Abstract
1. Introduction:
2. Results and Discussion
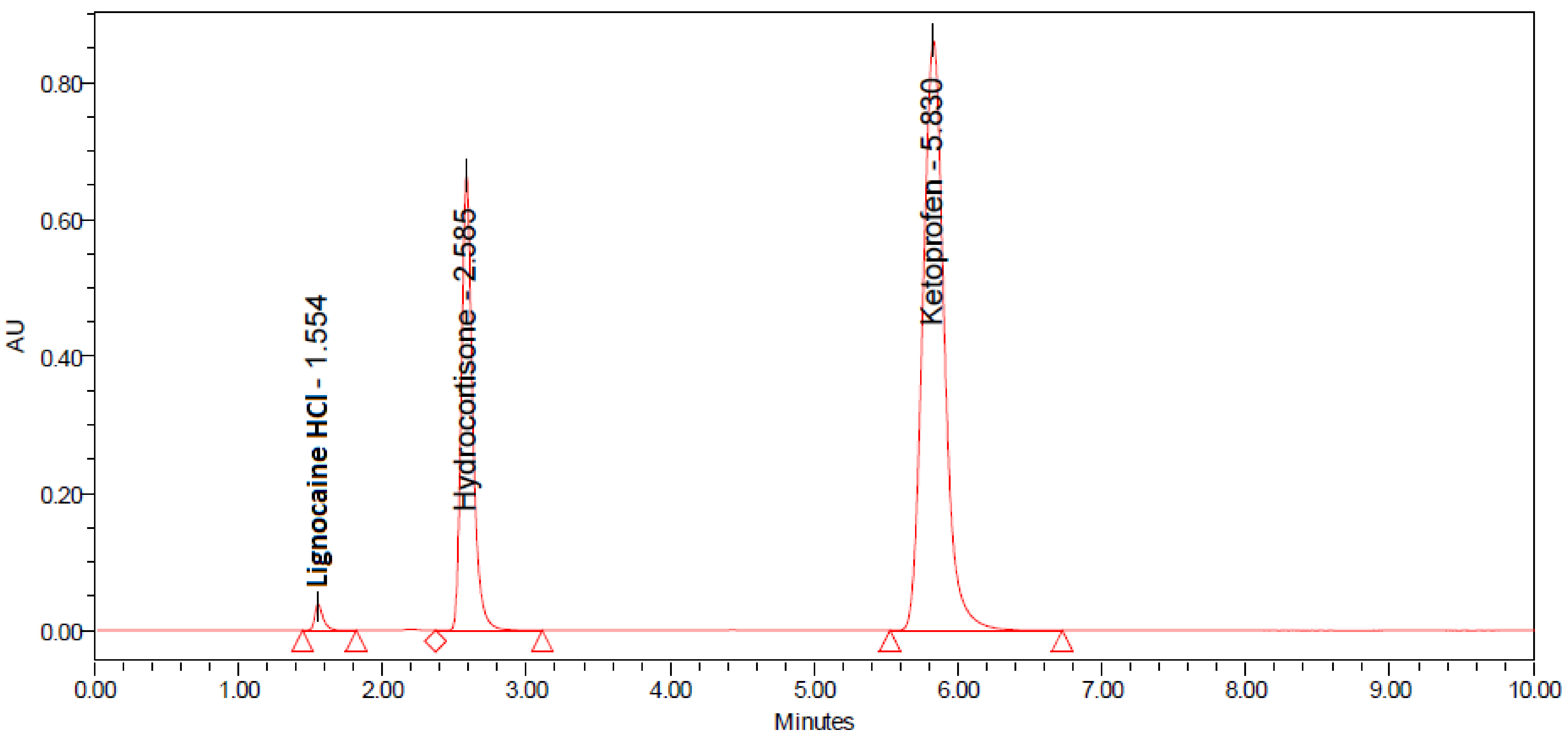
2.1. Development and Optimization of Method
2.2. System Suitability
2.3. Method Validation
2.3.1. Linearity
2.3.2. Accuracy
2.3.3. Precision
2.3.4. Robustness
2.3.5. Specificity
2.3.6. LOD and LOQ
2.4. Greenness Evaluation Using AGREE® Score
2.5. Application to Transdermal Gels
3. Materials and Methods
3.1. Materials
3.2. HPLC Instrumental Conditions
3.3. Preparation of Mobile Phase and Standard Solution
3.4. System Suitability Parameters
3.5. ICH Validation Parameters
3.5.1. Linearity
3.5.2. Accuracy
3.5.3. Precision
3.5.4. Robustness
3.5.5. LOD and LOQ
3.5.6. Specificity
3.5.7. Method Application to Transdermal Gels
3.5.8. Statistical Analysis
3.5.9. Greenness Estimation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Byrne, J.; Velasco-Torrijos, T.; Reinhardt, R. An RP-HPLC method for the stability-indicating analysis of impurities of both fusidic acid and betamethasone-17-valerate in a semi-solid pharmaceutical dosage form. J. Chromatogr. Sci. 2015, 53, 1498–1503. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nawaz, M.; Arayne, M.S.; Sultana, N.; Haider, A.; Hisaindee, S. Simultaneous determination of fusidic acid and steroids from bulk drugs and human plasma by reversed phase HPLC. Acta Chromatogr. 2014, 26, 57–66. [Google Scholar] [CrossRef]
- Byrne, J.; Velasco-Torrijos, T.; Reinhardt, R. Development and validation of a novel stability-indicating HPLC method for the simultaneous assay of betamethasone-17-valerate, fusidic acid, potassium sorbate, methylparaben and propylparaben in a topical cream preparation. J. Pharm. Biomed. Anal. 2014, 96, 111–117. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hanif, S.; Sarfraz, R.M.; Syed, M.A.; Mahmood, A.; Hussain, Z. Smart mucoadhesive buccal chitosan/HPMC scaffold for sore throat: In vitro, ex vivo and pharmacokinetic profiling in humans. J. Drug Deliv. Sci. Technol. 2022, 71, 103271. [Google Scholar] [CrossRef]
- Hanif, S.; Sarfraz, R.M.; Syed, M.A.; Mahmood, A.; Minhas, M.U.; Irfan, M. Development and optimization of tibezonium iodide and lignocaine hydrochloride containing novel mucoadhesive buccal tablets: A pharmacokinetic investigation among healthy humans. Drug Dev. Ind. Pharm. 2021, 47, 1209–1222. [Google Scholar] [CrossRef]
- Gandla, K.; Kumar, D.S.; Praveen, J.; Suman, E. RP-HPLC Method Development and Validation for Simultaneous Estimation of Lignocaine Hydrochloride and Clotrimazole Hydrochloride in Ear Drops. Asian J. Pharm. Anal. 2017, 7, 163. [Google Scholar] [CrossRef]
- Ter Weijden, E.; Van den Broek, M.P.H.; Ververs, F.F.T. Easy and fast LC–MS/MS determination of lidocaine and MEGX in plasma for therapeutic drug monitoring in neonates with seizures. J. Chromatogr. B 2012, 881, 111–114. [Google Scholar] [CrossRef]
- Ingle, S.; Tegeli, V.; Birajdar, A.; Matole, V.; Adlinge, S.; Nangare, G. UV Spectrophotometric Method Development and Validation of Lignocaine Hydrochloride in Bulk and Semisolid Dosage Form. Res. J. Pharm. Technol. 2021, 14, 5280–5282. [Google Scholar] [CrossRef]
- Chik, Z.; Johnston, A.; Tucker, A.T.; Burn, R.T.; Perrett, D. Validation and application of capillary electrophoresis for the analysis of lidocaine in a skin tape stripping study. Biomed. Chromatogr. 2007, 21, 775–779. [Google Scholar] [CrossRef]
- Isik, I.B.; Tekin, N.; Sagdinc, S.G. The analyses of solvent effects on infrared spectra and thermodynamic parameters, Hirshfeld surface, reduced density gradient and molecular docking of ketoprofen as a member of nonsteroidal anti-inflammatory drugs. J. Mol. Struct. 2022, 1250, 131861. [Google Scholar] [CrossRef]
- Sarzi-Puttini, P.; Atzeni, F.; Lanata, L.; Bagnasco, M.; Colombo, M.; Fischer, F.; D’Imporzano, M. Pain and ketoprofen: What is its role in clinical practice? Reumatismo 2010, 62, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Cardosa, M.; Inbasegaran, K. Morphine-sparing effect of ketoprofen after abdominal surgery. Anaesth. Intensive Care 2000, 28, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkova, B.; Peikova, L. HPLC determination of ketoprofen in tablet dosage forms. Trakia J. Sci. 2013, 11, 55. [Google Scholar]
- Pomykalski, A.; Hopkała, H. Applications of derivative UV spectrophotometry for the determinations of fenbufen and ketoprofen in pharmaceutical preparations. Acta Pol. Pharm. 2005, 62, 171–176. [Google Scholar] [PubMed]
- Kormosh, Z.; Hunka, I.; Basel, Y. Spectrophotometric determination of ketoprofen and its application in pharmaceutical analysis. Acta Pol. Pharm. 2009, 66, 3–9. [Google Scholar] [PubMed]
- Nováková, L.; Matysová, L.; Solichová, D.; Koupparis, M.A.; Solich, P. Comparison of performance of C18 monolithic rod columns and conventional C18 particle-packed columns in liquid chromatographic determination of Estrogel and Ketoprofen gel. J. Chromatogr. B 2004, 813, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Labbozzetta, S.; Valvo, L.; Bertocchi, P.; Alimonti, S.; Gaudiano, M.C.; Manna, L. Focused microwave-assisted extraction and LC determination of ketoprofen in the presence of preservatives in a pharmaceutical cream formulation. Chromatographia 2009, 69, 365–368. [Google Scholar] [CrossRef]
- Šafra, J.; Pospíšilová, M. Separation and determination of ketoprofen, methylparaben and propylparaben in pharmaceutical preparation by micellar electrokinetic chromatography. J. Pharm. Biomed. Anal. 2008, 48, 452–455. [Google Scholar] [CrossRef]
- Blanco, M.; Coello, J.; Iturriaga, H.; Maspoch, S.; Perez-Maseda, C. Chiral and nonchiral determination of ketoprofen in pharmaceuticals by capillary zone electrophoresis. J. Chromatogr. A 1998, 799, 301–307. [Google Scholar] [CrossRef]
- Emara, K.M.; Ali, A.M.M.; Abo-El Maali, N. The polarographic behaviour of ketoprofen and assay of its capsules using spectrophotometric and voltammetric methods. Talanta 1994, 41, 639–645. [Google Scholar] [CrossRef]
- Van Overbeke, A.; Baeyens, W.; Van den Bossche, W. Quantitative Fourier transform infrared/attenuated total reflectance analysis of ketoprofen in some pharmaceutical formulations. Vib. Spectrosc. 1995, 9, 121–130. [Google Scholar] [CrossRef]
- Kormosh, Z.; Hunka, I.; Bazel, Y.; Matviychuk, O. Potentiometric determination of ketoprofen and piroxicam at a new PVC electrode based on ion associates of Rhodamine 6G. Mater. Sci. Eng. C 2010, 30, 997–1002. [Google Scholar] [CrossRef]
- Dołowy, M.; Kulpińska-Kucia, K.; Pyka, A. Validation of a Thin-Layer Chromatography for the Determination of Hydrocortisone Acetate and Lidocaine in a Pharmaceutical Preparation. Sci. World J. 2014, 2014, 107879. [Google Scholar] [CrossRef] [PubMed]
- Rasool, N.; Kanwal, Q.; Waseem, M.; Khan, M.I. Analytical method development and determination of hydrocortisone acetate and fusidic acid simultaneously in cream formulation, by reversed-phase HPLC. Biomed. Chromatogr. 2021, 35, e4997. [Google Scholar] [CrossRef] [PubMed]
- Nalkiashary, S.M.S.; Nezhati, M.N.; Panahi, H.A. Development and validation of simultaneous HPLC method for determination of lidocaine, hydrocortisone acetate and methyl paraben in Anti Hemorrhoid ointment. Pharmacophore 2020, 11, 35–42. [Google Scholar]
- Katakam, L.N.R.; Dongala, T.; Ettaboina, S.K. Novel stability indicating UHPLC method development and validation for simultaneous quantification of hydrocortisone acetate, pramoxine hydrochloride, potassium sorbate and sorbic acid in topical cream formulation. Talanta Open 2020, 1, 100004. [Google Scholar] [CrossRef]
- Abbas, N.; Arshad, M.S.; Hussain, A.; Irfan, M.; Ahsan, M.; Rasool, M.F.; ur Rehman, M.H. Development and validation of a spectroscopic method for the simultaneous analysis of miconazole nitrate and hydrocortisone acetate in pharmaceutical dosage form. Trop. J. Pharm. Res. 2017, 16, 413–420. [Google Scholar] [CrossRef]
- Widiretani, D.; Luailia, I.; Indrayanto, G. Simultaneous densitometric determination of hydrocortisone acetate and 2-phenoxyethanol in creams. JPC-J. Planar Chromatogr.-Mod. TLC 2013, 26, 37–42. [Google Scholar] [CrossRef]
- Gorman, G.; Sokom, S.; Coward, L.; Arnold, J.J. Development and Application of a High-Performance Liquid Chromatography Stability-Indicating Assay for Beyond-Use Date Determination of Compounded Topical Gels Containing Multiple Active Drugs. Int. J. Pharm. Compd. 2017, 21, 164–170. [Google Scholar]
- Swartz, M.E.; Krull, I.S. Analytical Method Development and Validation; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Williams, S. Ghost peaks in reversed-phase gradient HPLC: A review and update. J. Chromatogr. A 2004, 1052, 1–11. [Google Scholar] [CrossRef]
- Ermer, J.; Miller, J.H.M. Method Validation in Pharmaceutical Analysis: A Guide to Best Practice; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Titosky, J.; Momenimovahed, A.; Corbin, J.; Thomson, K.; Smallwood, G.; Olfert, J.S. Repeatability and intermediate precision of a mass concentration calibration system. Aerosol Sci. Technol. 2019, 53, 701–711. [Google Scholar] [CrossRef]
- Mandrioli, M.; Tura, M.; Scotti, S.; Gallina Toschi, T.J.M. Fast detection of 10 cannabinoids by RP-HPLC-UV method in Cannabis sativa L. Molecules 2019, 24, 2113. [Google Scholar] [CrossRef] [PubMed]
- Wenzl, T.; Haedrich, J.; Schaechtele, A.; Piotr, R.; Stroka, J.; Eppe, G.; Scholl, G. Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Food and Feed; Institute for Reference Materials and Measurements (IRMM): Geel, Belgium, 2016. [Google Scholar]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness metric approach and software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef] [PubMed]
- Rainville, P.; Plumb, R.S. Transfer of the USP assay for simvastatin to UPLC. Ultraperfomance LC 2007, 80, 9–11. [Google Scholar]
- Mullick, P.; Mutalik, S.P.; Hegde, A.R.; Pandey, A.; Jagadish, P.C.; Kini, S.G.; Jain, S.; Mutalik, S. Simultaneous estimation of Apremilast and betamethasone Dipropionate in microsponge-based topical formulation using a stability indicating RP-HPLC method: A quality-by-design approach. J. Chromatogr. Sci. 2021, 59, 928–940. [Google Scholar] [CrossRef]
- Borman, P.; Elder, D. Q2 (R1) validation of analytical procedures. ICH Qual. Guidel. 2017, 5, 127–166. [Google Scholar]
- Syed, M.A.; Khan, I.U.; Iqbal, M.S.; Syed, H.K.; Irfan, M. Development of a novel, fast, simple, non-derived RP-HPLC method for simultaneous estimation of benzocaine and tibezonium iodide from mucoadhesive dosage form as well as human saliva and its validation. Lat. Am. J. Pharm. 2021, 40, 1281–1287. [Google Scholar]
- Raza, A.; Murtaza, S.H.; Hanif, S.; Iqbal, J.; Ali, I.; Aftab, T.; Shakir, R.; Bedar, R.; Syed, M.A. Validation of a rapid and economical RP-HPLC method for simultaneous determination of metformin hydrochloride and sitagliptin phosphate monohydrate: Greenness evaluation using AGREE score. Pak. J. Pharm. Sci. 2022, 35, 15–21. [Google Scholar]



{kind=link}
{kind=link}
| Parameters | LIG | HYD | KET |
|---|---|---|---|
| System suitability | |||
| Retention time (min) | 1.54 | 2.57 | 5.78 |
| Tailing factor | 1.39 | 1.19 | 1.10 |
| Plate count (USP) | 6167 | 6094 | 6003 |
| Resolution (USP) | - | 9.4 | 15.7 |
| Linearity | |||
| Linear function | y = 3175.2x + 116.67 | y = 1 × 106x + 526,211 | y = 6 × 106x + 3 × 106 |
| Coefficient of linear regression (r2) | 0.9994 | 0.9990 | 0.9997 |
| Linearity range (ppm) | 0.6–56 | 0.6–56 | 0.2–100 |
| LOD (ppm) | 0.563 | 0.611 | 0.223 |
| LOQ (ppm) | 1.690 | 1.833 | 0.669 |
| Parameters | LIG | HYD | KET | |||
|---|---|---|---|---|---|---|
| Dose Level (%) | Theoretical Contents (ppm) | Mean Area (uA) | Theoretical Contents (ppm) | Mean Area (uA) | Theoretical Contents (ppm) | Mean Area (uA) |
| 80 | 16 | 105,831.08 | 16 | 3,411,719 | 40 | 5,949,786 |
| 100 | 20 | 132,123.7 | 20 | 4,259,328 | 60 | 7,446,541 |
| 120 | 24 | 158,540.0 | 24 | 5,111,193 | 80 | 8,861,380 |
| Accuracy | LIG | HYD | KET | |||
|---|---|---|---|---|---|---|
| Concentration Level (%) | Amount (ppm) | Recovery % ± RSD | Amount (ppm) | Recovery % ± RSD | Amount (ppm) | Recovery % ± RSD |
| 80 | 15.91 | 99.38 ± 0.62 | 16.0 | 100.00 ± 1.16 | 40.25 | 100.50 ± 1.44 |
| 100 | 20.0 | 100.0 ± 0.46 | 20.14 | 100.50 ± 0.91 | 60.30 | 100.20 ± 1.58 |
| 120 | 23.93 | 99.58 ± 0.51 | 24.01 | 100.00 ± 1.29 | 79.86 | 99.83 ± 0.38 |
| Machine/Day 1 & 2 | Analyst 1 & 2 | LIG | HYD | KET | |||
|---|---|---|---|---|---|---|---|
| Concentration Level (%) | Amount (ppm) | Recovery % ± RSD | Amount (ppm) | Recovery % ± RSD | Amount (ppm) | Recovery % ± RSD | |
| 80 | 15.84 | 99.01 ± 1.28 | 16.05 | 100.32 ± 1.76 | 39.63 | 99.08 ± 1.08 | |
| 100 | 19.87 | 99.39 ± 1.47 | 19.16 | 99.58 ± 0.73 | 59.68 | 99.47± 0.97 | |
| 120 | 24.18 | 100.76 ± 1.10 | 23.80 | 99.19 ± 0.90 | 80.08 | 100.10 ± 0.58 | |
| Inter day precision | |||||||
| Machine/Day 3 & 4 | Analyst 3 & 4 | LIG | HYD | KET | |||
| Concentration level (%) | Content recovered (ppm) | Recovery % ± RSD | Content recovered (ppm) | Recovery % ± RSD | Content recovered (ppm) | Recovery % ± RSD | |
| 80 | 15.99 | 99.95 ± 0.92 | 16.09 | 100.11± 0.37 | 39.89 | 99.74 ± 1.18 | |
| 100 | 19.94 | 99.71 ± 1.46 | 19.85 | 99.27 ± 1.77 | 60.18 | 100.30 ± 0.75 | |
| 120 | 23.87 | 99.47 ± 0.60 | 23.96 | 99.85 ± 0.91 | 79.34 | 99.18 ± 0.31 | |
| Parameters | LIG % ± RSD | HYD % ± RSD | KET % ± RSD |
|---|---|---|---|
| Optimized conditions at 100% analytical dose = pH 3.0, 30 °C and 1.00 mL/min | |||
| Flow rate | |||
| 0.85 mL/min | 100.33 ± 0.95 | 100.21 ± 1.67 | 99.15 ± 1.24 |
| 1.15 mL/min | 98.89 ± 1.36 | 99.57 ± 1.18 | 99.90 ± 0.66 |
| Temperature range | |||
| 20 °C | 100.26 ± 1.74 | 100.49 ± 1.14 | 100.14 ± 0.86 |
| 30 °C | 100.58 ± 1.09 | 99.41 ± 0.68 | 98.69 ± 0.70 |
| Mobile phase | |||
| pH 2.5 | 99.51 ± 1.38 | 98.47 ± 1.62 | 99.35 ± 1.41 |
| pH 3.5 | 98.86 ± 0.91 | 100.01 ± 0.75 | 99.74 ± 0.58 |
| Active | Brand | Labelled Claim | Amount Found (%) | USP Interpretation |
|---|---|---|---|---|
| Lignocaine HCl | Lignocaine Gel® | 2.0% w/v | 99.80 ± 0.83 | in limits |
| Ketoprofen | Profenid Gel® | 2.5% w/v | 100.09 ± 1.07 | in limits |
| Hydrocortisone | Hydrocortisone 1% Gel | 1.0% w/v | 97.38 ± 2.15 | in limits |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehmood, T.; Hanif, S.; Azhar, F.; Ali, I.; Alafnan, A.; Hussain, T.; Moin, A.; Alamri, M.A.; Syed, M.A. HPLC Method Validation for the Estimation of Lignocaine HCl, Ketoprofen and Hydrocortisone: Greenness Analysis Using AGREE Score. Int. J. Mol. Sci. 2023, 24, 440. https://doi.org/10.3390/ijms24010440
Mehmood T, Hanif S, Azhar F, Ali I, Alafnan A, Hussain T, Moin A, Alamri MA, Syed MA. HPLC Method Validation for the Estimation of Lignocaine HCl, Ketoprofen and Hydrocortisone: Greenness Analysis Using AGREE Score. International Journal of Molecular Sciences. 2023; 24(1):440. https://doi.org/10.3390/ijms24010440
Chicago/Turabian StyleMehmood, Tariq, Sana Hanif, Faiza Azhar, Ijaz Ali, Ahmed Alafnan, Talib Hussain, Afrasim Moin, Mubarak A. Alamri, and Muhammad Ali Syed. 2023. "HPLC Method Validation for the Estimation of Lignocaine HCl, Ketoprofen and Hydrocortisone: Greenness Analysis Using AGREE Score" International Journal of Molecular Sciences 24, no. 1: 440. https://doi.org/10.3390/ijms24010440
APA StyleMehmood, T., Hanif, S., Azhar, F., Ali, I., Alafnan, A., Hussain, T., Moin, A., Alamri, M. A., & Syed, M. A. (2023). HPLC Method Validation for the Estimation of Lignocaine HCl, Ketoprofen and Hydrocortisone: Greenness Analysis Using AGREE Score. International Journal of Molecular Sciences, 24(1), 440. https://doi.org/10.3390/ijms24010440