
Targeting RNA Structure to Inhibit Editing in Trypanosomes

Abstract
:1. Introduction
2. Results
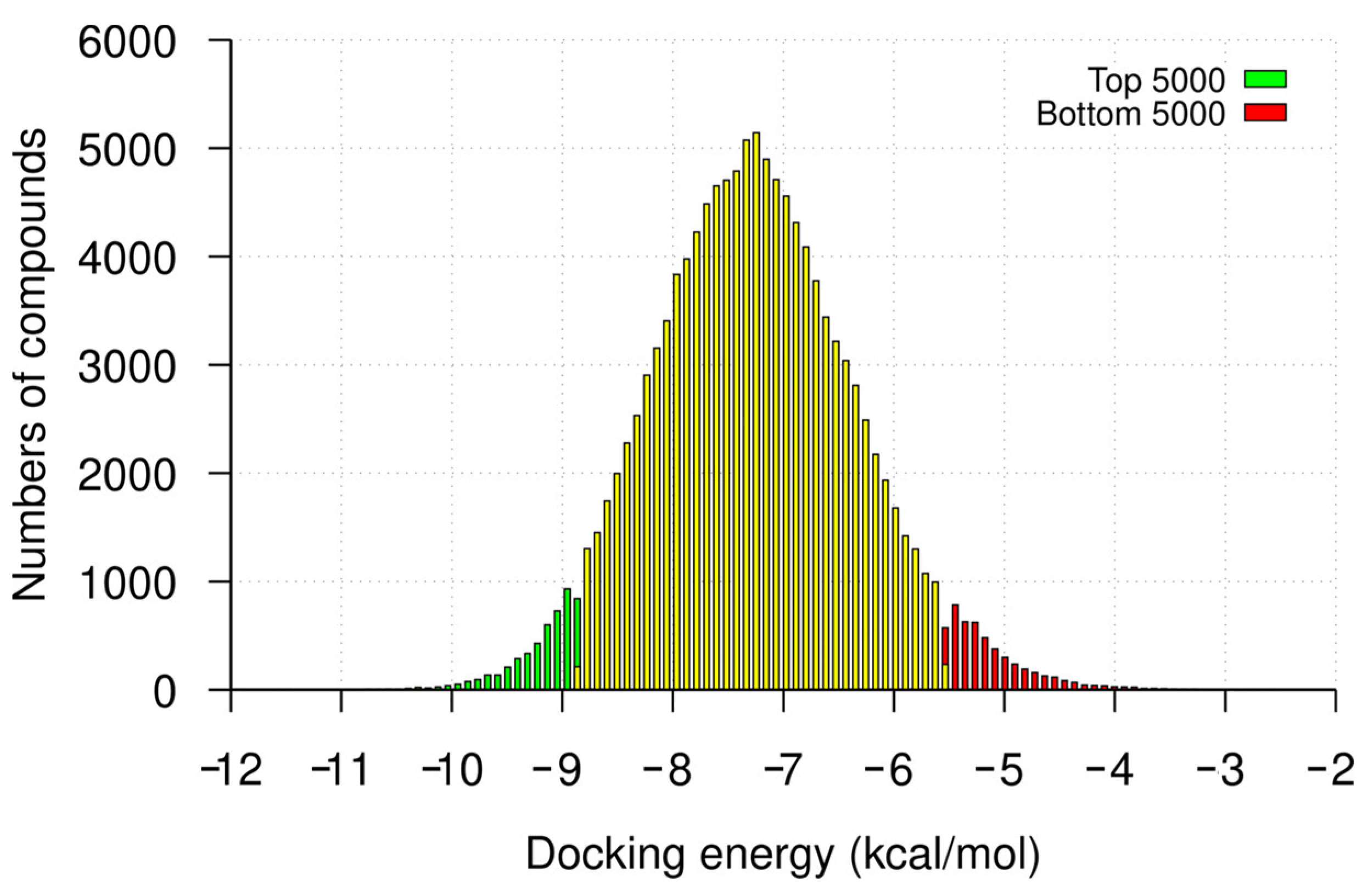
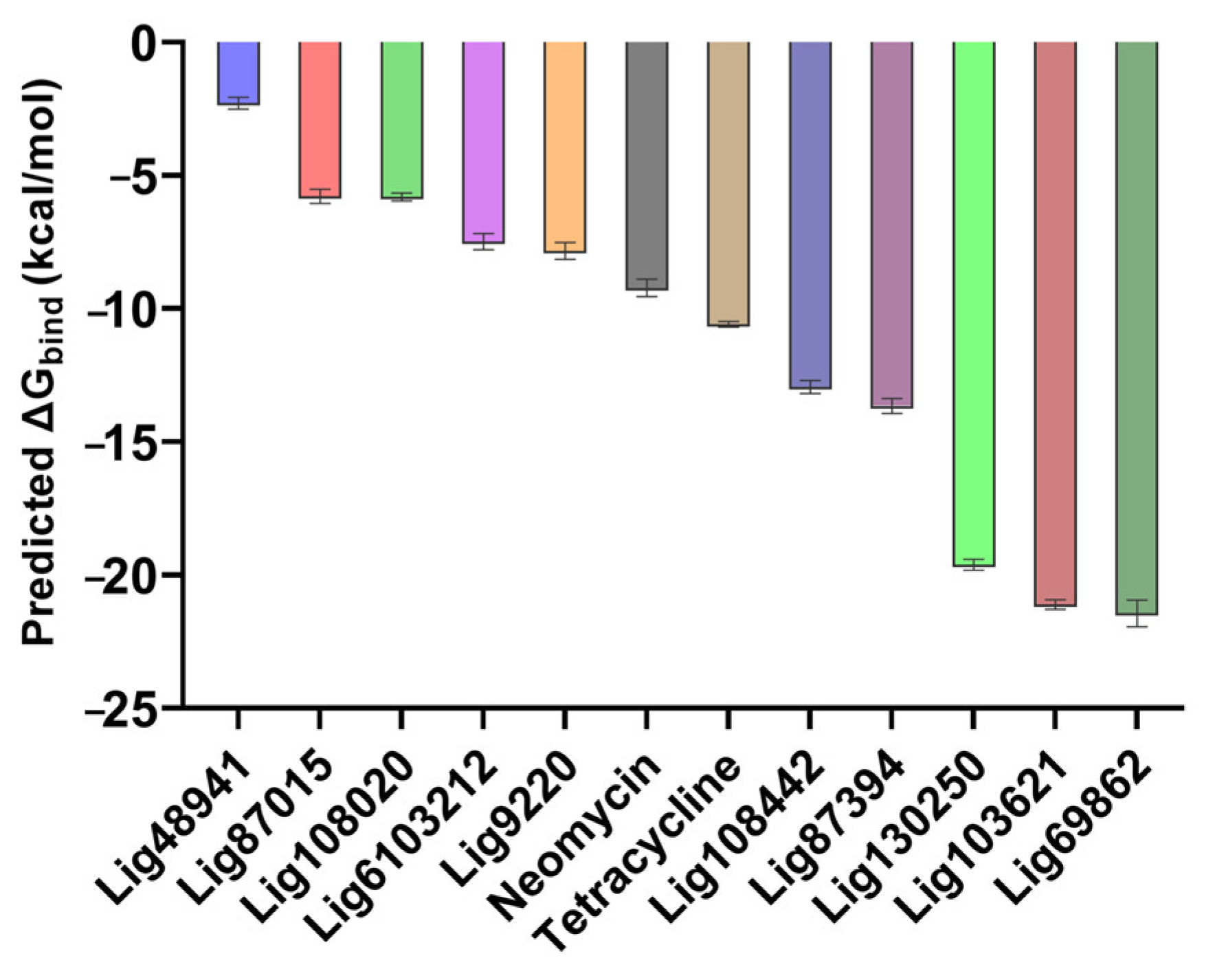
2.1. Hit Identification by Virtual Screening
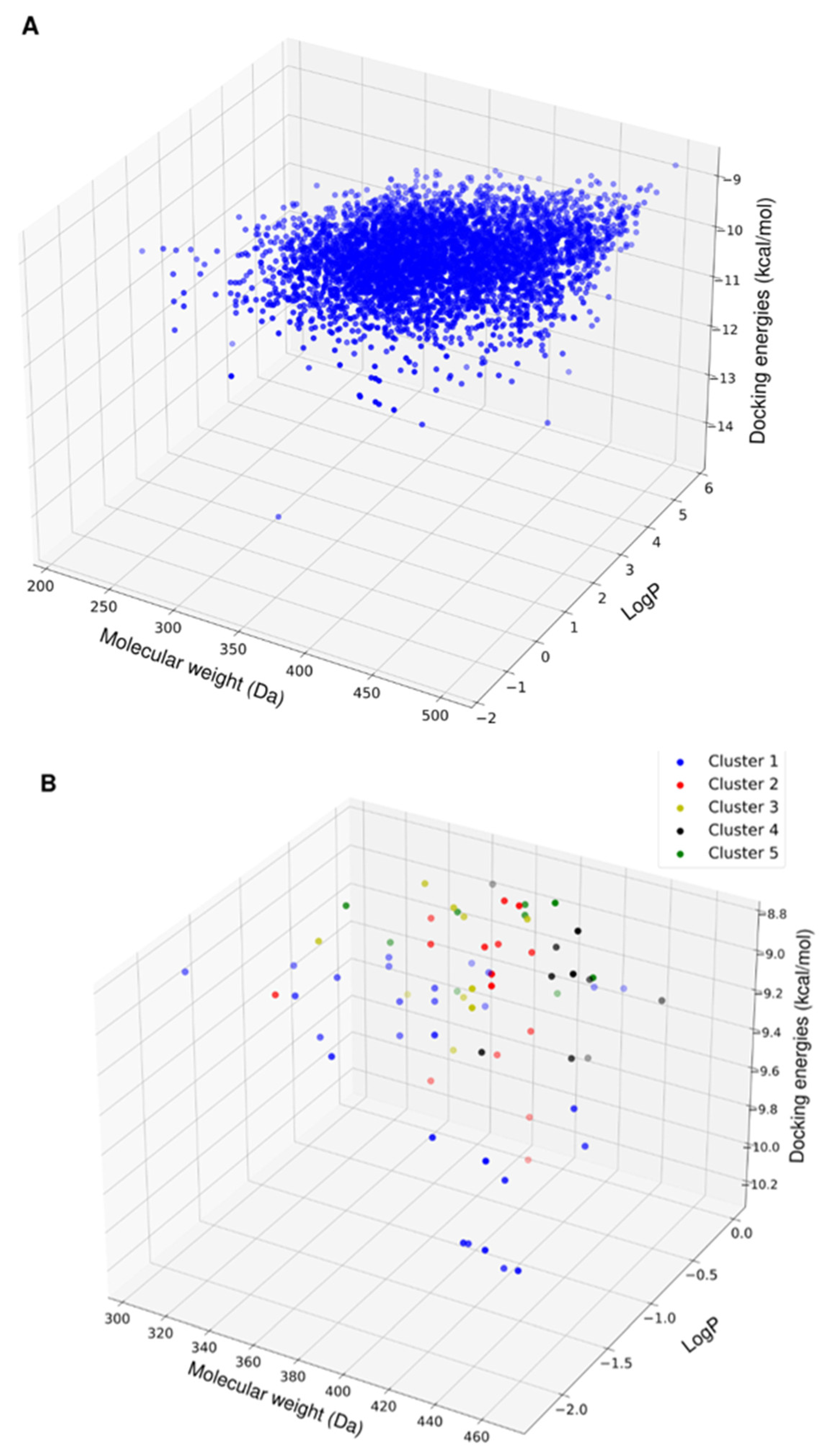
2.2. Chemoinformatic Analysis
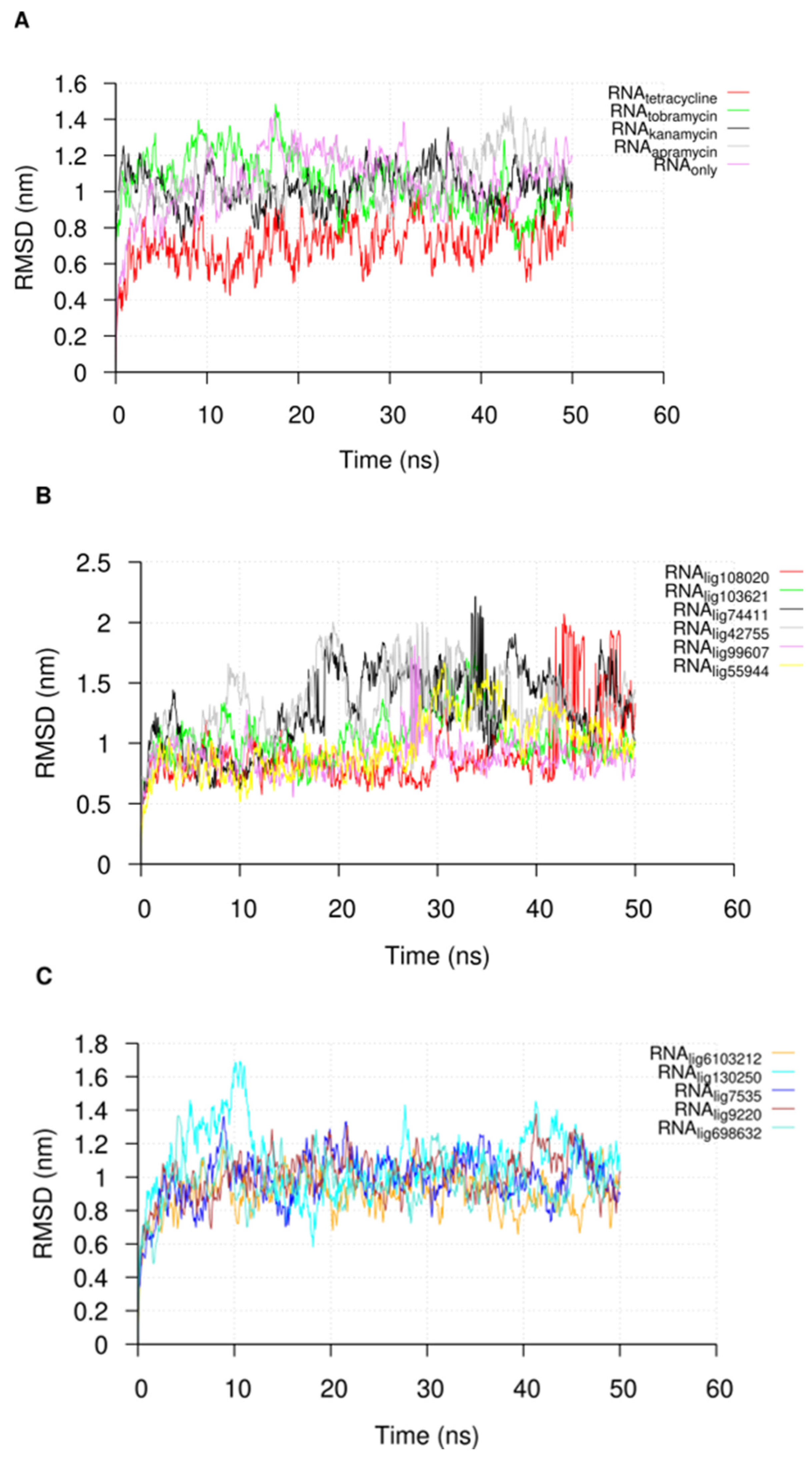
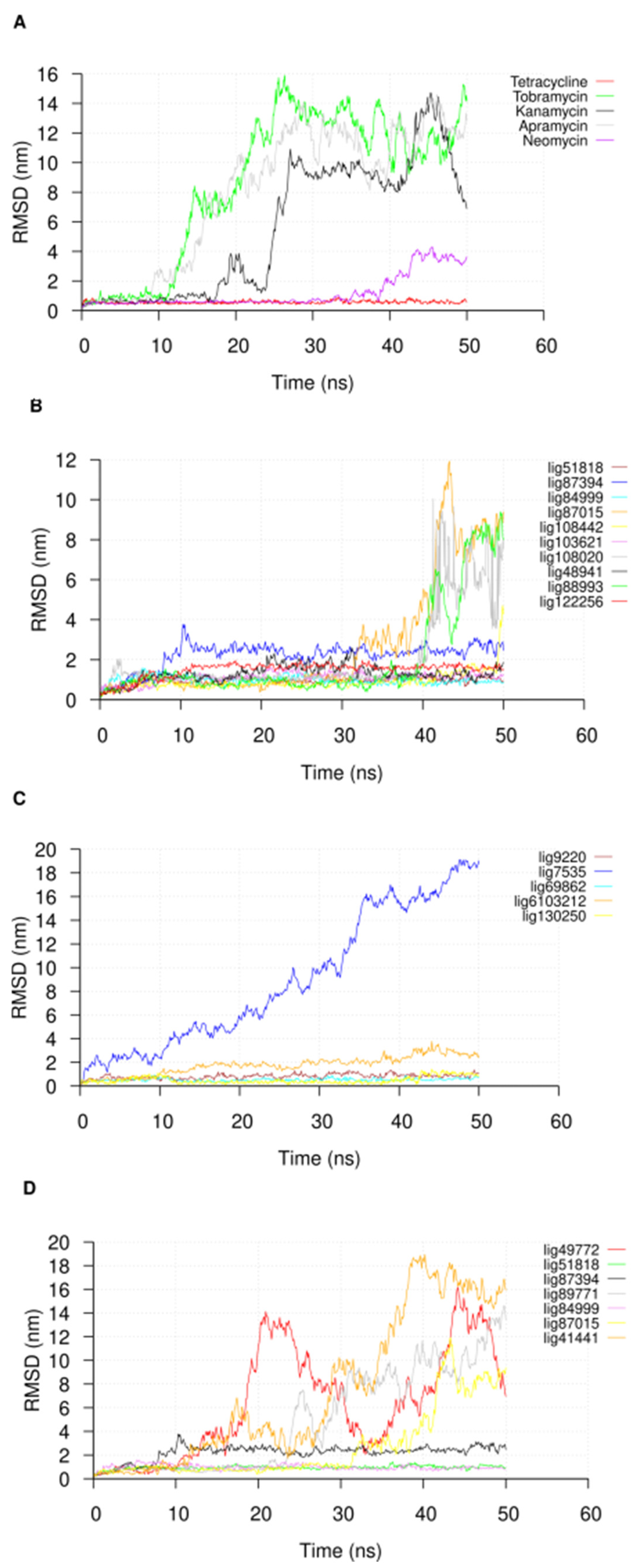
2.3. Molecular Dynamic Simulations
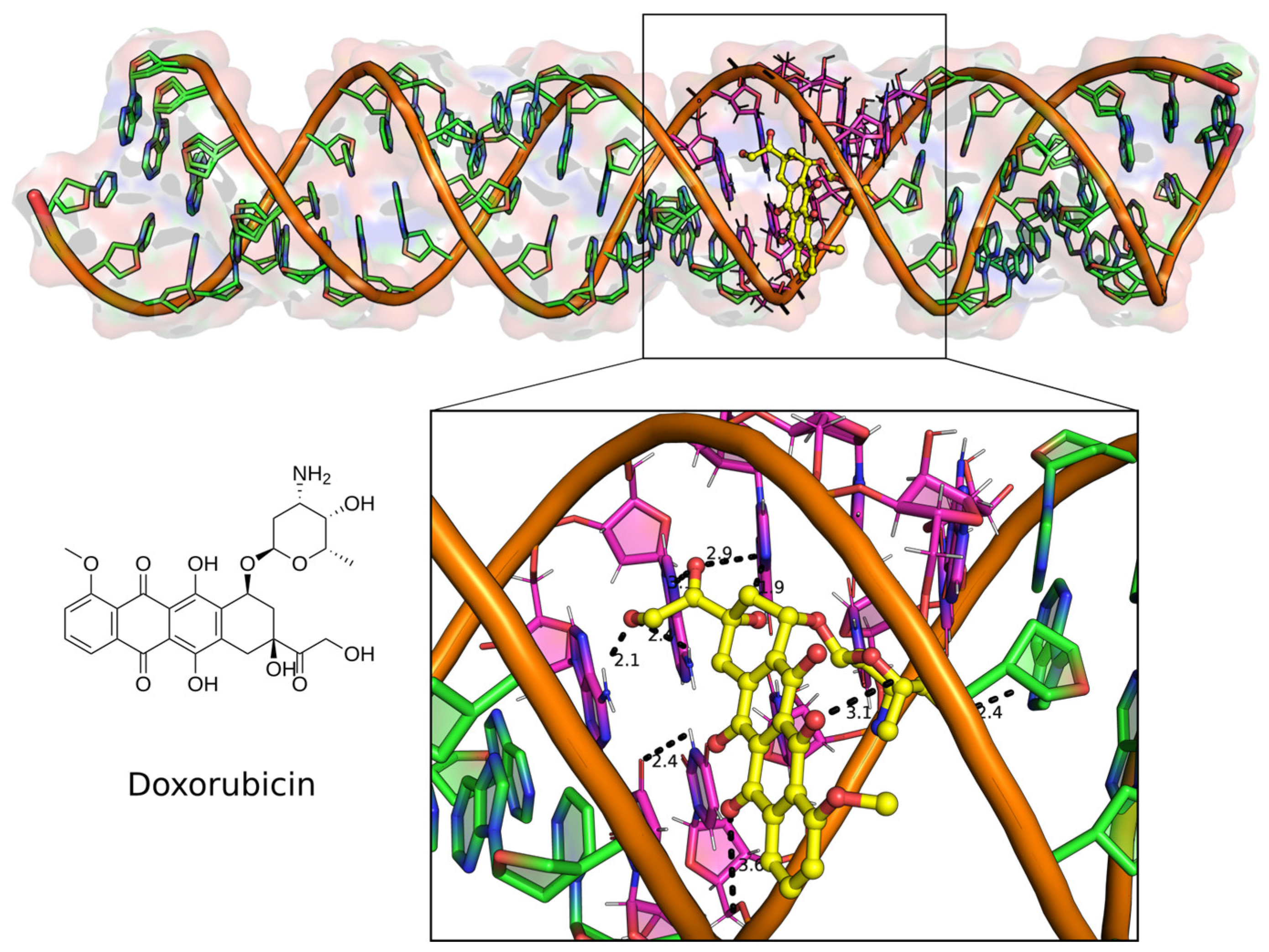
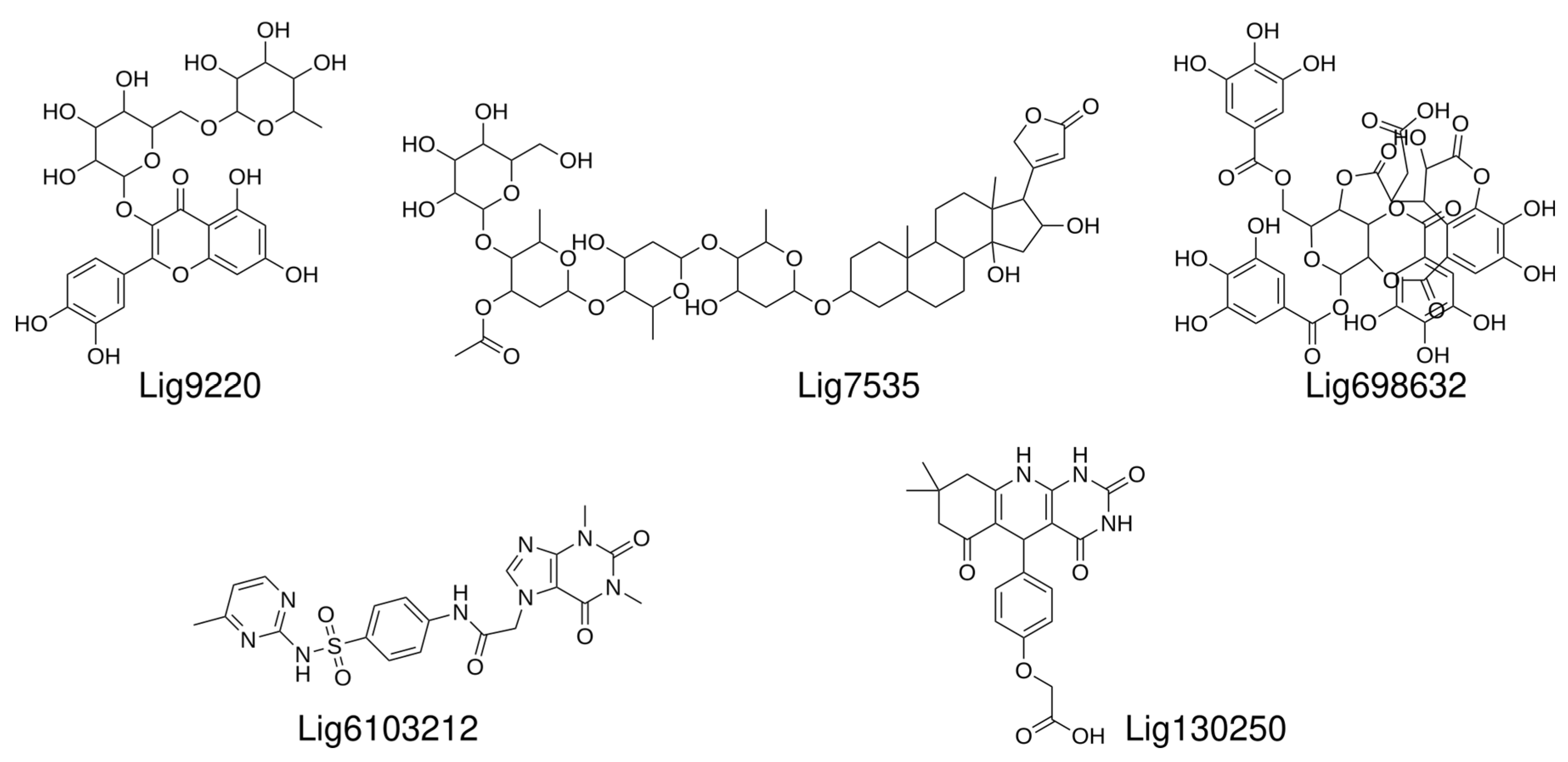
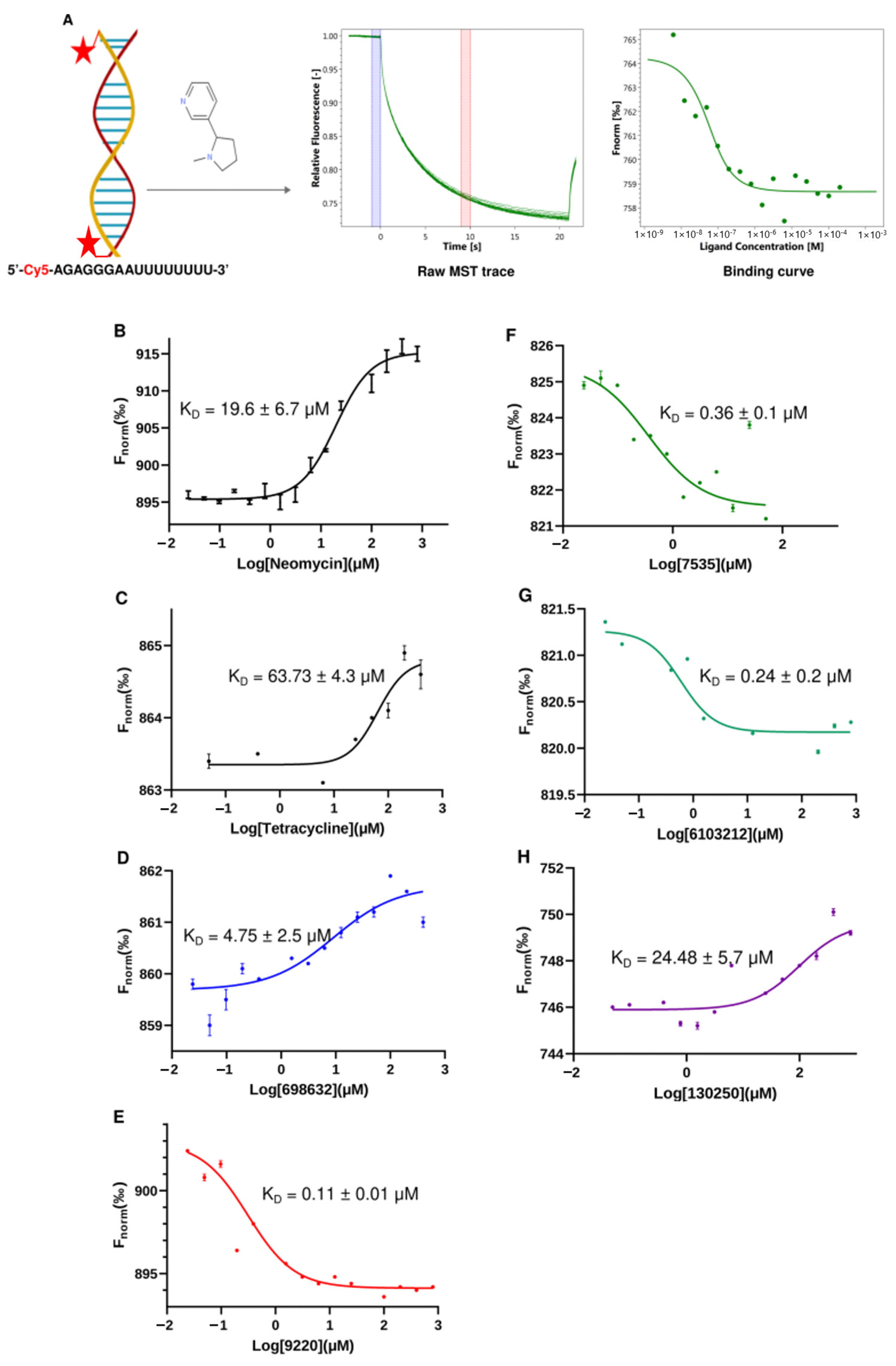
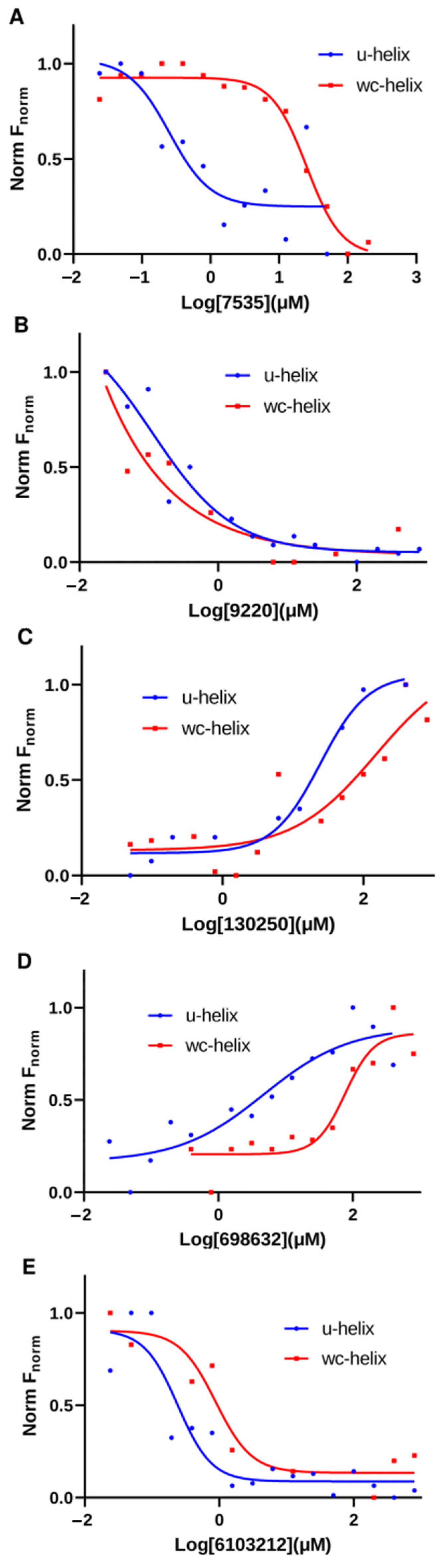
2.4. Top Hit Compounds Show Direct Binding to U-Helix with High Affinities
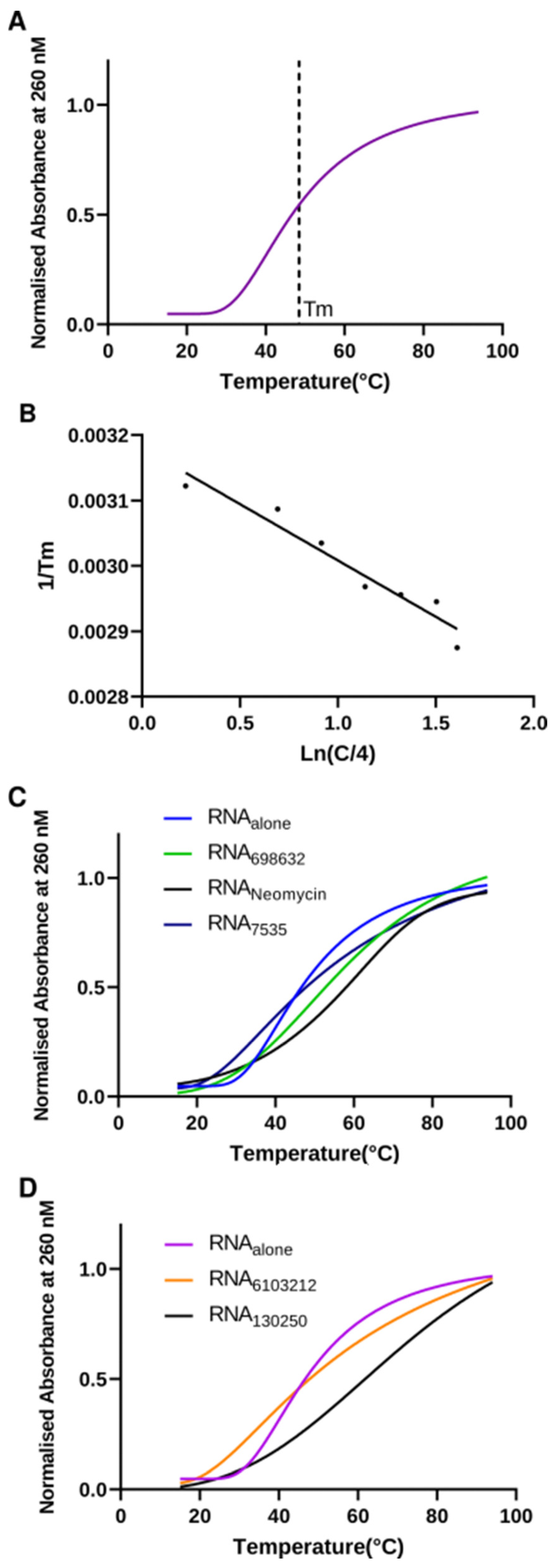
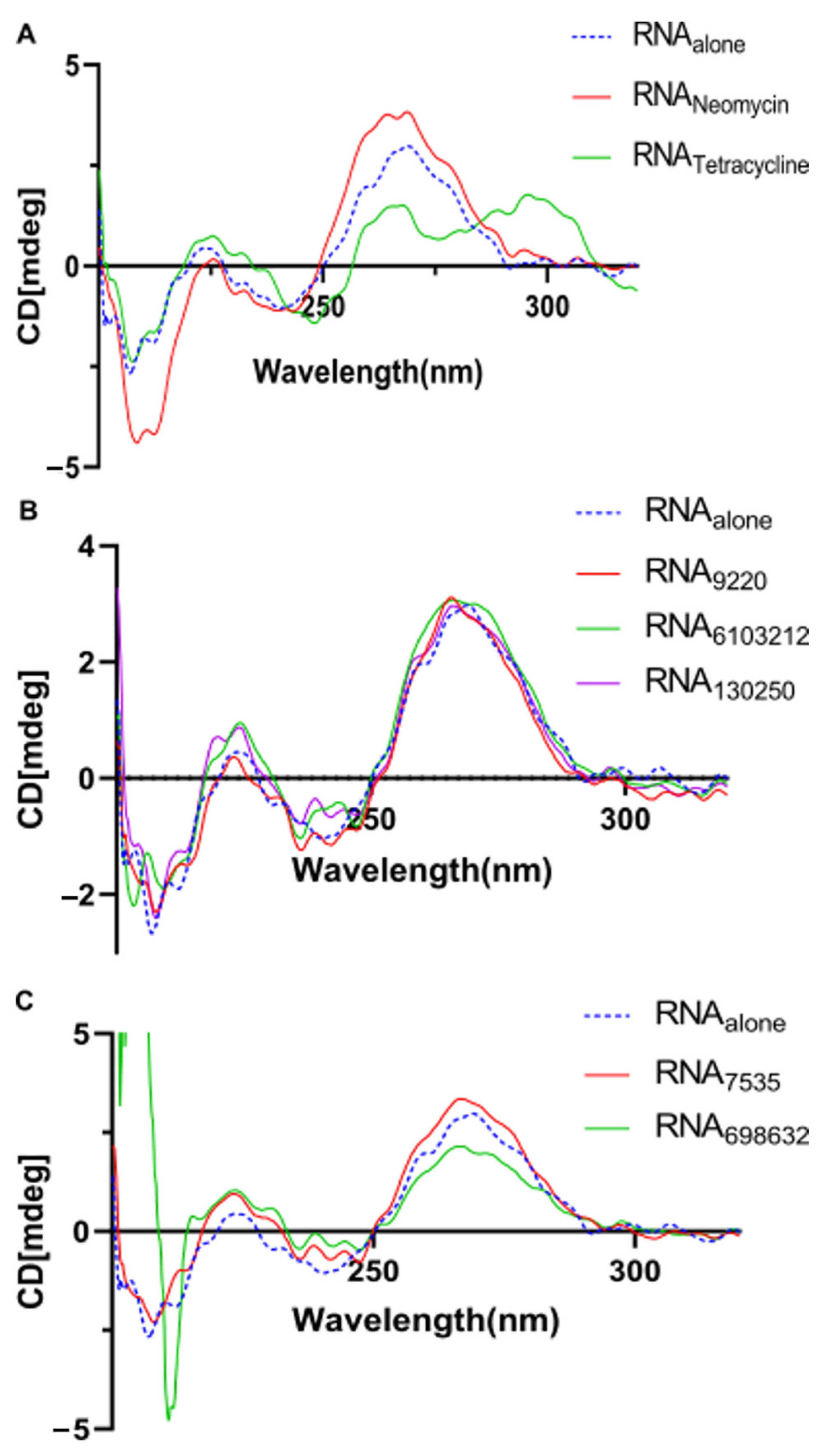
2.5. Lead Compounds Stabilize U-Helix RNA without Affecting Helical Conformation
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. Labeled and Unlabeled RNA
4.3. Virtual Screening
4.4. Cheminformatic Analysis
4.5. Molecular Dynamic Simulations
4.6. Microscale Thermophoresis
4.7. UV Melting Experiments
4.8. Circular Dichroism Spectroscopy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | AutoDock Tools |
| CD | Circular Dichroism |
| CT | Strand Concentration |
| Cy5 | Sulfo-Cyanine5 |
| DTP | Developmental Therapeutics Program |
| H | Enthalpy Change |
| EDTA | Ethylenediaminetetraacetic Acid |
| fs | Femtosecond |
| HEPES | 4-1-Piperazineethanesulfonic Acid |
| ITC | Isothermal Titration Calorimetry |
| LINCS | Linear Constraint Solver for Molecular Simulations |
| MST | Microscale Thermophoresis |
| MD | Molecular Dynamics |
| MMGBSA | Molecular Mechanics-Generalized Born Surface Area |
| RMSD | Root Mean Square Deviation |
| nm | Nanometer |
| ns | Nanosecond |
| NCI | National Cancer Institute |
| pre-mRNA | Pre-Edited mRNA |
| S | Entropy Change |
| SPR | Surface Plasmon Resonance |
| Tm | Melting Temperature |
| UV | Ultra Violet |
References
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meymandi, S.; Hernandez, S.; Park, S.; Sanchez, D.R.; Forsyth, C. Treatment of Chagas Disease in the United States. Curr. Treat. Options Infect. Dis. 2018, 10, 373–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, C.H.; Welburn, S.C. The long wait for a new drug for Human African Trypanosomiasis. Trends Parasitol. 2018, 34, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; de Koning, H.P.; Maser, P.; Horn, D. Drug resistance in African trypanosomiasis: The melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Knoop, V. When you can’t trust the DNA: RNA editing changes transcript sequences. Cell. Mol. Life Sci. 2011, 68, 567–586. [Google Scholar] [CrossRef]
- Benne, R.; Van den Burg, J.; Brakenhoff, J.P.; Sloof, P.; Van Boom, J.H.; Tromp, M.C. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell 1986, 46, 819–826. [Google Scholar] [CrossRef]
- Amaro, R.E.; Schnaufer, A.; Interthal, H.; Hol, W.; Stuart, K.D.; McCammon, J.A. Discovery of drug-like inhibitors of an essential RNA-editing ligase in Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 2008, 105, 17278–17283. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Connell, G.J. Identification of specific inhibitors for a trypanosomatid RNA editing reaction. RNA 2010, 16, 2435–2441. [Google Scholar] [CrossRef] [Green Version]
- Blum, B.; Simpson, L. Guide RNAs in kinetoplastid mitochondria have a nonencoded 3′ Oligo(U) tail involved in recognition of the preedited region. Cell 1990, 62, 391–397. [Google Scholar] [CrossRef]
- Leung, S.S.; Koslowsky, D.J. Interactions of mRNAs and gRNAs involved in trypanosome mitochondrial RNA editing: Structure probing of an mRNA bound to its cognate gRNA. RNA 2001, 7, 1803–1816. [Google Scholar] [CrossRef] [Green Version]
- Reifur, L.; Yu, L.E.; Cruz-Reyes, J.; Vanhartesvelt, M.; Koslowsky, D.J. The impact of mRNA structure on guide RNA targeting in kinetoplastid RNA editing. PLoS ONE 2010, 5, e12235. [Google Scholar] [CrossRef] [Green Version]
- Mooers, B.H.; Singh, A. The crystal structure of an oligo(U):pre-mRNA duplex from a trypanosome RNA editing substrate. RNA 2011, 17, 1870–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criswell, A.; Mooers, B.H. Structural studies of a double-stranded RNA from trypanosome RNA editing by small-angle X-ray scattering. In RNA-RNA Interactions; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1240, pp. 165–189. [Google Scholar]
- Seeman, N.C.; Rosenberg, J.M.; Rich, A. Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl. Acad. Sci. USA 1976, 73, 804–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15—ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butina, D. Unsupervised database clustering based on Daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Entzian, C.; Schubert, T. Studying small molecule-aptamer interactions using microscale thermophoresis (MST). Methods 2016, 97, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Leeder, W.M.; Reuss, A.J.; Brecht, M.; Kratz, K.; Wachtveitl, J.; Goringer, H.U. Charge reduction and thermodynamic stabilization of substrate RNAs inhibit RNA editing. PLoS ONE 2015, 10, e0118940. [Google Scholar] [CrossRef]
- Plum, G.E.; Pilch, D.S.; Singleton, S.F.; Breslauer, K.J. Nucleic acid hybridization: Triplex stability and energetics. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 319–350. [Google Scholar] [CrossRef]
- Hermann, T. Rational ligand design for RNA: The role of static structure and conformational flexibility in target recognition. Biochimie 2002, 84, 869–875. [Google Scholar] [CrossRef]
- Wu, P.; Nakano, S.; Sugimoto, N. Temperature dependence of thermodynamic properties for DNA/DNA and RNA/DNA duplex formation. Eur. J. Biochem. 2002, 269, 2821–2830. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.; Cruz, P.; Tinoco, I.; Jovin, T.M.; Vandesande, J.H. Z-RNA—A left-handed RNA double helix. Nature 1984, 311, 584–586. [Google Scholar] [CrossRef]
- Miyahara, T.; Nakatsuji, H.; Sugiyama, H. Similarities and differences between RNA and DNA double-helical structures in circular dichroism spectroscopy: A SAC-CI study. J. Phys. Chem. A 2016, 120, 9008–9018. [Google Scholar] [CrossRef]
- Salavati, R.; Moshiri, H.; Kala, S.; Najafabadi, H.S. Inhibitors of RNA editing as potential chemotherapeutics against trypanosomatid pathogens. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Durrant, J.D.; Hall, L.; Swift, R.V.; Landon, M.; Schnaufer, A.; Amaro, R.E. Novel naphthalene-based inhibitors of Trypanosoma brucei RNA editing ligase 1. PLoS Negl. Trop. Dis. 2010, 4, e803. [Google Scholar] [CrossRef]
- Rocca, R.; Moraca, F.; Costa, G.; Nadai, M.; Scalabrin, M.; Talarico, C.; Distinto, S.; Maccioni, E.; Ortuso, F.; Artese, A.; et al. Identification of G-quadruplex DNA/RNA binders: Structure-based virtual screening and biophysical characterization. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1329–1340. [Google Scholar] [CrossRef] [Green Version]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do structurally similar molecules have similar biological activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef] [PubMed]
- Gamo, F.J.; Sanz, L.M.; Vidal, J.; de Cozar, C.; Alvarez, E.; Lavandera, J.L.; Vanderwall, D.E.; Green, D.V.; Kumar, V.; Hasan, S.; et al. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Sydow, D.; Rodriguez-Guerra, J.; Kimber, T.B.; Schaller, D.; Taylor, C.J.; Chen, Y.; Leja, M.; Misra, S.; Wichmann, M.; Ariamajd, A.; et al. TeachOpenCADD 2022: Open source and FAIR Python pipelines to assist in structural bioinformatics and cheminformatics research. Nucleic Acids Res. 2022, 50, W753–W760. [Google Scholar] [CrossRef] [PubMed]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef] [PubMed]
- Sponer, J.; Bussi, G.; Krepl, M.; Banas, P.; Bottaro, S.; Cunha, R.A.; Gil-Ley, A.; Pinamonti, G.; Poblete, S.; Jurecka, P.; et al. RNA structural dynamics as captured by molecular simulations: A comprehensive overview. Chem. Rev. 2018, 118, 4177–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaud, J.P.; Delsuc, M.A. Biophysical techniques for ligand screening and drug design. Curr. Opin. Pharmacol. 2009, 9, 622–628. [Google Scholar] [CrossRef]
- Chen, D.; Ranganathan, A.; AP, I.; Siegal, G.; Carlsson, J. Complementarity between in silico and biophysical screening approaches in fragment-based lead discovery against the A(2A) adenosine receptor. J. Chem. Inf. Model. 2013, 53, 2701–2714. [Google Scholar] [CrossRef]
- Trosset, J.Y.; Cavé, C. In silico drug-target profiling. Methods Mol. Biol. 2019, 1953, 89–103. [Google Scholar] [CrossRef]
- Le Grice, S.F. Targeting the HIV RNA genome: High-hanging fruit only needs a longer ladder. Curr. Top. Microbiol. Immunol. 2015, 389, 147–169. [Google Scholar] [CrossRef]
- Moon, M.H.; Hilimire, T.A.; Sanders, A.M.; Schneekloth, J.S., Jr. Measuring RNA-ligand interactions with microscale thermophoresis. Biochemistry 2018, 57, 4638–4643. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Gan, J.; Huang, Z. Structure-based DNA-targeting strategies with small molecule ligands for drug discovery. Med. Res. Rev. 2013, 33, 1119–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, A.; Levy, Y. Structure, stability and specificity of the binding of ssDNA and ssRNA with proteins. PLoS Comput. Biol. 2019, 15, e1006768. [Google Scholar] [CrossRef]
- Tan, X.; Jia, F.; Wang, P.; Zhang, K. Nucleic acid-based drug delivery strategies. J. Control Release 2020, 323, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, B.; Vancamp, L.; Trosoko, J.E.; Msnsour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Zegers, J.; Peters, M.; Albada, B. DNA G-quadruplex-stabilizing metal complexes as anticancer drugs. J. Biol. Inorg. Chem. 2023, 28, 117–138. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Chen, C.C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins 2004, 55, 288–304. [Google Scholar] [CrossRef]
- Khan, T.; Ahmad, R.; Azad, I.; Raza, S.; Joshi, S.; Khan, A.R. Computer-aided drug design and virtual screening of targeted combinatorial libraries of mixed-ligand transition metal complexes of 2-butanone thiosemicarbazone. Comput. Biol. Chem. 2018, 75, 178–195. [Google Scholar] [CrossRef]
- Seiwert, S.D.; Heidmann, S.; Stuart, K. Direct visualization of uridylate deletion in vitro suggests a mechanism for kinetoplastid RNA editing. Cell 1996, 84, 831–841. [Google Scholar] [CrossRef] [Green Version]
- Mooers, B.H. Direct-methods structure determination of a trypanosome RNA-editing substrate fragment with translational pseudosymmetry. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Delano, W. The PyMOL Molecular Graphics System, Version 2.5.1; Schrodinger, Ltd.: New York, NY, USA, 2021. [Google Scholar]
- Landrum, G. RDKit: A Software Suite for Cheminformatics, Computational Chemistry, and Predictive Modeling. 2013. Available online: http://rdkit.org/ (accessed on 30 April 2023).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Lee, J.; Hitzenberger, M.; Rieger, M.; Kern, N.R.; Zacharias, M.; Im, W. CHARMM-GUI supports the Amber force fields. J. Chem. Phys. 2020, 153, 035103. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single-crystals—A new molecular-dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Miller III, B.R.; McGee Jr, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Tresanco, M.S.; Valdes-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Jerabek-Willemsen, M.; Andre, T.; Wanner, R.; Roth, H.M.; Duhr, S.; Baaske, P.; Breitsprecher, D. MicroScale thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014, 1077, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Rainard, J.M.; Pandarakalam, G.C.; McElroy, S.P. Using microscale thermophoresis to characterize hits from high-throughput ccreening: A european lead factory perspective. Slas Discov. 2018, 23, 225–241. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, S.J.; Turner, D.H. Biophysical, chemical, and functional probes of RNA structure, interactions and folding: Part A. In Methods in Enzymology; Herschlag, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 468, pp. 371–387. [Google Scholar]
- Gladysz, M.; Andralojc, W.; Czapik, T.; Gdaniec, Z.; Kierzek, R. Thermodynamic and structural contributions of the 6-thioguanosine residue to helical properties of RNA. Sci. Rep. 2019, 9, 4385. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| U-Helix + Drug | |||
|---|---|---|---|
| (C) | (kJ/mol) | (eu) | |
| U-helix alone | 48.19 | 48.26 | 153.5 |
| Neomycin | 55.97 | 58.64 | 184.17 |
| Lig7535 | 66.82 | 47.68 | 156.42 |
| Lig698632 | 54.79 | 64.81 | 200.51 |
| Lig6103212 | 72.84 | 33.60 | 109.43 |
| Lig130250 | 74.63 | 32.43 | 104.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acquah, F.A.; Mooers, B.H.M. Targeting RNA Structure to Inhibit Editing in Trypanosomes. Int. J. Mol. Sci. 2023, 24, 10110. https://doi.org/10.3390/ijms241210110
Acquah FA, Mooers BHM. Targeting RNA Structure to Inhibit Editing in Trypanosomes. International Journal of Molecular Sciences. 2023; 24(12):10110. https://doi.org/10.3390/ijms241210110
Chicago/Turabian StyleAcquah, Francis A., and Blaine H. M. Mooers. 2023. "Targeting RNA Structure to Inhibit Editing in Trypanosomes" International Journal of Molecular Sciences 24, no. 12: 10110. https://doi.org/10.3390/ijms241210110