
In-Frame Deletion of Dystrophin Exons 8–50 Results in DMD Phenotype
 ,
,
Abstract
:1. Introduction
2. Results
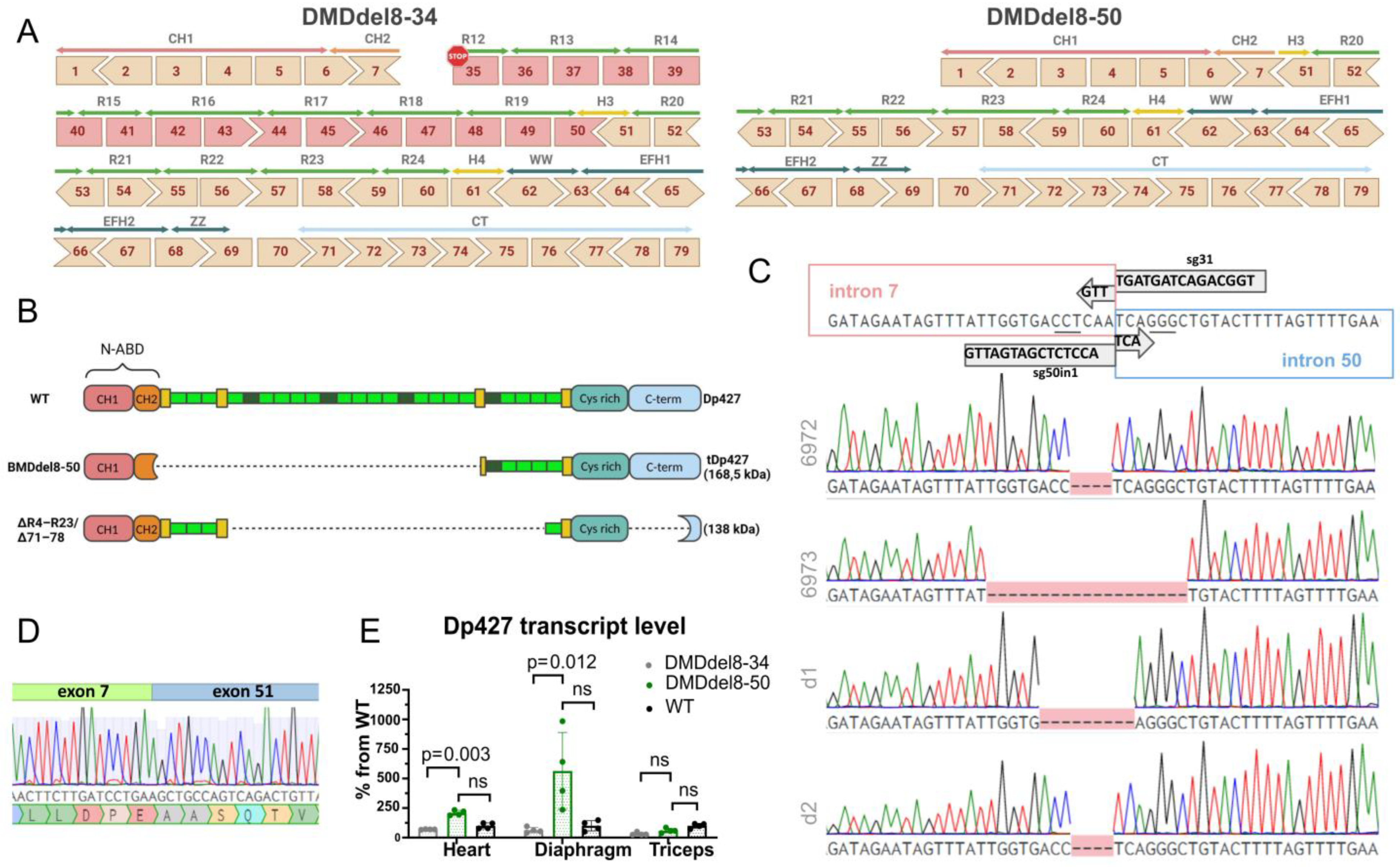
2.1. Exons 8 to 50 of the DMD Gene Are Deleted Using the CRISPR-Cas9 Genome Editing Technique
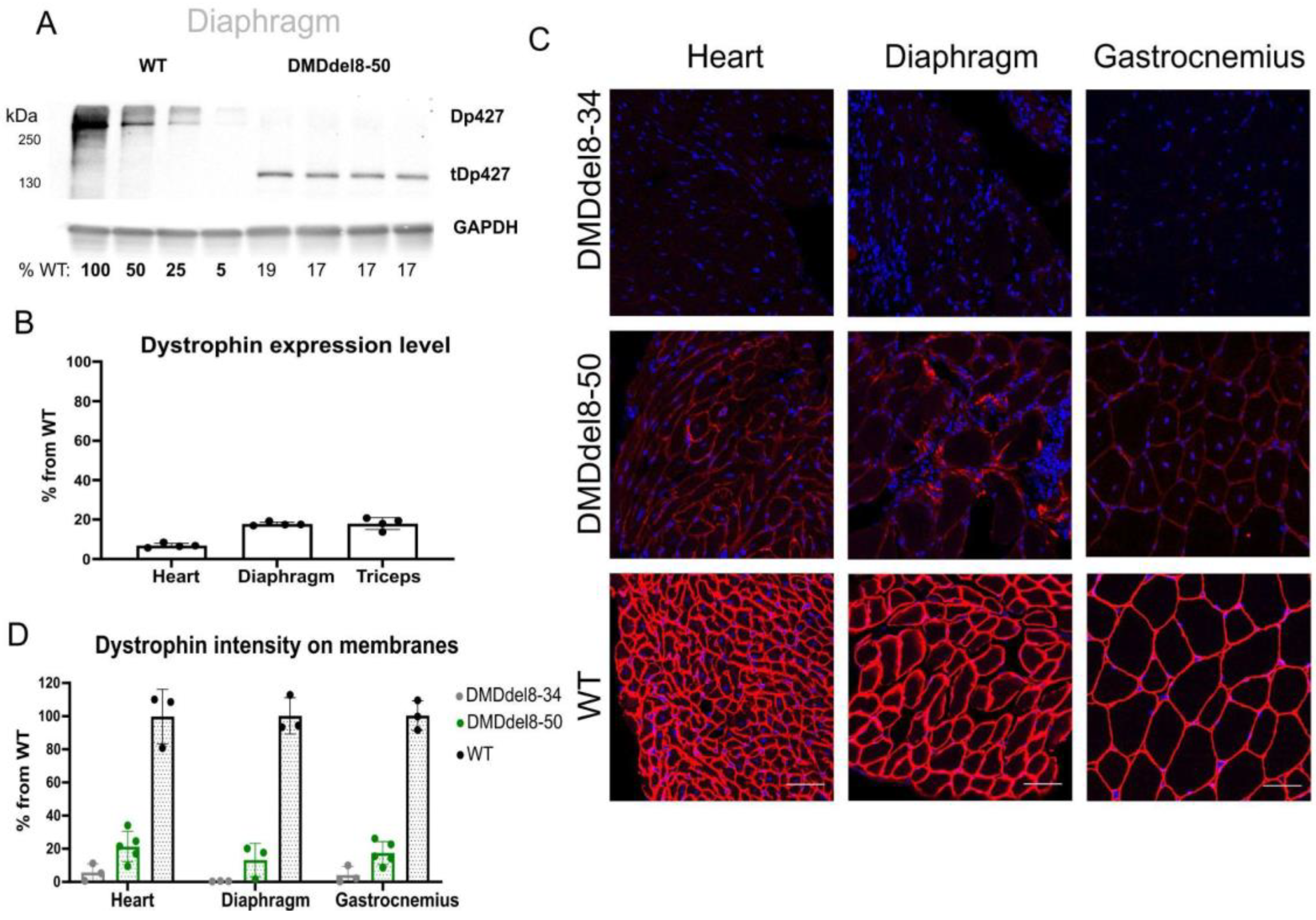
2.2. In BMDdel8-50 Muscles, Dystrophin Expression Is Below Normal Levels
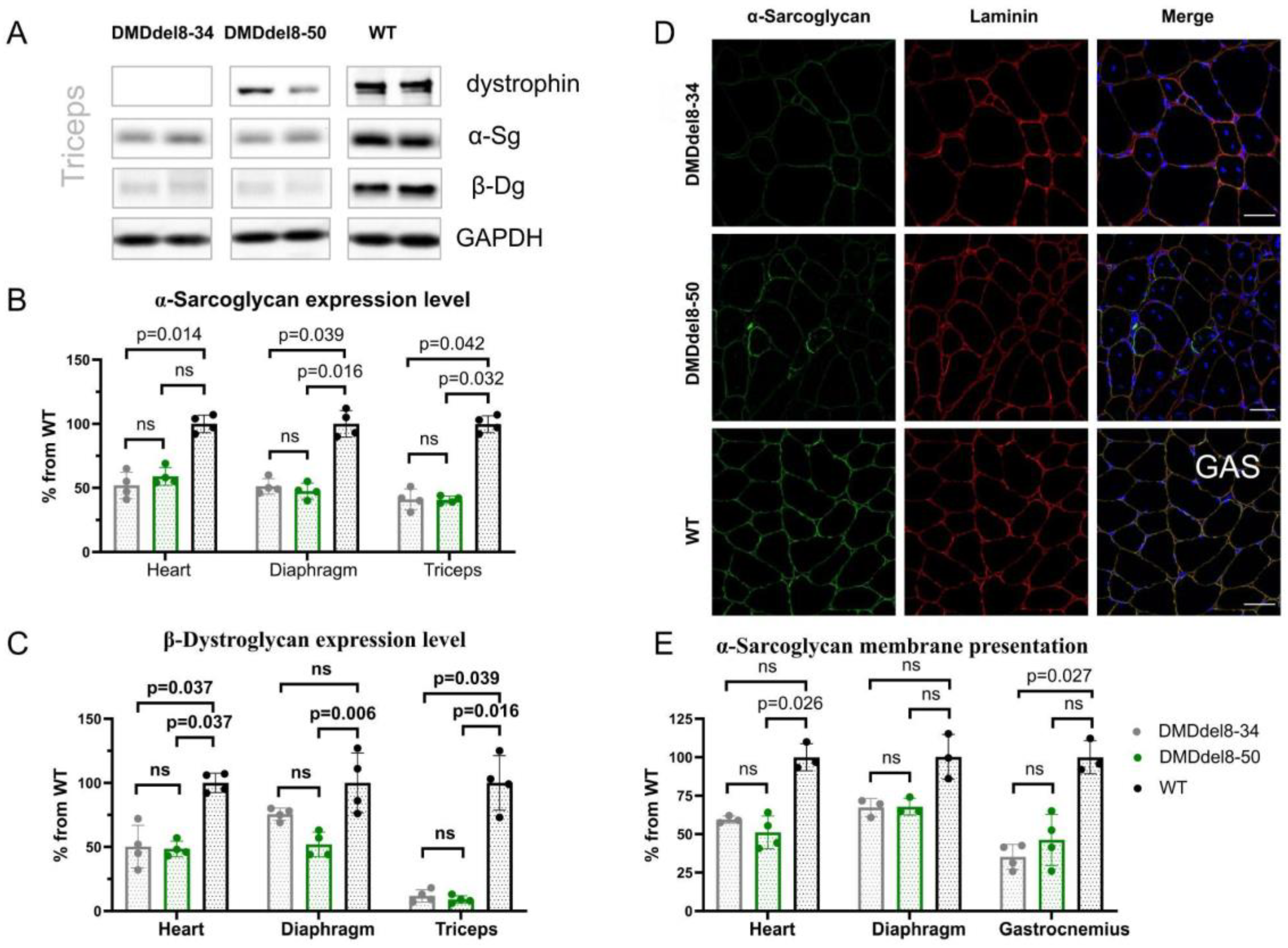
2.3. Components of DAGC Are Diminished in BMDdel8-50 Muscles
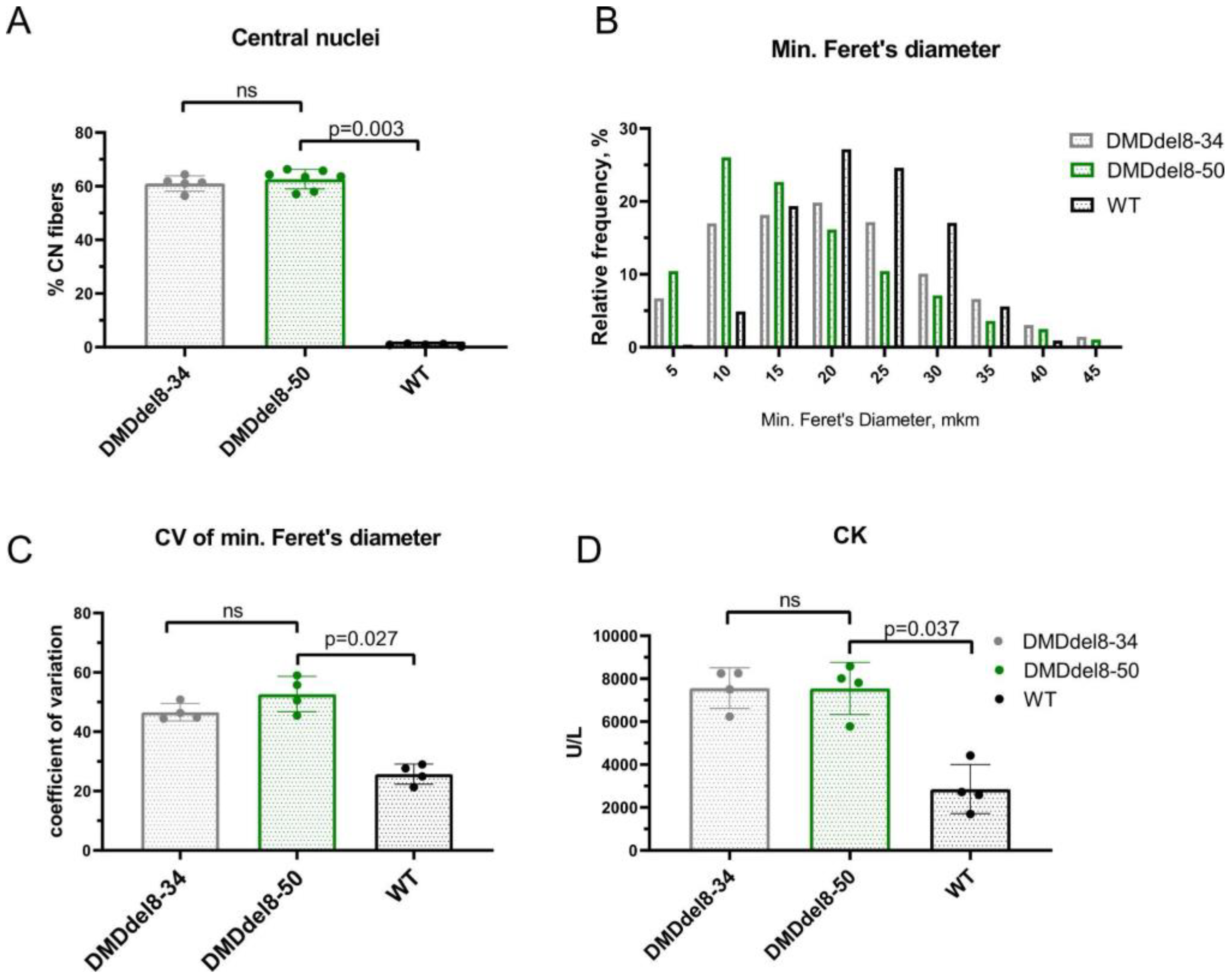
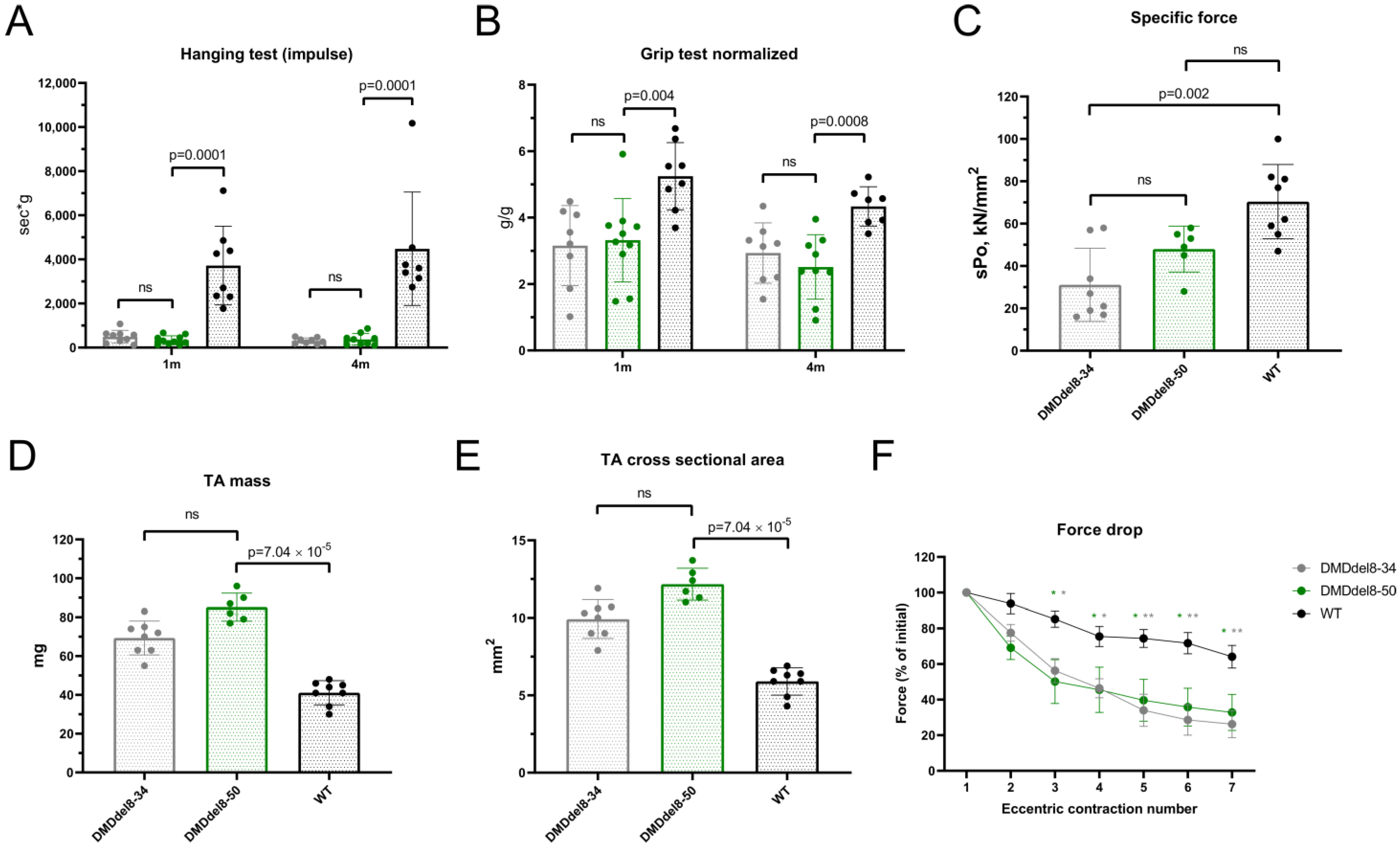
2.4. Skeletal Muscles from BMDdel8-50 Mice Exhibit DMD-Like Characteristics
2.5. Muscle Performance in DMDdel8-50 Mice Was Not Enhanced by Truncated Dystrophin
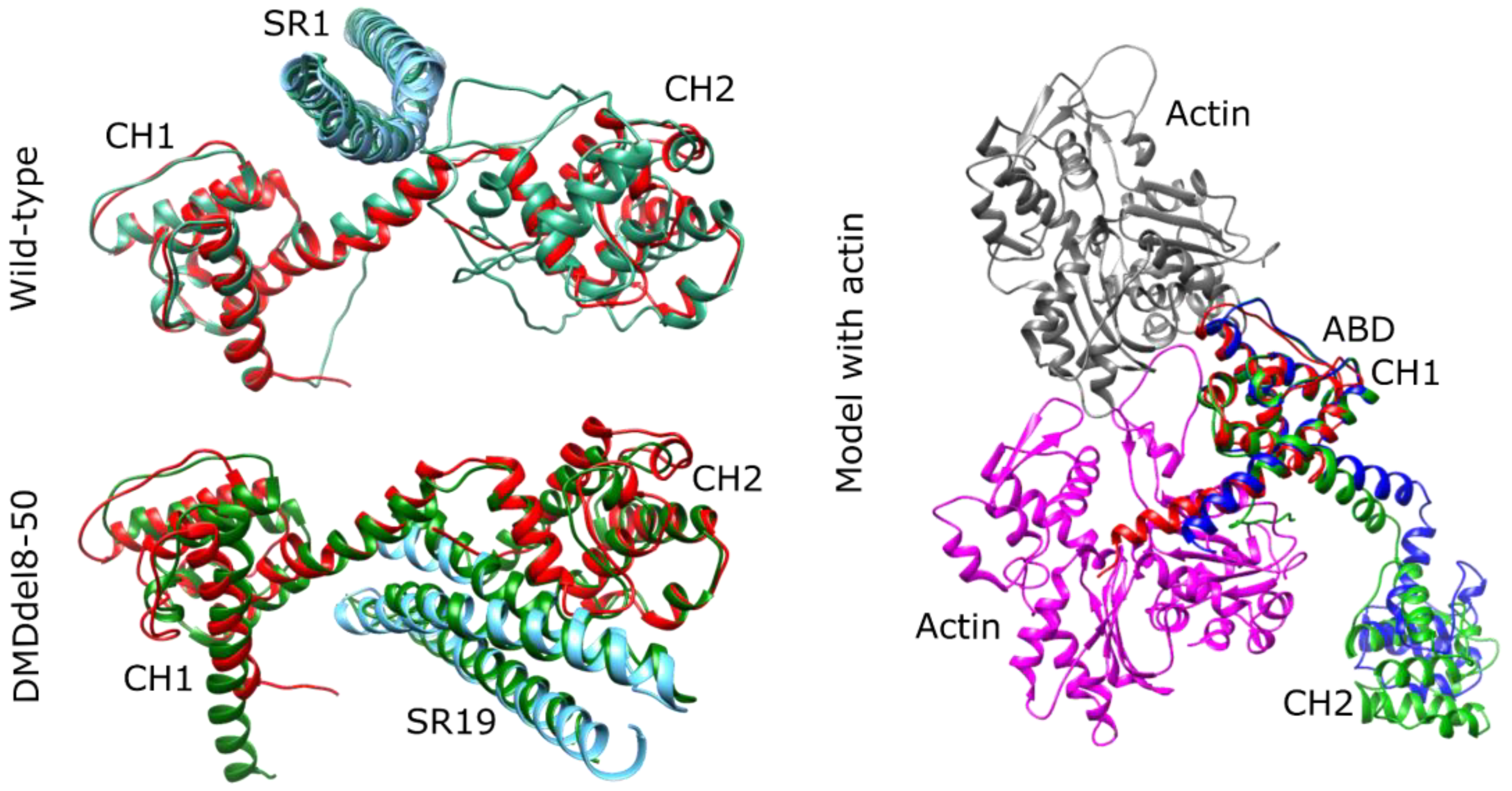
2.6. DMDdel8-50 Dystrophin 3D Structure In Silico Prediction
3. Discussion
4. Materials and Methods
4.1. Guide Design and Synthesis
4.2. Microinjections into the Pronucleus of Zygotes, Embryo Cultivation, and Transfer
4.3. Animal Studies
4.4. Creatine Kinase
4.5. Histopathology
4.6. Hanging Wire Test and Grip Strength Test
4.7. Measurement of Isometric Force and Muscle Susceptibility to Eccentric Contraction-Induced Injury
4.8. Genomic DNA Isolation, PCR, and Sequences Analysis
4.9. RT–qPCR
4.10. Western Blotting
4.11. Immunofluorescence
4.12. Dystrophin Protein Modeling
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An Explanation for the Phenotypic Differences between Patients Bearing Partial Deletions of the DMD Locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.M.; Salhany, R.M.; Deighton, A.; Harwood, M.; Mah, J.; Gooch, K.L. The Clinical Course of Duchenne Muscular Dystrophy in the Corticosteroid Treatment Era: A Systematic Literature Review. Orphanet J. Rare Dis. 2021, 16, 237. [Google Scholar] [CrossRef]
- Bushby, K.M.; Gardner-Medwin, D.; Nicholson, L.V.; Johnson, M.A.; Haggerty, I.D.; Cleghorn, N.J.; Harris, J.B.; Bhattacharya, S.S. The Clinical, Genetic and Dystrophin Characteristics of Becker Muscular Dystrophy. II. Correlation of Phenotype with Genetic and Protein Abnormalities. J. Neurol. 1993, 240, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.-J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne Muscular Dystrophy Mutation Database: An Overview of Mutation Types and Paradoxical Cases That Confirm the Reading-Frame Rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Anwar, S.; He, M.; Lim, K.R.Q.; Maruyama, R.; Yokota, T. A Genotype-Phenotype Correlation Study of Exon Skip-Equivalent In-Frame Deletions and Exon Skip-Amenable Out-of-Frame Deletions across the DMD Gene to Simulate the Effects of Exon-Skipping Therapies: A Meta-Analysis. J. Pers. Med. 2021, 11, 46. [Google Scholar] [CrossRef]
- van den Bergen, J.C.; Wokke, B.H.; Janson, A.A.; van Duinen, S.G.; Hulsker, M.A.; Ginjaar, H.B.; van Deutekom, J.C.; Aartsma-Rus, A.; Kan, H.E.; Verschuuren, J.J. Dystrophin Levels and Clinical Severity in Becker Muscular Dystrophy Patients. J. Neurol. Neurosurg. Psychiatry 2014, 85, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Freda, M.P.; Vitiello, L.; Danieli, G.A.; Pegoraro, E.; Angelini, C. Duchenne Phenotype with In-Frame Deletion Removing Major Portion of Dystrophin Rod: Threshold Effect for Deletion Size? Muscle Nerve 1996, 19, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Prim. 2021, 7, 1–19. [Google Scholar] [CrossRef]
- Singh, S.M.; Kongari, N.; Cabello-Villegas, J.; Mallela, K.M.G. Missense Mutations in Dystrophin That Trigger Muscular Dystrophy Decrease Protein Stability and Lead to Cross-β Aggregates. Proc. Natl. Acad. Sci. USA 2010, 107, 15069–15074. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Lucchetti-Miganeh, C.; Yaou, R.B.; Kaplan, J.-C.; Chelly, J.; Leturcq, F.; Barloy-Hubler, F.; Le Rumeur, E. Assessment of the Structural and Functional Impact of In-Frame Mutations of the DMD Gene, Using the Tools Included in the EDystrophin Online Database. Orphanet J. Rare Dis. 2012, 7, 45. [Google Scholar] [CrossRef]
- Bies, R.D.; Caskey, C.T.; Fenwick, R. An Intact Cysteine-Rich Domain Is Required for Dystrophin Function. J. Clin. Investig. 1992, 90, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef] [PubMed]
- Duan, D. Systemic AAV Micro-Dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Mol. Ther. 2018, 26, 2337–2356. [Google Scholar] [CrossRef]
- Wilton-Clark, H.; Yokota, T. Recent Trends in Antisense Therapies for Duchenne Muscular Dystrophy. Pharmaceutics 2023, 15, 778. [Google Scholar] [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Melo, D.; Bao, B.; Trieu, N.; Mizobe, Y.; Maruyama, R.; Mamchaoui, K.; Tanihata, J.; Aoki, Y.; et al. Exons 45–55 Skipping Using Mutation-Tailored Cocktails of Antisense Morpholinos in the DMD Gene. Mol. Ther. 2019, 27, 2005–2017. [Google Scholar] [CrossRef] [PubMed]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A Single CRISPR-Cas9 Deletion Strategy That Targets the Majority of DMD Patients Restores Dystrophin Function in HiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef]
- Duchêne, B.L.; Cherif, K.; Iyombe-Engembe, J.-P.; Guyon, A.; Rousseau, J.; Ouellet, D.L.; Barbeau, X.; Lague, P.; Tremblay, J.P. CRISPR-Induced Deletion with SaCas9 Restores Dystrophin Expression in Dystrophic Models In Vitro and In Vivo. Mol. Ther. 2018, 26, 2604–2616. [Google Scholar] [CrossRef]
- Chey, Y.C.J.; Arudkumar, J.; Aartsma-Rus, A.; Adikusuma, F.; Thomas, P.Q. CRISPR Applications for Duchenne Muscular Dystrophy: From Animal Models to Potential Therapies. WIREs Mech. Dis. 2023, 15, e1580. [Google Scholar] [CrossRef]
- Delalande, O.; Molza, A.-E.; Dos Santos Morais, R.; Chéron, A.; Pollet, É.; Raguenes-Nicol, C.; Tascon, C.; Giudice, E.; Guilbaud, M.; Nicolas, A.; et al. Dystrophin’s Central Domain Forms a Complex Filament That Becomes Disorganized by in-Frame Deletions. J. Biol. Chem. 2018, 293, 6637–6646. [Google Scholar] [CrossRef]
- Niu, X.; Menhart, N. Structural Perturbations of Exon-Skipping Edits within the Dystrophin D20:24 Region. Biochemistry 2021, 60, 765–779. [Google Scholar] [CrossRef]
- Ruszczak, C.; Mirza, A.; Menhart, N. Differential Stabilities of Alternative Exon-Skipped Rod Motifs of Dystrophin. Biochim. Biophys. Acta 2009, 1794, 921–928. [Google Scholar] [CrossRef] [PubMed]
- McCourt, J.L.; Rhett, K.K.; Jaeger, M.A.; Belanto, J.J.; Talsness, D.M.; Ervasti, J.M. In Vitro Stability of Therapeutically Relevant, Internally Truncated Dystrophins. Skelet. Muscle 2015, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, R.W.; Allen, H.D.; Ray, W.C.; Alvarez, C.E.; Kissel, J.T.; Pestronk, A.; Weiss, R.B.; Flanigan, K.M.; Mendell, J.R.; Montanaro, F. Analysis of Dystrophin Deletion Mutations Predicts Age of Cardiomyopathy Onset in Becker Muscular Dystrophy. Circ. Cardiovasc. Genet. 2009, 2, 544–551. [Google Scholar] [CrossRef]
- Nicolas, A.; Raguénès-Nicol, C.; Ben Yaou, R.; Ameziane-Le Hir, S.; Chéron, A.; Vié, V.; Claustres, M.; Leturcq, F.; Delalande, O.; Hubert, J.-F.; et al. Becker Muscular Dystrophy Severity Is Linked to the Structure of Dystrophin. Hum. Mol. Genet. 2015, 24, 1267–1279. [Google Scholar] [CrossRef]
- Iyombe-Engembe, J.-P.; Ouellet, D.L.; Barbeau, X.; Rousseau, J.; Chapdelaine, P.; Lagüe, P.; Tremblay, J.P. Efficient Restoration of the Dystrophin Gene Reading Frame and Protein Structure in DMD Myoblasts Using the CinDel Method. Mol. Ther. Nucleic Acids 2016, 5, e283. [Google Scholar] [CrossRef]
- Egorova, T.V.; Zotova, E.D.; Reshetov, D.A.; Polikarpova, A.V.; Vassilieva, S.G.; Vlodavets, D.V.; Gavrilov, A.A.; Ulianov, S.V.; Buchman, V.L.; Deykin, A.V. CRISPR/Cas9-Generated Mouse Model of Duchenne Muscular Dystrophy Recapitulating a Newly Identified Large 430 Kb Deletion in the Human DMD Gene. Dis. Model. Mech. 2019, 12, dmm037655. [Google Scholar] [CrossRef]
- Juan-Mateu, J.; Gonzalez-Quereda, L.; Rodriguez, M.J.; Baena, M.; Verdura, E.; Nascimento, A.; Ortez, C.; Baiget, M.; Gallano, P. DMD Mutations in 576 Dystrophinopathy Families: A Step Forward in Genotype-Phenotype Correlations. PLoS ONE 2015, 10, e0135189. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.Q.; Hauser, M.A.; DelloRusso, C.; Duan, D.; Crawford, R.W.; Phelps, S.F.; Harper, H.A.; Robinson, A.S.; Engelhardt, J.F.; Brooks, S.V.; et al. Modular Flexibility of Dystrophin: Implications for Gene Therapy of Duchenne Muscular Dystrophy. Nat. Med. 2002, 8, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Labun, K.; Montague, T.G.; Gagnon, J.A.; Thyme, S.B.; Valen, E. CHOPCHOP v2: A Web Tool for the next Generation of CRISPR Genome Engineering. Nucleic Acids Res. 2016, 44, W272–W276. [Google Scholar] [CrossRef]
- Beggs, A.H.; Hoffman, E.P.; Snyder, J.R.; Arahata, K.; Specht, L.; Shapiro, F.; Angelini, C.; Sugita, H.; Kunkel, L.M. Exploring the Molecular Basis for Variability among Patients with Becker Muscular Dystrophy: Dystrophin Gene and Protein Studies. Am. J. Hum. Genet. 1991, 49, 54–67. [Google Scholar]
- de Feraudy, Y.; Ben Yaou, R.; Wahbi, K.; Stalens, C.; Stantzou, A.; Laugel, V.; Desguerre, I.; Servais, L.; Leturcq, F.; Amthor, H.; et al. Very Low Residual Dystrophin Quantity Is Associated with Milder Dystrophinopathy. Ann. Neurol. 2021, 89, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Ripolone, M.; Velardo, D.; Mondello, S.; Zanotti, S.; Magri, F.; Minuti, E.; Cazzaniga, S.; Fortunato, F.; Ciscato, P.; Tiberio, F.; et al. Muscle Histological Changes in a Large Cohort of Patients Affected with Becker Muscular Dystrophy. Acta Neuropathol. Commun. 2022, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yue, Y.; Duan, D. Preservation of Muscle Force in Mdx3cv Mice Correlates with Low-Level Expression of a near Full-Length Dystrophin Protein. Am. J. Pathol. 2008, 172, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yue, Y.; Duan, D. Marginal Level Dystrophin Expression Improves Clinical Outcome in a Strain of Dystrophin/Utrophin Double Knockout Mice. PLoS ONE 2010, 5, e15286. [Google Scholar] [CrossRef] [PubMed]
- van Putten, M.; Hulsker, M.; Nadarajah, V.D.; van Heiningen, S.H.; van Huizen, E.; van Iterson, M.; Admiraal, P.; Messemaker, T.; den Dunnen, J.T.; ’t Hoen, P.A.C.; et al. The Effects of Low Levels of Dystrophin on Mouse Muscle Function and Pathology. PLoS ONE 2012, 7, e31937. [Google Scholar] [CrossRef]
- Phelps, S.F.; Hauser, M.A.; Cole, N.M.; Rafael, J.A.; Hinkle, R.T.; Faulkner, J.A.; Chamberlain, J.S. Expression of Full-Length and Truncated Dystrophin Mini-Genes in Transgenic Mdx Mice. Hum. Mol. Genet. 1995, 4, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, C.; Muses, S.; McClorey, G.; Wells, K.E.; Coursindel, T.; Terry, R.L.; Betts, C.; Hammond, S.; O’Donovan, L.; Hildyard, J.; et al. How Much Dystrophin Is Enough: The Physiological Consequences of Different Levels of Dystrophin in the Mdx Mouse. Hum. Mol. Genet. 2015, 24, 4225–4237. [Google Scholar] [CrossRef]
- Heier, C.R.; McCormack, N.M.; Tully, C.B.; Novak, J.S.; Newell-Stamper, B.L.; Russell, A.J.; Fiorillo, A.A. The X-Linked Becker Muscular Dystrophy (Bmx) Mouse Models Becker Muscular Dystrophy via Deletion of Murine Dystrophin Exons 45–47. J. Cachexia Sarcopenia Muscle 2023, 14, 940–954. [Google Scholar] [CrossRef]
- Wells, D.J. What Is the Level of Dystrophin Expression Required for Effective Therapy of Duchenne Muscular Dystrophy? J. Muscle Res. Cell Motil. 2019, 40, 141–150. [Google Scholar] [CrossRef]
- Winnard, A.V.; Klein, C.J.; Coovert, D.D.; Prior, T.; Papp, A.; Snyder, P.; Bulman, D.E.; Ray, P.N.; McAndrew, P.; King, W.; et al. Characterization of Translational Frame Exception Patients in Duchenne/Becker Muscular Dystrophy. Hum. Mol. Genet. 1993, 2, 737–744. [Google Scholar] [CrossRef]
- Takeshima, Y.; Nishio, H.; Narita, N.; Wada, H.; Ishikawa, Y.; Ishikawa, Y.; Minami, R.; Nakamura, H.; Matsuo, M. Amino-Terminal Deletion of 53% of Dystrophin Results in an Intermediate Duchenne-Becker Muscular Dystrophy Phenotype. Neurology 1994, 44, 1648–1651. [Google Scholar] [CrossRef] [PubMed]
- Nevo, Y.; Muntoni, F.; Sewry, C.; Legum, C.; Kutai, M.; Harel, S.; Dubowitz, V. Large In-Frame Deletions of the Rod-Shaped Domain of the Dystrophin Gene Resulting in Severe Phenotype. Isr. Med. Assoc. J. 2003, 5, 94–97. [Google Scholar] [PubMed]
- Potter, R.A.; Griffin, D.A.; Heller, K.N.; Peterson, E.L.; Clark, E.K.; Mendell, J.R.; Rodino-Klapac, L.R. Dose-Escalation Study of Systemically Delivered RAAVrh74.MHCK7.Micro-Dystrophin in the Mdx Mouse Model of Duchenne Muscular Dystrophy. Hum. Gene Ther. 2021, 32, 375–389. [Google Scholar] [CrossRef]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of RAAVrh74.MHCK7.Micro-Dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020, 77, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Banks, G.B.; Gregorevic, P.; Allen, J.M.; Finn, E.E.; Chamberlain, J.S. Functional Capacity of Dystrophins Carrying Deletions in the N-Terminal Actin-Binding Domain. Hum. Mol. Genet. 2007, 16, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Carsana, A.; Frisso, G.; Tremolaterra, M.R.; Lanzillo, R.; Vitale, D.F.; Santoro, L.; Salvatore, F. Analysis of Dystrophin Gene Deletions Indicates That the Hinge III Region of the Protein Correlates with Disease Severity. Ann. Hum. Genet. 2005, 69, 253–259. [Google Scholar] [CrossRef]
- Wasala, L.P.; Watkins, T.B.; Wasala, N.B.; Burke, M.J.; Yue, Y.; Lai, Y.; Yao, G.; Duan, D. The Implication of Hinge 1 and Hinge 4 in Micro-Dystrophin Gene Therapy for Duchenne Muscular Dystrophy. Hum. Gene. Ther. 2023. [CrossRef]
- Harper, S.Q.; Crawford, R.W.; DelloRusso, C.; Chamberlain, J.S. Spectrin-like Repeats from Dystrophin and Alpha-Actinin-2 Are Not Functionally Interchangeable. Hum. Mol. Genet. 2002, 11, 1807–1815. [Google Scholar] [CrossRef]
- Teramoto, N.; Sugihara, H.; Yamanouchi, K.; Nakamura, K.; Kimura, K.; Okano, T.; Shiga, T.; Shirakawa, T.; Matsuo, M.; Nagata, T.; et al. Pathological Evaluation of Rats Carrying In-Frame Mutations in the Dystrophin Gene: A New Model of Becker Muscular Dystrophy. Dis. Model. Mech. 2020, 13, dmm044701. [Google Scholar] [CrossRef]
- Niba, E.T.E.; Awano, H.; Lee, T.; Takeshima, Y.; Shinohara, M.; Nishio, H.; Matsuo, M. Dystrophin Dp71 Subisoforms Localize to the Mitochondria of Human Cells. Life 2021, 11, 978. [Google Scholar] [CrossRef]
- Wieneke, S.; Heimann, P.; Leibovitz, S.; Nudel, U.; Jockusch, H. Acute Pathophysiological Effects of Muscle-Expressed Dp71 Transgene on Normal and Dystrophic Mouse Muscle. J. Appl. Physiol. 2003, 95, 1861–1866. [Google Scholar] [CrossRef]
- Slaoui, M.; Bauchet, A.-L.; Fiette, L. Tissue Sampling and Processing for Histopathology Evaluation. Methods Mol. Biol. 2017, 1641, 101–114. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; van Putten, M. Assessing Functional Performance in the Mdx Mouse Model. J. Vis. Exp. 2014, 85, e51303. [Google Scholar] [CrossRef]
- Starikova, A.V.; Skopenkova, V.V.; Polikarpova, A.V.; Reshetov, D.A.; Vassilieva, S.G.; Velyaev, O.A.; Shmidt, A.A.; Savchenko, I.M.; Soldatov, V.O.; Egorova, T.V.; et al. Therapeutic Potential of Highly Functional Codon-Optimized Microutrophin for Muscle-Specific Expression. Sci. Rep. 2022, 12, 848. [Google Scholar] [CrossRef]
- Benchling [Biology Software]. 2023. Available online: https://benchling.com (accessed on 9 April 2023).
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy Quantitative Assessment of Genome Editing by Sequence Trace Decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, H.; Ni, J.; Pan, J.; Hua, H.; Wang, Y. Identification of Suitable Reference Genes for Gene Expression Studies in Rat Skeletal Muscle Following Sciatic Nerve Crush Injury. Mol. Med. Rep. 2019, 19, 4377–4387. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Jones, T.R.; Kang, I.H.; Wheeler, D.B.; Lindquist, R.A.; Papallo, A.; Sabatini, D.M.; Golland, P.; Carpenter, A.E. CellProfiler Analyst: Data Exploration and Analysis Software for Complex Image-Based Screens. BMC Bioinform. 2008, 9, 482. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly Accurate Protein Structure Prediction for the Human Proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | DMD Intron | Sequence, 5′-3′ (PAM) | Coordinates |
|---|---|---|---|
| sg31 1 | 7 | GTGGCAGACTAGTAGTTTG(AGG) | X:82534746-82534764 |
| sg50in1 | 50 | GGTTAGTAGCTCTCCATCA(GGG) | X:83480495-83480513 |
| sg50in2 | 50 | GACATCGGCACAACAATCA(AGG) | X:83504960-83504977 |
| sg50in3 | 50 | GGTCCAAACCTATCTGTGA(GGG) | X:83488341-83488359 |
| Deletion | Guide Combination | Embryos | Transplanted | Recipients | Newborn Pups | Founders |
|---|---|---|---|---|---|---|
| 8–50 | sg31 + sg50.3 | 106 | 28 | 2 | 7 | 0 |
| 8–50 | sg31 + sg50.1 | 330 | 67 | 6 | 10 | 2 + 2 1 |
| Site | Sequence, 5′-3′ (PAM) | Chr |
|---|---|---|
| Sg50.1 | GTTAGTAGCTCTCCATCA (GGG) | X |
| Sg50.1 OT1 (Asb18) | GTTtaTAGCTCTCCATCA(TGG) | 1 |
| Sg50.1 OT2 (Malrd1) | GTaAGTAGCTCTtCATCA(AGG) | 2 |
| Sg50.1 OT3 (Ift88) | aTTAGTAGCTCTCCAcCA(AGG) | 14 |
| Sg31 target | GTGGCAGACTAGTAGTTTG(AGG) | X |
| Sg31 OT1 (Tff3) | cTGcCAGACTAcTAGTTTG(GGG) | 17 |
| Sg31 OT2 (Unc5c) | cTGGCAGACTAaTAGcTTG(AGG) | 3 |
| Sg31 OT3 (Fmn2) | tTGGaAtACTAGTAGTTTG(TGG) | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egorova, T.V.; Galkin, I.I.; Velyaev, O.A.; Vassilieva, S.G.; Savchenko, I.M.; Loginov, V.A.; Dzhenkova, M.A.; Korshunova, D.S.; Kozlova, O.S.; Ivankov, D.N.; et al. In-Frame Deletion of Dystrophin Exons 8–50 Results in DMD Phenotype. Int. J. Mol. Sci. 2023, 24, 9117. https://doi.org/10.3390/ijms24119117
Egorova TV, Galkin II, Velyaev OA, Vassilieva SG, Savchenko IM, Loginov VA, Dzhenkova MA, Korshunova DS, Kozlova OS, Ivankov DN, et al. In-Frame Deletion of Dystrophin Exons 8–50 Results in DMD Phenotype. International Journal of Molecular Sciences. 2023; 24(11):9117. https://doi.org/10.3390/ijms24119117
Chicago/Turabian StyleEgorova, Tatiana V., Ivan I. Galkin, Oleg A. Velyaev, Svetlana G. Vassilieva, Irina M. Savchenko, Vyacheslav A. Loginov, Marina A. Dzhenkova, Diana S. Korshunova, Olga S. Kozlova, Dmitry N. Ivankov, and et al. 2023. "In-Frame Deletion of Dystrophin Exons 8–50 Results in DMD Phenotype" International Journal of Molecular Sciences 24, no. 11: 9117. https://doi.org/10.3390/ijms24119117
APA StyleEgorova, T. V., Galkin, I. I., Velyaev, O. A., Vassilieva, S. G., Savchenko, I. M., Loginov, V. A., Dzhenkova, M. A., Korshunova, D. S., Kozlova, O. S., Ivankov, D. N., & Polikarpova, A. V. (2023). In-Frame Deletion of Dystrophin Exons 8–50 Results in DMD Phenotype. International Journal of Molecular Sciences, 24(11), 9117. https://doi.org/10.3390/ijms24119117