
Effect of Organic Selenium on the Homeostasis of Trace Elements, Lipid Peroxidation, and mRNA Expression of Antioxidant Proteins in Mouse Organs

 ,
,  ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
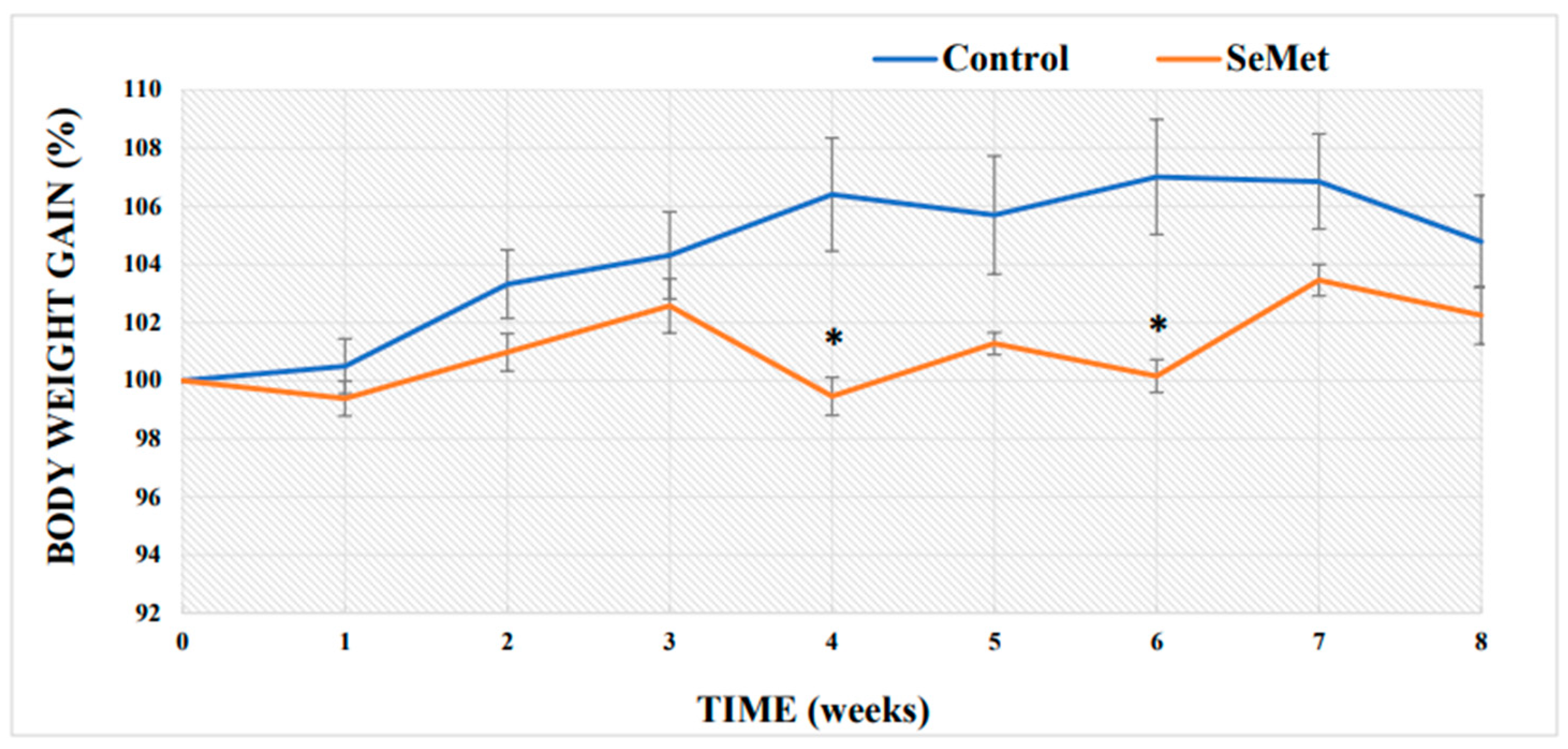
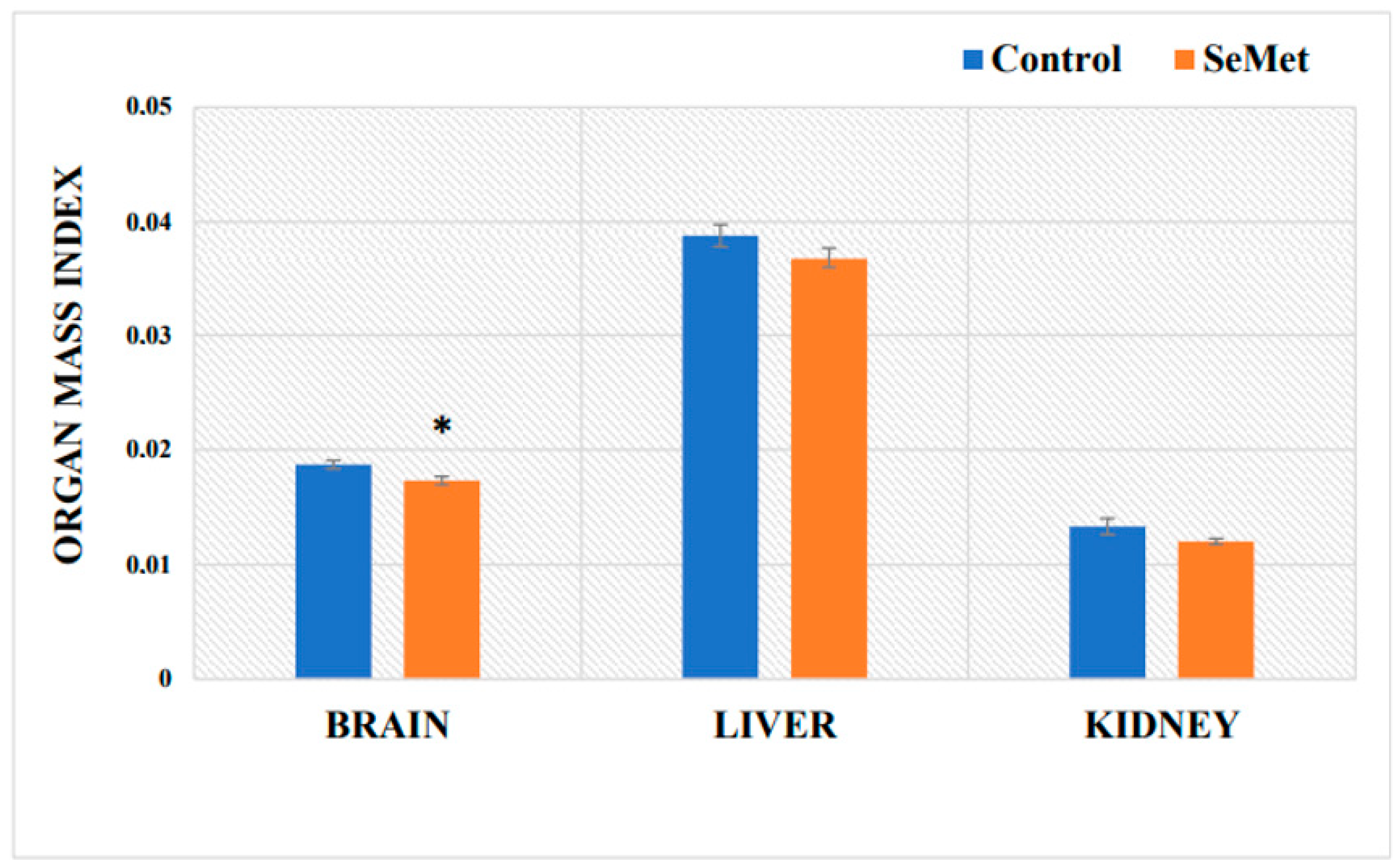
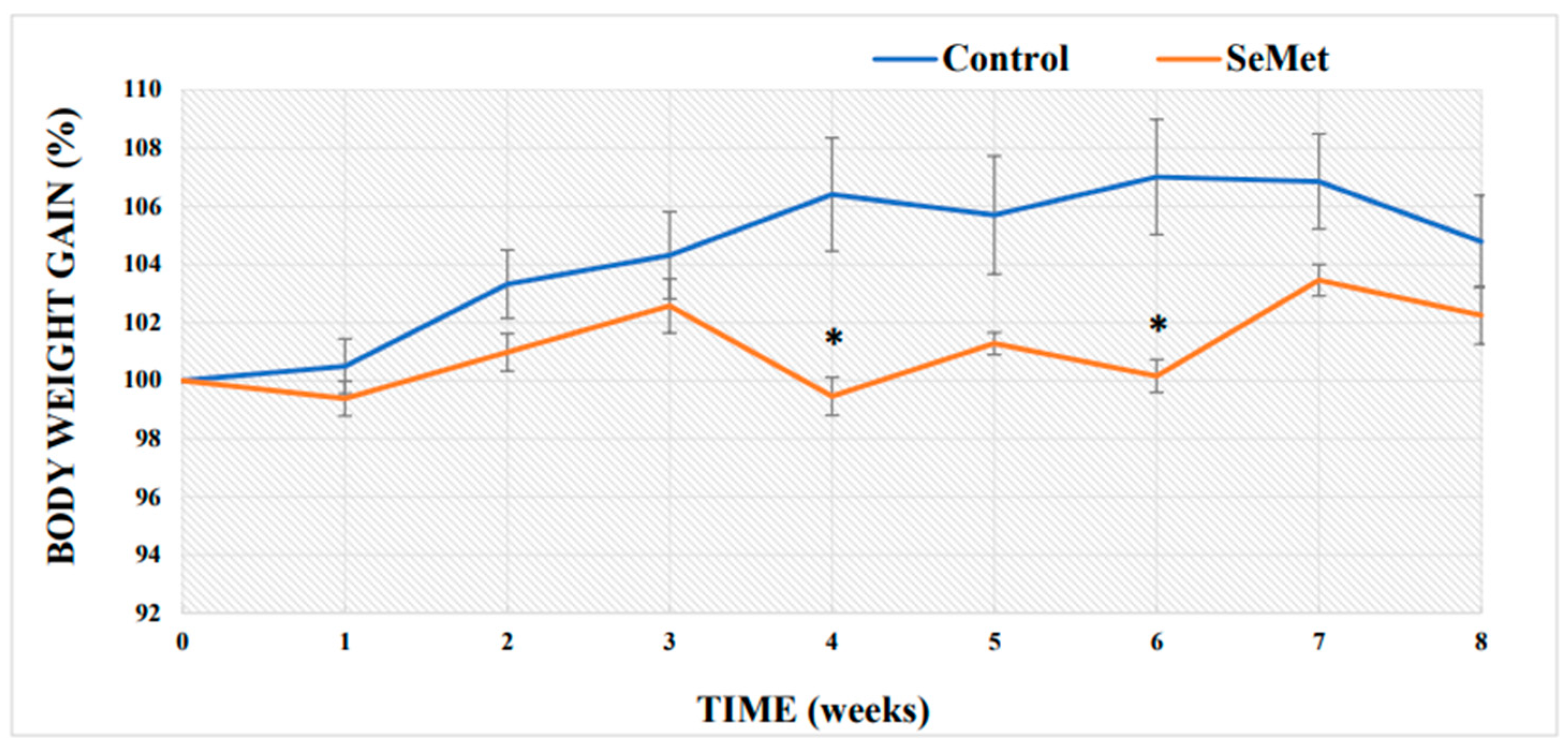
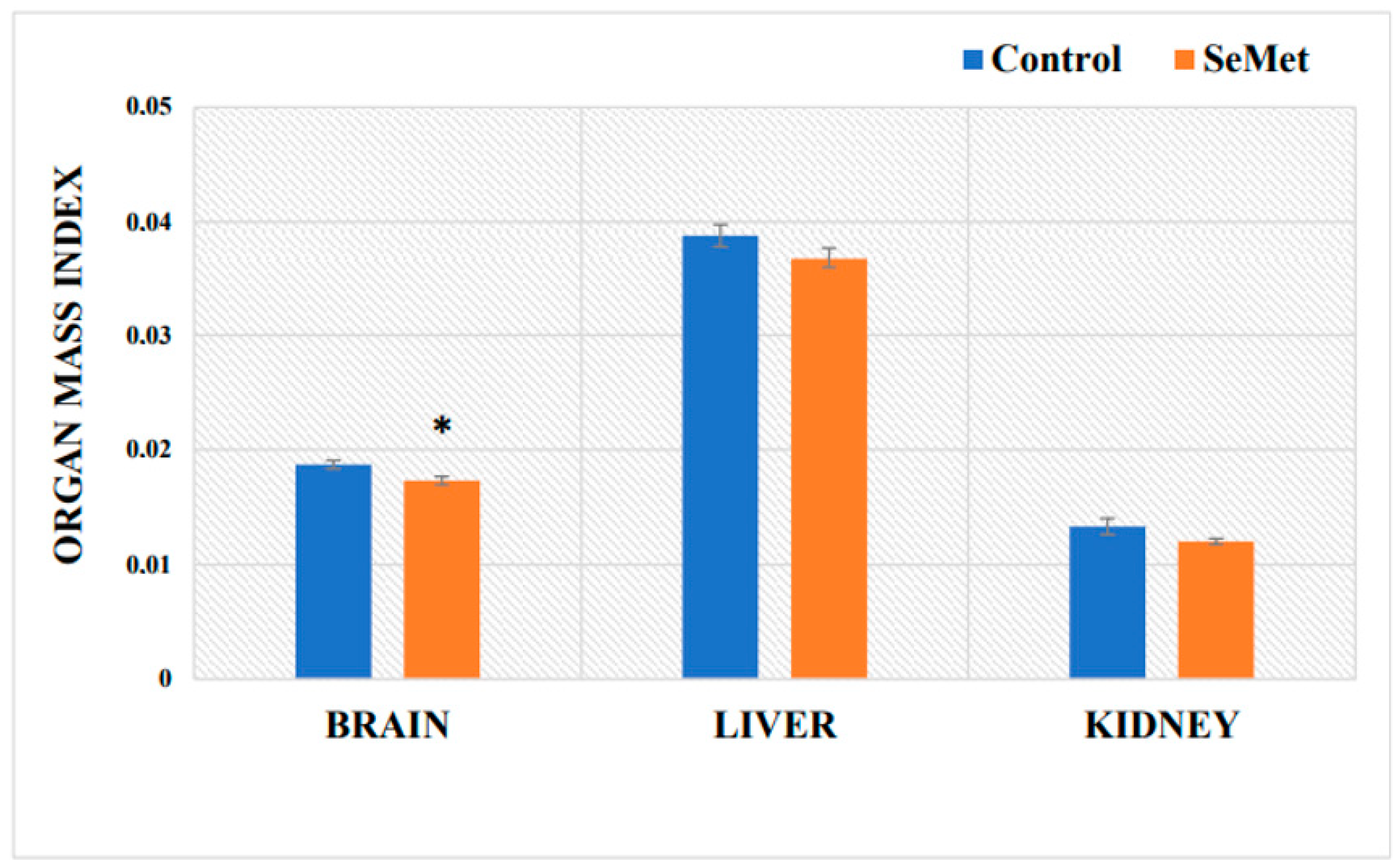
2.1. Evaluation of Selenium Effect on Body Weight and Relative Organ Mass Index of Mice
2.2. Evaluation of Selenium, Iron, Zinc, and Copper Concentrations in Mouse Tissues
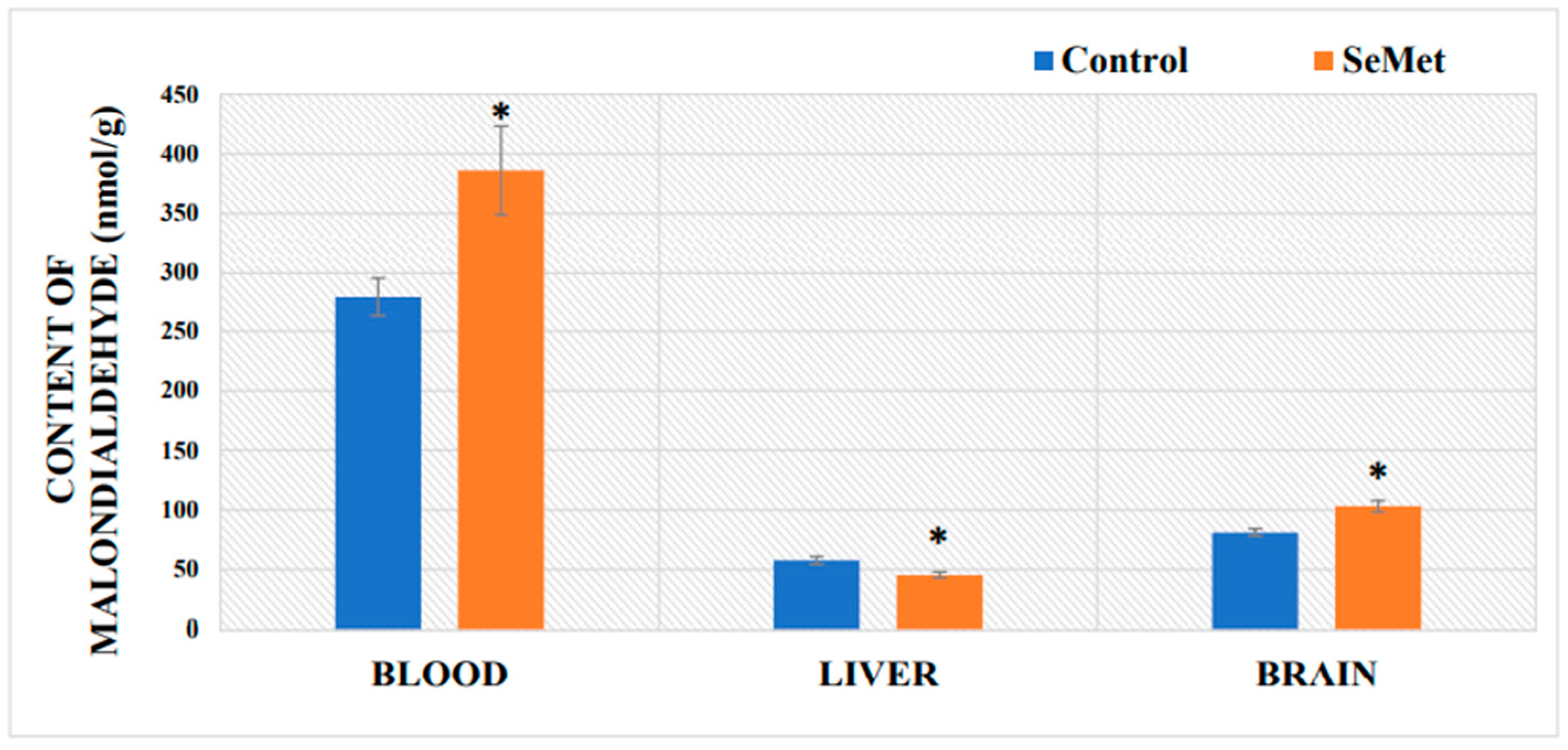
2.3. Evaluation of Malondialdehyde Content in Mouse Tissues after Exposure to Selenomethionine
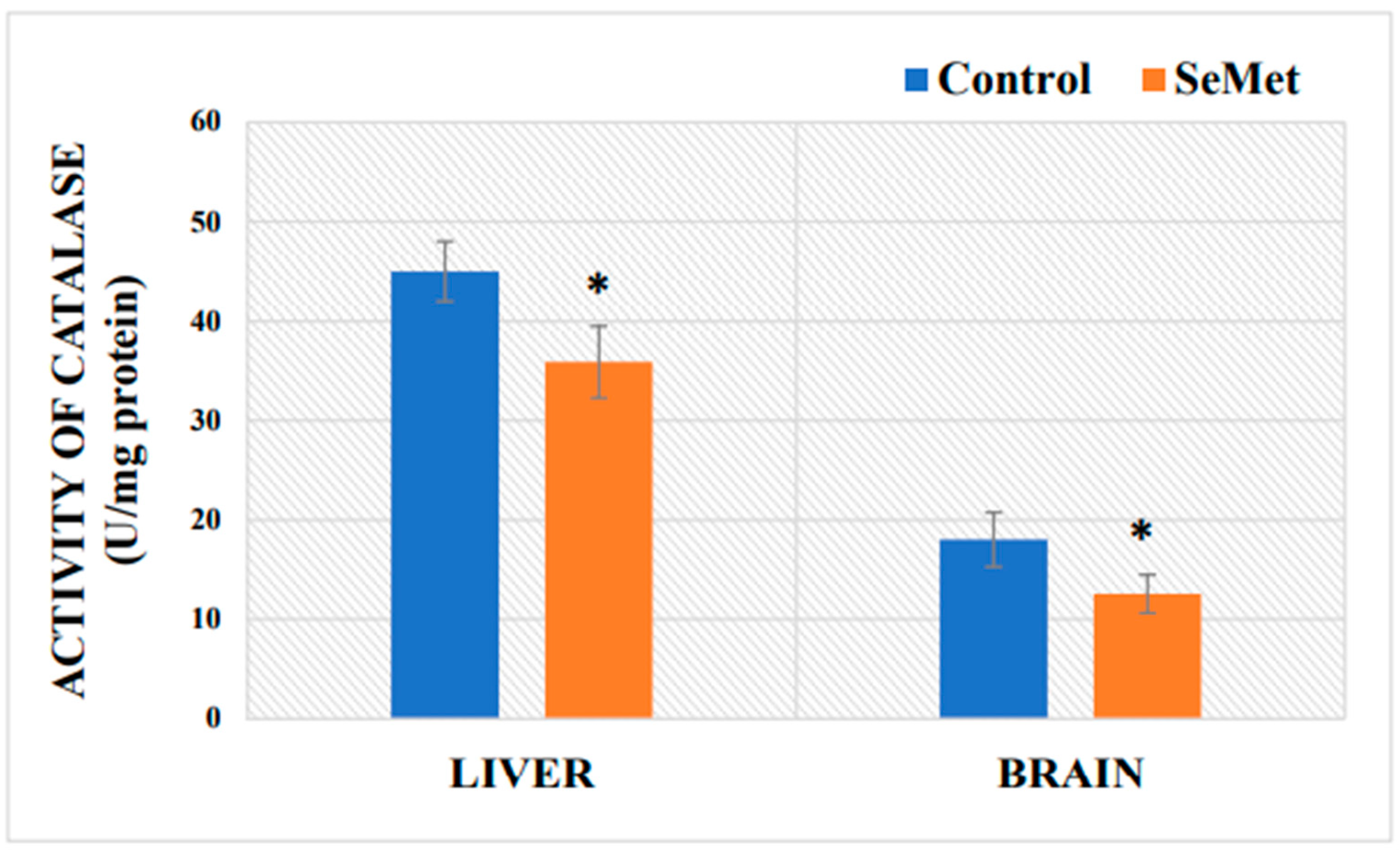
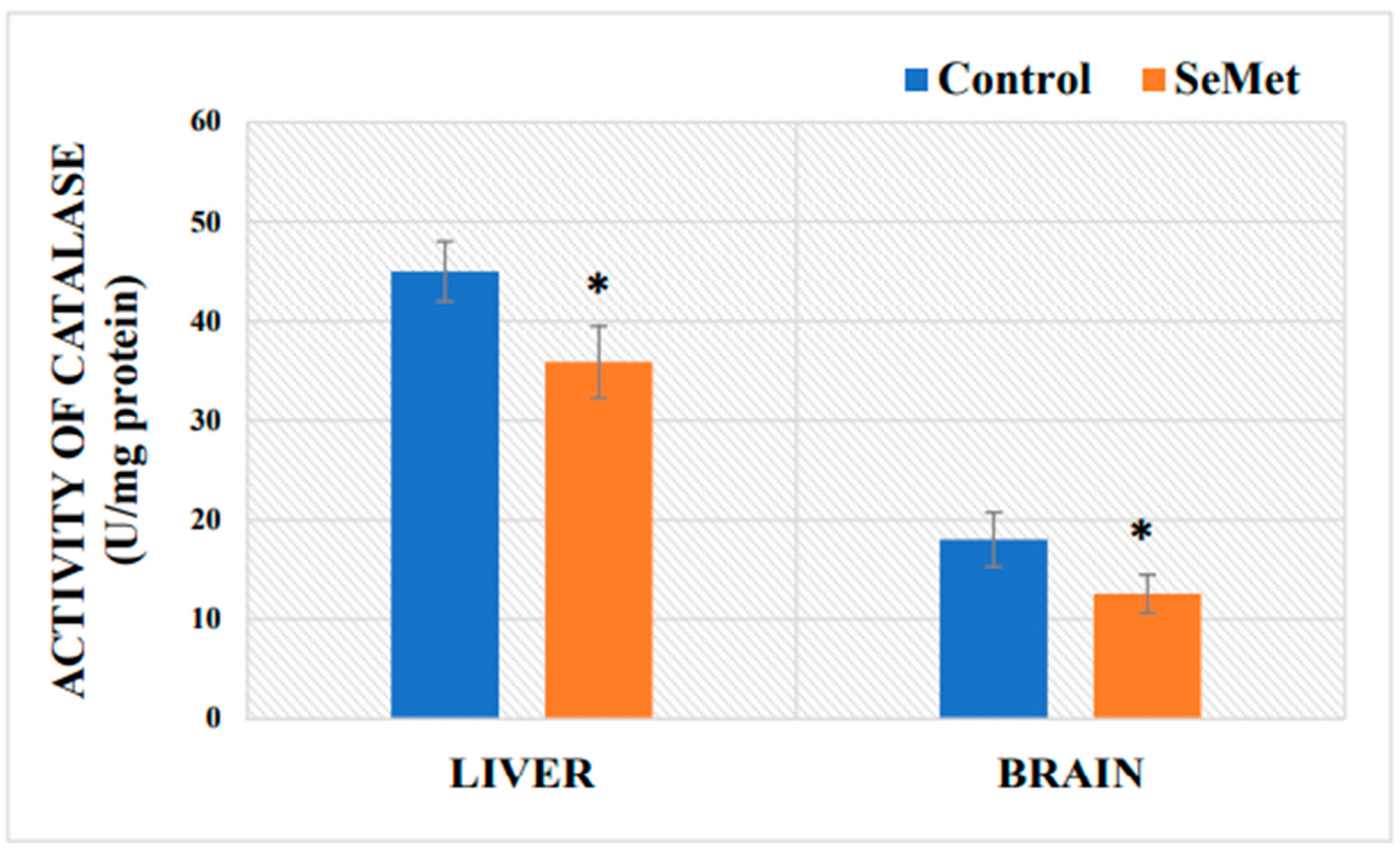
2.4. Determination of Mouse Brain and Liver Catalase Activity after Exposure to Selenomethionine
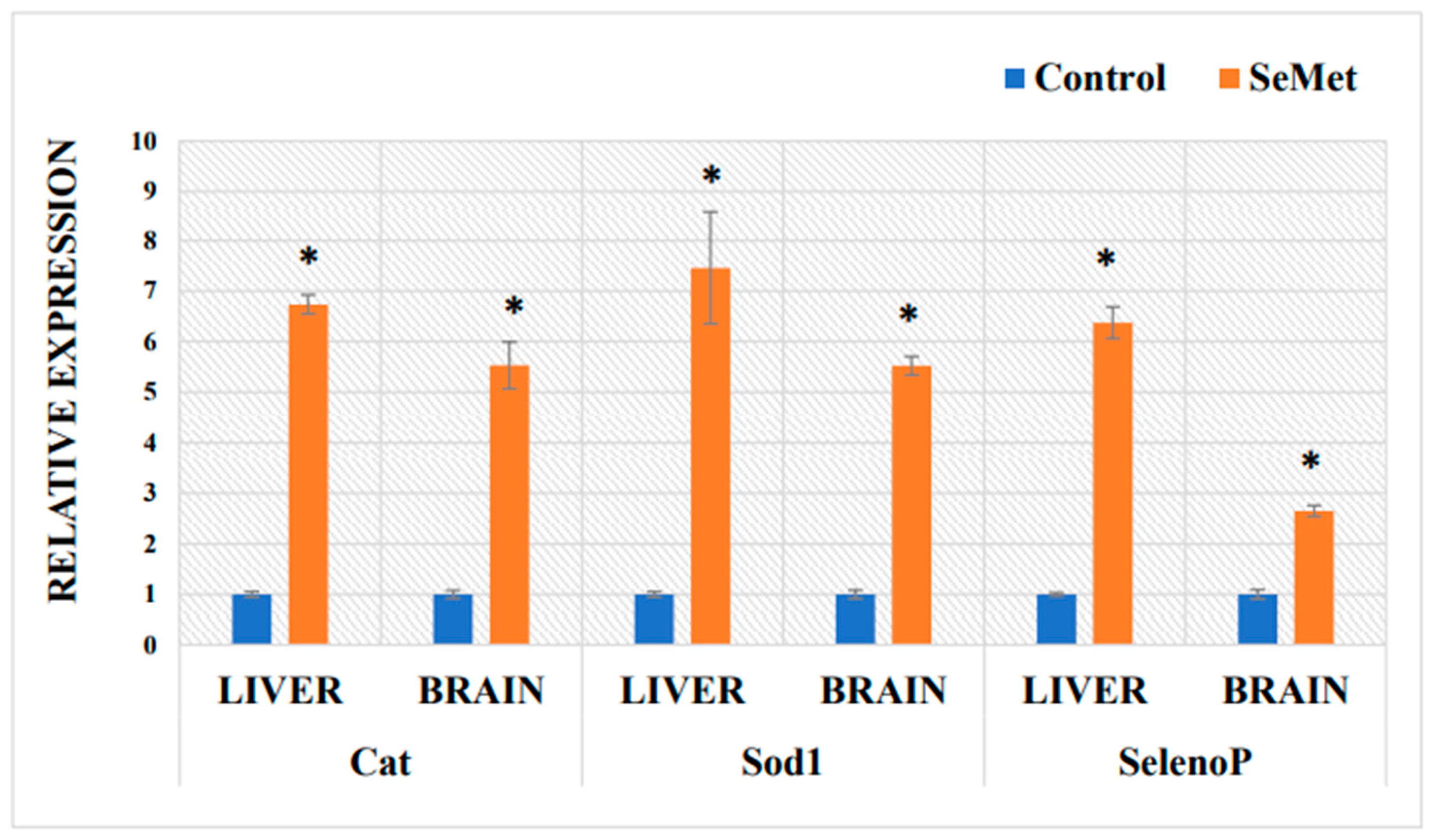
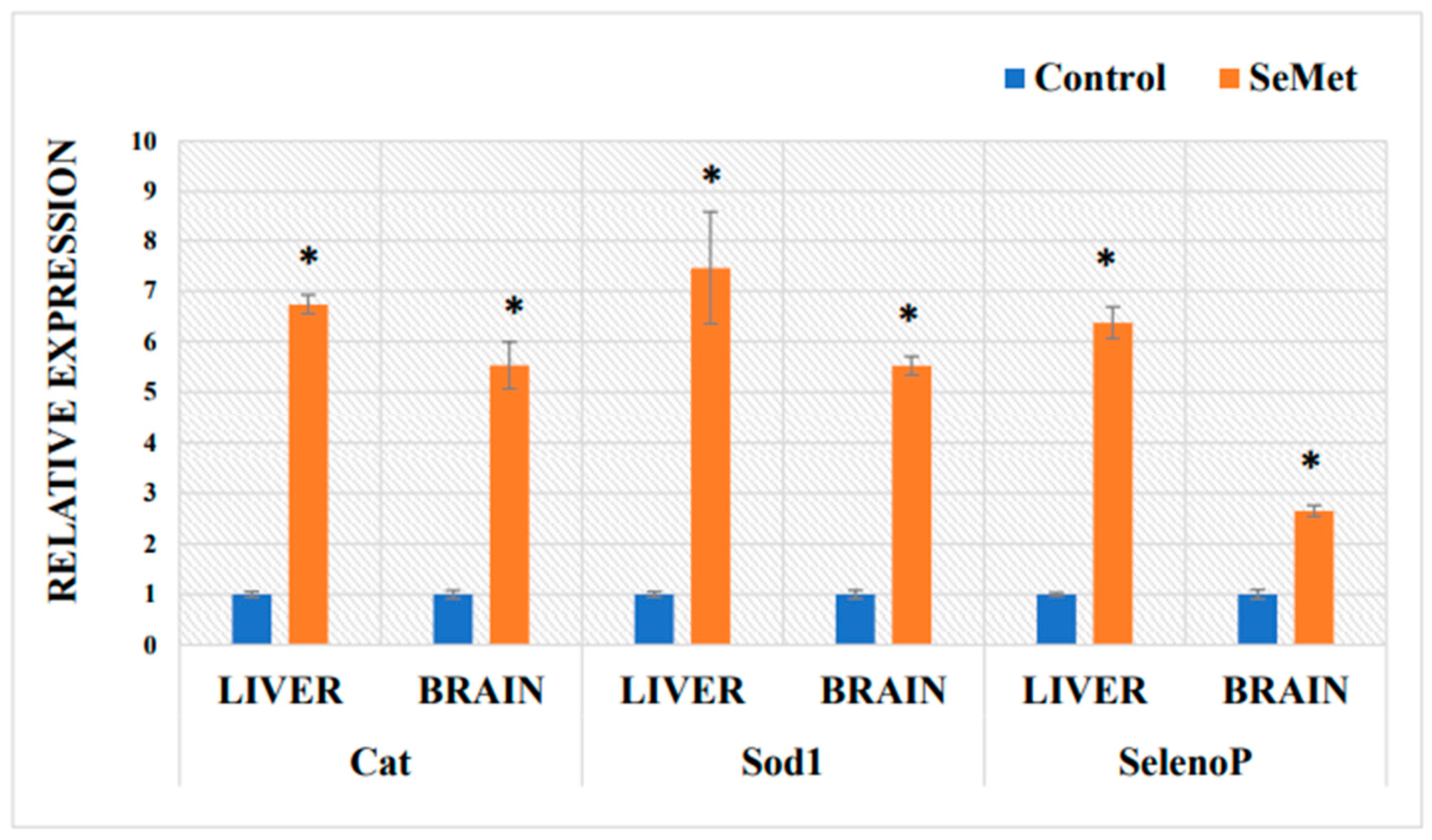
2.5. Determination of mRNA Expression in Mouse Liver and Brain of Antioxidative Proteins after Exposure to Selenomethionine
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Animals and Experimental Procedure
4.3. Determination of Malondialdehyde Content
4.4. Determination of Trace Element Concentrations
4.5. Preparation of the Brain and Liver Homogenates
4.6. Protein Concentration Assay
4.7. Measurement of Enzyme Catalase Activity
4.8. RNA Extraction and Real-Time Quantitative Reverse Transcription (RT) PCR (RT-qPCR)
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kieliszek, M.; Bano, I.; Zare, H. A comprehensive review on selenium and its effects on human health and distribution in Middle Eastern countries. Biol. Trace Elem. Res. 2022, 200, 971–987. [Google Scholar] [CrossRef]
- Kieliszek, M.; Błazejak, S. Current knowledge on the importance of selenium in food for living organisms: A review. Molecules 2016, 21, 609. [Google Scholar] [CrossRef] [Green Version]
- Burk, R.F.; Hill, K.E. Regulation of selenium metabolism and transport. Annu. Rev. Nutr. 2015, 35, 109–134. [Google Scholar] [CrossRef]
- Saito, Y. Selenium transport mechanism via selenoprotein P—Its physiological role and related diseases. Front. Nutr. 2021, 8, 685517. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryukov, G.; Castellano, S.; Novoselov, S.; Lobanov, A.; Zentab, O.; Guigo, R.; Gladyshev, R. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitts, M.W.; Hoffmann, P.R. Endoplasmic reticulum-resident selenoproteins as regulators of calcium signalling and homeostasis. Cell Calcium 2018, 70, 76–86. [Google Scholar] [CrossRef]
- Sreelatha, A.; Yee, S.S.; Lopez, V.A.; Park, B.C.; Kinch, L.N.; Pilch, S.; Servage, K.A.; Zhang, J.; Jiou, J.; Karasiewicz-Urbańska, M.; et al. Protein AMPylation by an evolutionarily conserved pseudokinase. Cell 2018, 175, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Gladyshev, V.N.; Arnér, E.S.; Berry, M.J.; Brigelius-Flohé, R.; Bruford, E.A.; Burk, R.F.; Carlson, B.A.; Castellano, S.; Chavatte, L.; Conrad, M.; et al. Selenoprotein gene nomenclature. J. Biol. Chem. 2016, 291, 24036–24040. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.; Lee, J.; Wu, C.; Guo, X.; Lee, B.J.; Chun, J.-S.; Kim, J.-H. The role of selenium metabolism and selenoproteins in cartilage homeostasis and arthropathies. Exp. Mol. Med. 2020, 52, 1198–1208. [Google Scholar] [CrossRef]
- Dos Santos, M.; da Silva, F.M.; Muccillo-Baisch, A.L. Selenium content of Brazilian foods: A review of the literature values. J. Food Compos. Anal. 2017, 58, 10–15. [Google Scholar] [CrossRef]
- Solovyev, N.; Drobyshev, E.; Blume, B.; Michalke, B. Selenium at the neural barriers: A review. Front. Neurosci. 2021, 15, 630016. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, L.; Schweizer, U. Hierarchical regulation of selenoprotein expression and sex-specific effects of selenium. Biochim. Biophys. Acta. 2009, 1790, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.E.; Wu, S.; Motley, A.K.; Stevenson, T.D.; Winfrey, V.P.; Capecchi, M.R.; Atkins, J.F.; Burk, R.F. Production of selenoprotein P (Sepp1) by hepatocytes is central to selenium homeostasis. J. Biol. Chem. 2012, 287, 40414–40424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodin, O.; Hackler, J.; Misra, S.; Wendt, S.; Sun, Q.; Laaf, E.; Stoppe, C.; Björnstedt, M.; Schomburg, L. Selenoprotein P as biomarker of selenium status in clinical trials with therapeutic dosages of selenite. Nutrients 2020, 12, 1067. [Google Scholar] [CrossRef] [Green Version]
- Lamarche, J.; Ronga, L.; Szpunar, J.; Lobinski, R. Characterization and quantification of selenoprotein P: Challenges to Mass Spectrometry. Int. J. Mol. Sci. 2021, 22, 6283. [Google Scholar] [CrossRef]
- Kipp, A.P.; Strohm, D.; Brigelius-Flohé, R.; Schomburg, L.; Bechthold, A.; Leschik-Bonnet, E.; Heseker, H.; (German Nutrition Society, DGE). Revised reference values for selenium intake. J. Trace Elem. Med. Biol. 2015, 32, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Stolwijk, J.M.; Garje, R.; Sieren, J.C.; Buettner, G.R.; Zakharia, Y. Understanding the redox biology of selenium in the search of targeted cancer therapies. Antioxidants 2020, 9, 420. [Google Scholar] [CrossRef]
- Saito, Y. Selenoprotein P as an in vivo redox regulator: Disorders related to its deficiency and excess. J. Clin. Biochem. Nutr. 2020, 66, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Rock, C.; Moos, P.J. Selenoprotein P protects cells from lipid hydroperoxides generated by 15-LOX-1. Prostaglandins Leukot Essent Fatty Acids 2010, 83, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Takebe, G.; Yarimizu, J.; Saito, Y.; Hayashi, T.; Nakamura, H.; Yodoi, J.; Nagasawa, S.; Takahashi, K. A comparative study on the hydroperoxide and thiol specificity of the glutathione peroxidase family and selenoprotein P. J. Biol. Chem. 2002, 277, 41254–41258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Arteel, G.E. Interaction of peroxynitrite with selenoproteins and glutathione peroxidase mimics. Free Radic. Biol. Med. 2000, 28, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Toyama, T.; Kaneko, T.; Arisawa, K.; Saito, Y. Metal-binding properties of selenoprotein P–its relation to structure and function. Met. Res. 2022, 3, 18–27. [Google Scholar]
- Pitts, M.W.; Byrns, C.N.; Ogawa-Wong, A.N.; Kremer, P.; Berry, M.J. Selenoproteins in nervous system development and function. Biol. Trace Elem. Res. 2014, 161, 231–245. [Google Scholar] [CrossRef] [Green Version]
- Schomburg, L. Selenium deficiency due to diet, pregnancy, severe illness, or COVID-19—A preventable trigger for autoimmune disease. Int. J. Mol. Sci. 2021, 22, 8532. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, Q.; Zhang, K.; Sun, W.; Jis, X.; Yang, Y.; Yin, J.; Tang, C.; Zhang, J. Se deficiency induces renal pathological changes by regulating selenoprotein expression, disrupting redox balance, and activating inflammation. Metallomics 2020, 12, 1576–1584. [Google Scholar] [CrossRef]
- Gupta, S.; Read, S.A.; Shackel, N.A.; Hebbard, L.; George, J.; Ahlenstiel, G. The role of micronutrients in the infection and subsequent response to hepatitis c virus. Cells 2019, 8, 603. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Lu, Y.; Wang, N.; Feng, Y. The role and mechanisms of selenium supplementation on fatty liver-associated disorder. Antioxidants 2022, 11, 922. [Google Scholar] [CrossRef]
- Kyung, H.L.; Jeong, D. Bimodal actions of selenium essential for antioxidant and toxic pro-oxidant activities: The selenium paradox (Review). Mol. Med. Rep. 2012, 5, 299–304. [Google Scholar]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [Green Version]
- Barchielli, G.; Capperucci, A.; Tanini, D. The role of selenium in pathologies: An updated review. Antioxidants 2022, 11, 251. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Alarcon, M.; Cabrera-Vique, C. Selenium in food and the human body: A review. Sci. Total Environ. 2008, 400, 115–141. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Min, J.; Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal. Transduct. Target. Ther. 2022, 7, 378. [Google Scholar] [CrossRef]
- Levenson, C.W.; Morris, D. Zinc and neurogenesis: Making new neurons from development to adulthood. Adv. Nutr. 2011, 2, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Fukada, T.; Yamasaki, S.; Nishida, K.; Murakami, M.; Hirano, T. Zinc homeostasis and signalling in health and diseases. J. Biol. Inorg. Chem. 2011, 16, 1123–1134. [Google Scholar] [CrossRef] [Green Version]
- Kwik-Uribe, C.L.; Gietzen, D.; German, J.B.; Golub, M.S.; Keen, C.L. Chronic marginal iron intakes during early development in mice result in persistent changes in dopamine metabolism and myelin composition. J. Nutr. 2000, 130, 2821–2830. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Li, S.; Huang, X.; Chen, X.; Shan, H.; Zhang, M. The role of copper homeostasis in brain disease. Int. J. Mol. Sci. 2022, 23, 13850. [Google Scholar] [CrossRef]
- Georgieff, M.K. The role of iron in neurodevelopment: Fetal iron deficiency and the developing hippocampus. BioChem. Soc Trans. 2008, 36, 1267–1271. [Google Scholar] [CrossRef] [Green Version]
- DiGirolamo, A.M.; Ramirez-Zea, M. Role of zinc in maternal and child mental health. Am. J. Clin. Nutr. 2009, 89, 940S–945S. [Google Scholar] [CrossRef] [Green Version]
- Bredholt, M.; Frederiksen, J.L. Zinc in multiple sclerosis. ASN Neuro. 2016, 8, 175909141665151. [Google Scholar] [CrossRef]
- Prasad, A.S. Clinical, immunological, anti-inflammatory and antioxidant roles of zinc. Exp. Gerontol. 2008, 43, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in neurodegeneration—Cause or consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Monnot, A.D. Regulation of brain iron and copper homeostasis by brain barrier systems: Implication in neurodegenerative diseases. Pharmacol. Ther. 2012, 133, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlowski, H.; Luczkowski, M.; Remelli, M.; Valensin, D. Copper, zinc and iron in neurodegenerative diseases (Alzheimer’s, Parkinson’s and prion diseases). Coord. Chem. Rev. 2012, 256, 2129–2141. [Google Scholar] [CrossRef]
- Gammella, E.; Correnti, M.; Cairo, G.; Recalcati, S. Iron availability in tissue microenvironment: The key role of ferroportin. Int. J. Mol. Sci. 2021, 22, 2986. [Google Scholar] [CrossRef]
- Solovyev, N.D. Importance of selenium and selenoprotein for brain function: From antioxidant protection to neuronal signalling. J. Inorg. Biochem. 2015, 153, 1–12. [Google Scholar] [CrossRef]
- Kieliszek, M.; Lipinski, B. Selenium supplementation in the prevention of coronavirus infections (COVID-19). Med. Hypotheses 2020, 143, 109878. [Google Scholar] [CrossRef]
- Chen, Y.C.; Prabhu, K.; Mastro, A. Is selenium a potential treatment for cancer metastasis? Nutrients 2013, 5, 1149–1168. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Qian, Z.Y.; Zhou, P.H.; Zhou, X.; Zhang, D.L.; He, N.; Zhang, J.; Liu, Y.H.; Gu, Q. Effects of oral selenium and magnesium co-supplementation on lipid metabolism, antioxidative status, histopathological lesions, and related gene expression in rats fed a high-fat diet. Lipids. Health Dis. 2018, 17, 165. [Google Scholar] [CrossRef] [Green Version]
- Lammi, M.J.; Qu, C. Selenium-related transcriptional regulation of gene expression. Int. J. Mol. Sci. 2018, 19, 2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crissman, J.W.; Goodman, D.G.; Hildebrandt, P.K.; Maronpot, R.R.; Prater, D.A.; Riley, J.H.; Seaman, W.J.; Thake, D.C. Best practices guideline: Toxicologic histopathology. Toxicol. Pathol. 2004, 32, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Kuršvietienė, L.; Mongirdienė, A.; Bernatonienė, J.; Šulinskienė, J.; Stanevičienė, I. Selenium anticancer properties and impact on cellular redox status. Antioxidants 2020, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallenberg, M.; Misra, S.; Björnstedt, M. Selenium cytotoxicity in cancer. Basic Clin. Pharmacol. Toxicol. 2014, 114, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Q.; Xiao, Y.; Li, P.; Tian, F.; Zhao, J.; Zhang, H.; Chen, W. Varied doses and chemical forms of selenium supplementation differentially affect mouse intestinal physiology. Food Funct. 2019, 10, 5398–5412. [Google Scholar] [CrossRef]
- Huang, Z.; Rose, A.H.; Hoffmann, P.R. The role of selenium in inflammation and immunity: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox. Signal. 2012, 16, 705–743. [Google Scholar] [CrossRef] [Green Version]
- Edens, F.W.; Sefton, A.E. Organic selenium in animal nutrition—Utilisation, metabolism, storage and comparison with other selenium sources. Anim. Nutr. 2016, 4, E9. [Google Scholar] [CrossRef]
- Moreno-Reyes, R.; Egrise, D.; Ne`ve, J.; Ne`ve, N.; Pasteels, J.L.; Schoutens, A. Selenium deficiency-induced growth retardation is associated with an impaired bone metabolism and osteopenia. J. Bone Min. Res. 2001, 16, 1556–1563. [Google Scholar] [CrossRef]
- Gorini, F.; Sabatino, L.; Pingitore, A.; Vassalle, C. Selenium: An element of life essential for thyroid function. Molecules 2021, 26, 7084. [Google Scholar] [CrossRef]
- Raines, A.M.; Sunde, R.A. Selenium toxicity but not deficient or super-nutritional selenium status vastly alters the transcriptome in rodents. BMC Genom. 2011, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Chen, L.; Guo, K.; Zheng, L.; Liu, B.; Yu, W.; Guo, C.; Liu, Z.; Chen, Y.; Tang, Z. Effects of different selenium levels on gene expression of a subset of selenoproteins and antioxidative capacity in mice. Biol. Trace Elem. Res. 2013, 154, 255–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attia, Y.A.; Abdalah, A.A.; Zeweil, H.S.; Bovera, F.; Tag El-Din, A.A.; Araft, M.A. Effect of inorganic or organic selenium supplementation on productive performance, egg quality and some physiological traits of dual-purpose breeding hens. Czech. J. Anim. Sci. 2010, 55, 505–519. [Google Scholar] [CrossRef] [Green Version]
- Tos-Luty, S.; Obuchowska-Przebirowska, D.; Latuszynska, J.; Musik, I.; Tokarska-Rodak, M. Comparison of histological and ultrastructural changes in mice organs after supplementation with inorganic and organic selenium. Ann. Agric. Environ. Med. 2003, 10, 87–91. [Google Scholar] [PubMed]
- Li, K.X.; Wang, J.S.; Yuan, D.; Zhao, R.X.; Wang, Y.X.; Zhan, X.A. Effects of different selenium sources and levels on antioxidant status in broiler breeders. Asian-Australas J. Anim. Sci. 2018, 31, 1939–1945. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Li, R.; Jin, H.; You, H.J.; Ji, G.E. Effects of selenium- and zinc-enriched Lactobacillus plantarum SeZi on antioxidant capacities and Gut microbiome in an ICR mouse model. Antioxidants 2020, 9, 1028. [Google Scholar] [CrossRef]
- Grotto, D.; Carneiro, M.F.H.; de Castro, M.M.; Garcia, S.C.; Junior, F.B. Long-term excessive selenium supplementation induces hypertension in rats. Biol. Trace Elem. Res. 2018, 182, 70–77. [Google Scholar] [CrossRef]
- Staneviciene, I.; Sulinskiene, J.; Sadauskiene, I.; Liekis, A.; Ruzgaite, A.; Naginiene, R.; Baranauskiene, D.; Simakauskiene, V.; Krusnauskas, R.; Viezeliene, D. Effect of selenium on the iron homeostasis and oxidative damage in brain and liver of mice. Antioxidants 2022, 11, 1216. [Google Scholar] [CrossRef]
- Łanocha-Arendarczyk, N.; Baranowska-Bosiacka, I.; Kot, K.; Pilarczyk, B.; Tomza-Marciniak, A.; Kabat-Koperska, J.; Kosik-Bogacka, D. Biochemical profile, liver and kidney selenium (Se) status during acanthamoebiasis in a mouse model. Folia. Biol. 2018, 66, 33–40. [Google Scholar] [CrossRef]
- Fairweather-Tait, S.J.; Collings, R.; Hurst, R. Selenium bioavailability: Current knowledge and future research requirements. Am. J. Clin. Nutr. 2010, 91, 1484S–1491S. [Google Scholar] [CrossRef] [Green Version]
- Patterson, B.H.; Combs, G.F., Jr.; Taylor, P.R.; Patterson, K.Y.; Moler, J.E.; Wastney, M.E. Selenium kinetics in humans change following 2 years of supplementation with selenomethionine. Front. Endocrinol. 2021, 12, 621687. [Google Scholar] [CrossRef]
- Akahoshi, N.; Anan, Y.; Hashimoto, Y.; Tokoro, N.; Mizuno, R.; Hayashi, S.; Yamamoto, S.; Shimada, K.I.; Kamata, S.; Ishii, I. Dietary selenium deficiency or selenomethionine excess drastically alters organ selenium contents without altering the expression of most selenoproteins in mice. J. Nutr. Biochem. 2019, 69, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Touat-Hamici, Z.; Legrain, Y.; Bulteau, A.-L.; Chavatte, L. Selective up-regulation of human selenoproteins in response to oxidative stress. J. Biol. Chem. 2014, 289, 14750–14761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, N.; Bansal, M.P. Diminished reproductive potential of male mice in response to selenium-induced oxidative stress: Involvement of HSP70, HSP70-2, and MSJ-1. J. BioChem. Mol. Toxicol. 2009, 23, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Liu, Q.; Xu, H.B.; Zhu, Y.S.; Yang, X.L. Effects of selenium overexposure on glutathione peroxidase and thioredoxin reductase gene expressions and activities. Biol. Trace Elem. Res. 2002, 89, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Zoidis, E.; Pappas, A.C.; Georgiou, C.A.; Komaitis, E.; Feggeros, K. Selenium affects the expression of GPx4 and catalase in the liver of chicken. Comp. BioChem. Physiol. B BioChem. Mol. Biol. 2010, 155, 294–300. [Google Scholar] [CrossRef]
- Zhou, J.C.; Zhao, H.; Li, J.G.; Xia, X.J.; Wang, K.N.; Zhang, Y.J.; Liu, Y.; Zhao, Y.; Lei, X.G. Selenoprotein gene expression in thyroid and pituitary of young pigs is not affected by dietary selenium deficiency or excess. J. Nutr. 2009, 139, 1061–1066. [Google Scholar] [CrossRef] [Green Version]
- Schomburg, L. Selenoprotein P—Selenium transport protein, enzyme and biomarker of selenium status. Free Radic Biol. Med. 2022, 191, 150–163. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Y.; Schweizer, U.; Savaskan, N.E.; Hua, D.; Kipnis, J.; Hatfield, D.L.; Gladyshev, V.N. Comparative analysis of selenocysteine machinery and selenoproteome gene expression in mouse brain identifies neurons as key functional sites of selenium in mammals. J. Biol. Chem. 2008, 283, 2427–2438. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-H.; Song, G.-L. Roles of selenoproteins in brain function and the potential mechanism of selenium in Alzheimer’s disease. Front. Neurosci. 2021, 15, 646518. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, V.; Diederich, L.; Keller, T.C.S.; Kramer, C.M.; Luckstadt, W.; Panknin, C.; Suvorava, T.; Isakson, B.E.; Kelm, M.; Cortese-Krott, M.M. Red blood cell function and dysfunction: Redox regulation, nitric oxide metabolism, anemia. Antioxid. Redox. Signal. 2017, 26, 718–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcelik, D.; Ozaras, R.; Gurel, Z.; Uzun, H.; Aydin, S. Copper-mediated oxidative stress in rat liver. Biol. Trace Elem. Res. 2003, 96, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Zhang, Z.; Wen, L.; Chen, C.; Shi, Q.; Zhang, Y.; Ni, J.; Liu, Q. Selenomethionine ameliorates cognitive decline, reduces tau hyperphosphorylation, and reverses synaptic deficit in the triple transgenic mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 41, 85–99. [Google Scholar] [CrossRef]
- Shen, H.-M.; Yang, C.-F.; Ding, W.X.; Liu, J.; Ong, C.-N. Dual role of glutathione in selenite-induced oxidative stress and apoptosis in human hepatoma cells. Free Radic Biol. Med. 2000, 28, 1115–1124. [Google Scholar] [CrossRef]
- Genchi, G.; Lauria, G.; Catalano, A.; Sinicropi, M.S.; Carocci, A. Biological activity of selenium and its impact on human health. Int. J. Mol. Sci. 2023, 24, 2633. [Google Scholar] [CrossRef]
- Li, Z.; Liu, Y.; Wei, R.; Yong, V.W.; Xue, M. The important role of zinc in neurological diseases. Biomolecules 2022, 13, 28. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of oxidative stress. BioChem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef]
- Ward, R.J.; Dexter, D.T.; and Crichton, R.R. Iron, neuroinflammation and neurodegeneration. Int. J. Mol. Sci. 2022, 23, 7267. [Google Scholar] [CrossRef]
- Amin, S.B.; Myers, G.; Wang, H. Association between neonatal iron overload and early human brain development in premature infants. Early Hum. Dev. 2012, 88, 583–587. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Haldar, S.; Tripathi, A.K.; Horback, K.; Wong, J.; Sharma, D.; Beserra, A.; Suda, S.; Anbalagan, C.; Dev, S.; et al. Brain iron homeostasis: From molecular mechanisms to clinical significance and therapeutic opportunities. Antioxid. Redox. Signal. 2014, 20, 1324–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urrutia, P.J.; Bórquez, D.A.; Núñez, M.T. Inflaming the brain with iron. Antioxidants 2021, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, Y.; Li, T.; Wang, X.; Zhu, C. Iron metabolism and brain development in premature infants. Front. Physiol. 2019, 10, 463. [Google Scholar] [CrossRef]
- Hwang, I.; Lee, J.; Huh, J.Y.; Park, J.; Lee, H.B.; Ho, Y.S.; Ha, H. Catalase deficiency accelerates diabetic renal injury through peroxisomal dysfunction. Diabetes 2012, 61, 728–738. [Google Scholar] [CrossRef] [Green Version]
- Mita, Y.; Nakayama, K.; Inari, S.; Nishito, Y.; Yoshioka, Y.; Sakai, N.; Sotani, K.; Nagamura, T.; Kuzuhara, Y.; Inagaki, K.; et al. Selenoprotein P-neutralizing antibodies improve insulin secretion and glucose sensitivity in type 2 diabetes mouse models. Nat. Commun. 2017, 8, 1658. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R. Critical role of zinc as either an antioxidant or a prooxidant in cellular systems. Oxid. Med. Cell Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Marreiro, D.; Cruz, K.; Morais, J.; Beserra, J.; Severo, J.; de Oliveira, A. Zinc and oxidative stress: Current mechanisms. Antioxidants 2017, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, S.E.; Rehuman, N.A.; Harilal, S.; Vincent, A.; Rajamma, R.G.; Behl, T.; Uddin, M.S.; Ashraf, G.M.; Mathew, B. Molecular mechanism of zinc neurotoxicity in Alzheimer’s disease. Environ. Sci. Pollu. Res. 2020, 27, 43542–43552. [Google Scholar] [CrossRef]
- Prasad, A.S. Discovery of human zinc deficiency: Its impact on human health and disease. Adv. Nutr. 2013, 4, 176–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maret, W.; Sandstead, H.H. Zinc requirements and the risks and benefits of zinc supplementation. J. Trace Elem. Med. Biol. 2006, 20, 3–18. [Google Scholar] [CrossRef]
- Wang, L.; Yin, Y.L.; Liu, X.Z.; Shen, P.; Zheng, Y.G.; Lan, X.R.; Lu, C.B.; Wang, J.Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapia, L.; González-Agüero, M.; Cisternas, M.F.; Suazo, M.; Cambiazo, V.; Uauy, R.; González, M. Metallothionein is crucial for safe intracellular copper storage and cell survival at normal and supra-physiological exposure levels. Biochem. J. 2004, 378, 617–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malosio, M.L.; Tecchio, F.; Squitti, R. Molecular mechanisms underlying copper function and toxicity in neurons and their possible therapeutic exploitation for Alzheimer’s disease. Aging. Clin. Exp. Res. 2021, 33, 2027–2030. [Google Scholar] [CrossRef]
- Rossi, L.; Squitti, R.; Calabrese, L.; Rotilio, G.; Rossini, P.M. Alteration of peripheral markers of copper homeostasis in Alzheimer’s disease patients: Implications in aetiology and therapy. J. Nutr. Health Aging 2007, 11, 408–417. [Google Scholar]
- Osredkar, J. Copper and zinc, biological role and significance of copper/zinc imbalance. J. Clin. Toxicol. 2011, 2013, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, G. The role of zinc and copper in autism spectrum disorders. Acta Neurobiol. Exp. 2013, 73, 225–236. [Google Scholar]
- Yasuda, H.; Yoshida, K.; Yasuda, Y.; Tsutsui, T. Infantile zinc deficiency: Association with autism spectrum disorders. Sci. Rep. 2011, 1, 129. [Google Scholar] [CrossRef] [Green Version]
- Grabrucker, A.M. Environmental factors in autism. Front. Psychiatry 2013, 3, 118. [Google Scholar] [CrossRef] [Green Version]
- Maret, W. Zinc biochemistry: From a single zinc enzyme to a key element of life. Adv. Nutr. 2013, 4, 82–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide detoxification by brain cells. J. Neurosci. Res. 2005, 79, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.C.; Corsi, D.; Cavi, N.; Bruni, N.; Dosio, F. Superoxide dismutase administration: A review of proposed human uses. Molecules 2021, 26, 1844. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Rašković, A.; Milanović, I.; Pavlović, N.; Ćebović, T.; Vukmirović, S.; Mikov, M. Antioxidant activity of rosemary (Rosmarinus officinalis L.) essential oil and its hepatoprotective potential. BMC Complement Altern. Med. 2014, 14, 225. [Google Scholar] [CrossRef] [Green Version]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The chemistry of reactive oxygen species (ROS) revisited: Outlining their role in biological macromolecules (DNA, lipids and proteins) and induced pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Chelikani, P.; Fita, I.; Loewen, P.C. Diversity of structures and properties among catalases. Cell Mol. Life Sci. 2004, 61, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta Mol. Cell Res. 2006, 1763, 1755–1766. [Google Scholar] [CrossRef] [Green Version]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [Green Version]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alexandria J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, Y.; Balk, M.; Öğüş, İ.H.; Özer, N. The mechanism of inhibition of human erythrocyte catalase by Azide. Turk J. Biol. 2004, 28, 65–70. [Google Scholar]
- Khan, A.; Khan, I.; Khan, S.; Afzal, S.; Hamid, M.; Tariq, M.; Haq, I.U.; Ullah, N.; Khan, M.A.; Bilal, S.; et al. Selenium-enriched probiotics improve hepatic protection by regulating pro-inflammatory cytokines and antioxidant capacity in broilers under heat stress conditions. J. Adv. Vet. Anim. Res. 2019, 6, 355. [Google Scholar] [CrossRef] [PubMed]
- Nogales, F.; Ojeda, M.L.; Fenutría, M.; Murillo, M.L.; Carreras, O. Role of selenium and glutathione peroxidase on development, growth, and oxidative balance in rat offspring. Reproduction 2013, 146, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, M.; Mihara, M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal. Biochem. 1978, 86, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with polyphenol reagent. J. Biol. Chem. 1951, 193, 2652–2675. [Google Scholar] [CrossRef]
- Sadauskiene, I.; Liekis, A.; Bernotiene, R.; Sulinskiene, J.; Kasauskas, A.; Zekonis, G. The efects of Buckwheat leaf and flower extracts on antioxidant status in mice organs. Oxid. Med. Cell Longev. 2018, 2018, 6712407. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Group | Trace Element | Blood (µg/L Se) (mg/L Fe, Cu, Zn) | Brain (µg/g) | Liver (µg/g) |
|---|---|---|---|---|
| Control | Selenium (Se) | 210.508 ± 24.138 | 0.061 ± 0.012 | 0.615 ± 0.095 |
| SeMet | 2526.303 ± 181.058 * | 2.573 ± 0.147 * | 9.490 ± 0.331 * | |
| Control | Iron (Fe) | 706.097 ± 32.350 | 27.265 ± 1.895 | 119.068 ± 11.259 |
| SeMet | 497.121 ± 9.849 * | 38.803 ± 2.157 * | 272.894 ± 9.521 * | |
| Control | Copper (Cu) | 0.985 ± 0.064 | 3.715 ± 0.164 | 7.795 ± 0.416 |
| SeMet | 0.594 ± 0.026 * | 6.266 ± 0.581 * | 6.877 ±0.125 * | |
| Control | Zinc (Zn) | 6.640 ± 0.294 | 16.863 ± 0.505 | 32.453 ± 1.403 |
| SeMet | 6.307 ± 0.219 | 24.624 ± 2.389 * | 24.624 ± 2.389 * |
| Genes | Forward Primer, 5′-3′ | Reverse Primer, 5′-3′ |
|---|---|---|
| Sod1 | AGCATGGCGATGAAAGCGG | CCTGCACTGGTACAGCCTTGT |
| Cat | AAGATTGCCTTCTCCGGGTG | GACATCAGGTCTCTGCGAGG |
| SelenoP | GAAACTGTTCAGGGGCTTGC | CACATTGCTGAGGTTGTCCTCG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staneviciene, I.; Levinas, D.; Sadauskiene, I.; Liekis, A.; Viezeliene, D.; Kursvietiene, L.; Naginiene, R.; Baranauskiene, D.; Simakauskiene, V.; Vaitkiene, P.; et al. Effect of Organic Selenium on the Homeostasis of Trace Elements, Lipid Peroxidation, and mRNA Expression of Antioxidant Proteins in Mouse Organs. Int. J. Mol. Sci. 2023, 24, 9704. https://doi.org/10.3390/ijms24119704
Staneviciene I, Levinas D, Sadauskiene I, Liekis A, Viezeliene D, Kursvietiene L, Naginiene R, Baranauskiene D, Simakauskiene V, Vaitkiene P, et al. Effect of Organic Selenium on the Homeostasis of Trace Elements, Lipid Peroxidation, and mRNA Expression of Antioxidant Proteins in Mouse Organs. International Journal of Molecular Sciences. 2023; 24(11):9704. https://doi.org/10.3390/ijms24119704
Chicago/Turabian StyleStaneviciene, Inga, Dovydas Levinas, Ilona Sadauskiene, Arunas Liekis, Dale Viezeliene, Lolita Kursvietiene, Rima Naginiene, Dale Baranauskiene, Vaida Simakauskiene, Paulina Vaitkiene, and et al. 2023. "Effect of Organic Selenium on the Homeostasis of Trace Elements, Lipid Peroxidation, and mRNA Expression of Antioxidant Proteins in Mouse Organs" International Journal of Molecular Sciences 24, no. 11: 9704. https://doi.org/10.3390/ijms24119704
APA StyleStaneviciene, I., Levinas, D., Sadauskiene, I., Liekis, A., Viezeliene, D., Kursvietiene, L., Naginiene, R., Baranauskiene, D., Simakauskiene, V., Vaitkiene, P., Miniotaite, G., & Sulinskiene, J. (2023). Effect of Organic Selenium on the Homeostasis of Trace Elements, Lipid Peroxidation, and mRNA Expression of Antioxidant Proteins in Mouse Organs. International Journal of Molecular Sciences, 24(11), 9704. https://doi.org/10.3390/ijms24119704