


Bioengineered Bovine Papillomavirus L1 Protein Virus-like Particle (VLP) Vaccines for Enhanced Induction of CD8 T Cell Responses through Cross-Priming


Abstract
:1. Introduction
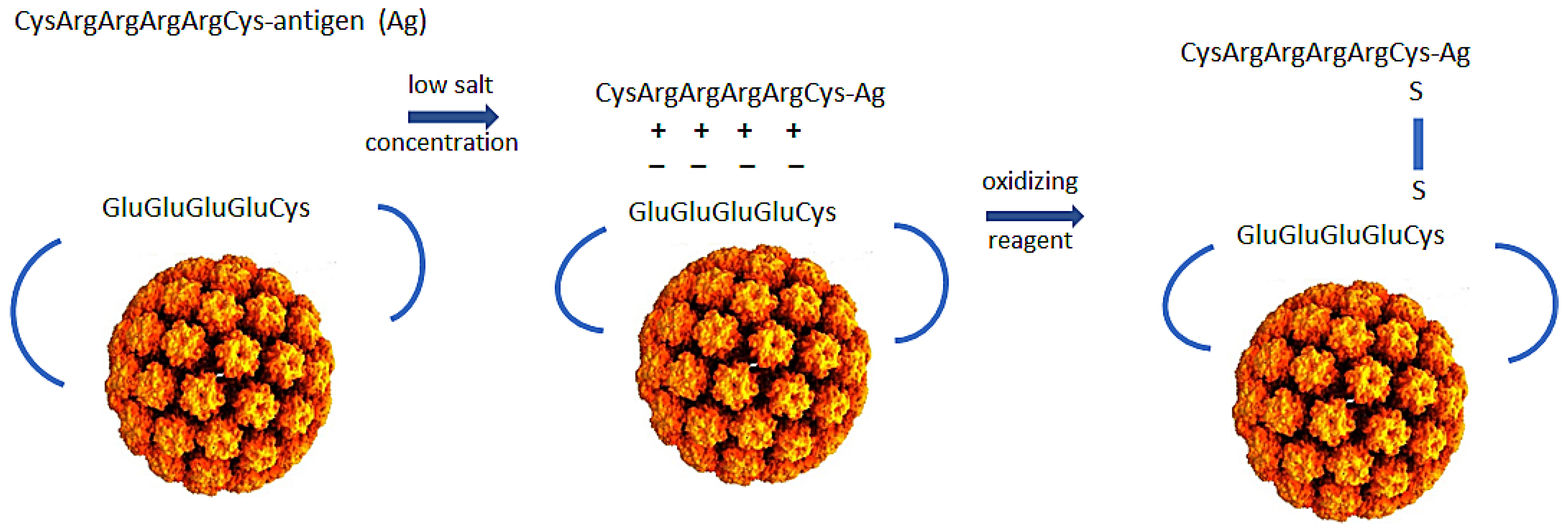
2. Production of Polyionic Bovine Papillomavirus L1 VLPs
3. Formulation of Polyionic VLP Vaccines
4. Immunological Properties of Papillomavirus VLPs
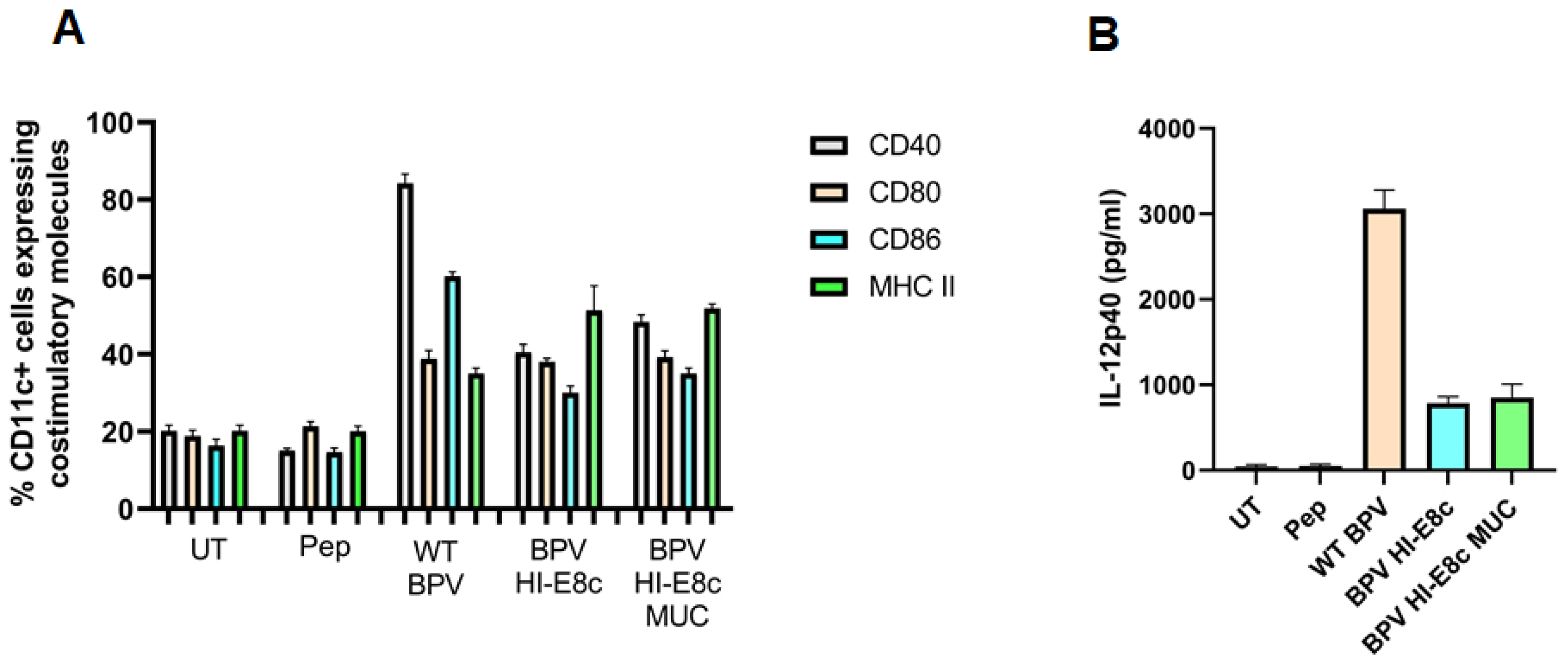
4.1. Activation and Maturation of Dendritic Cells
4.2. Stimulation of Macrophages, Monocytes, and Plasmacytoid Dendritic Cells
4.3. Activation of T Cells by Antigen-Specific Polyionic VLPs
5. Immunological Properties of Polyionic VLPs
6. Immunogenicity of Polyionic VLP Vaccines in Mice
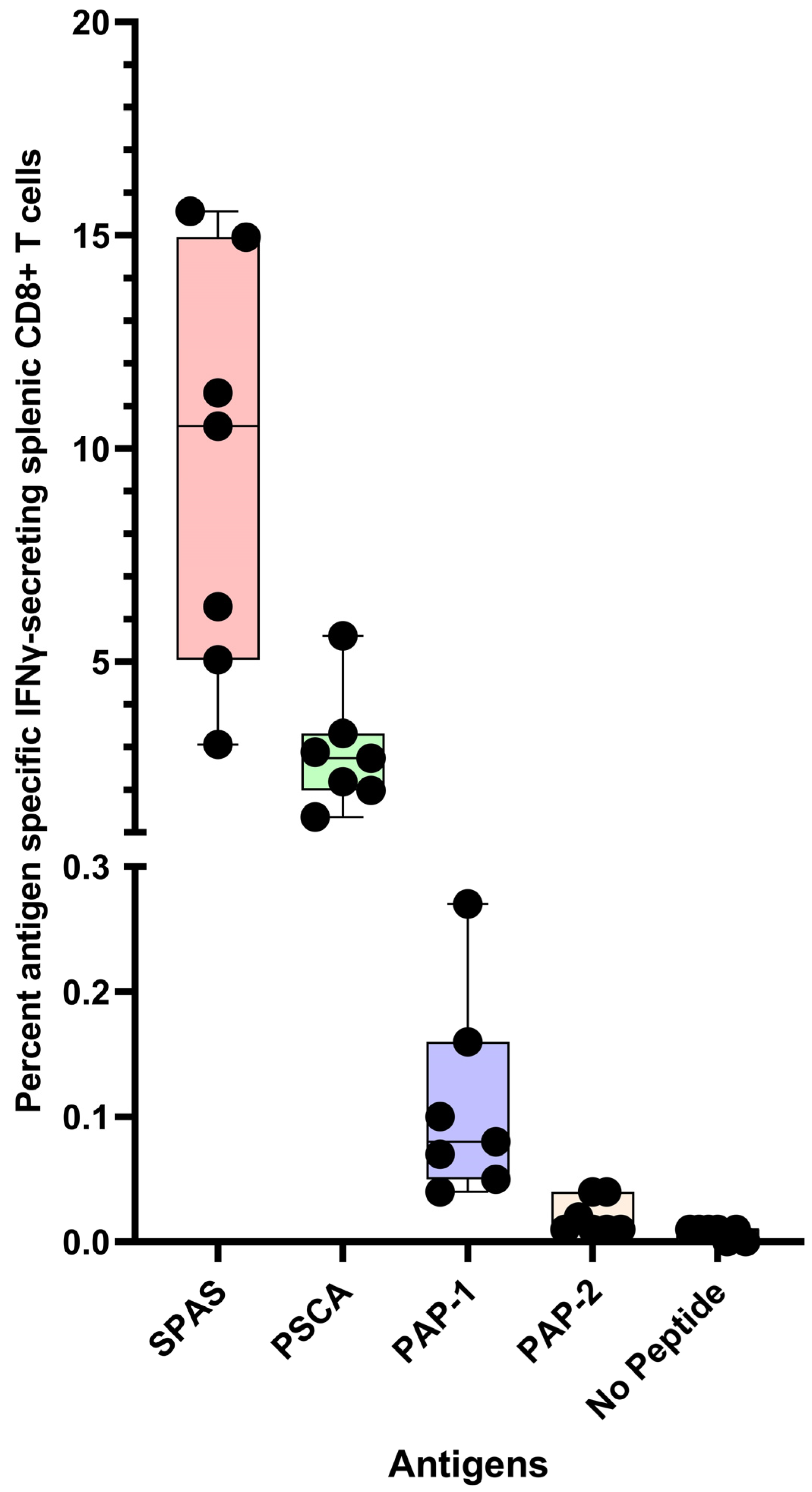
6.1. Immunogenicity of Polyionic VLPs Formulated with Tumor Antigens
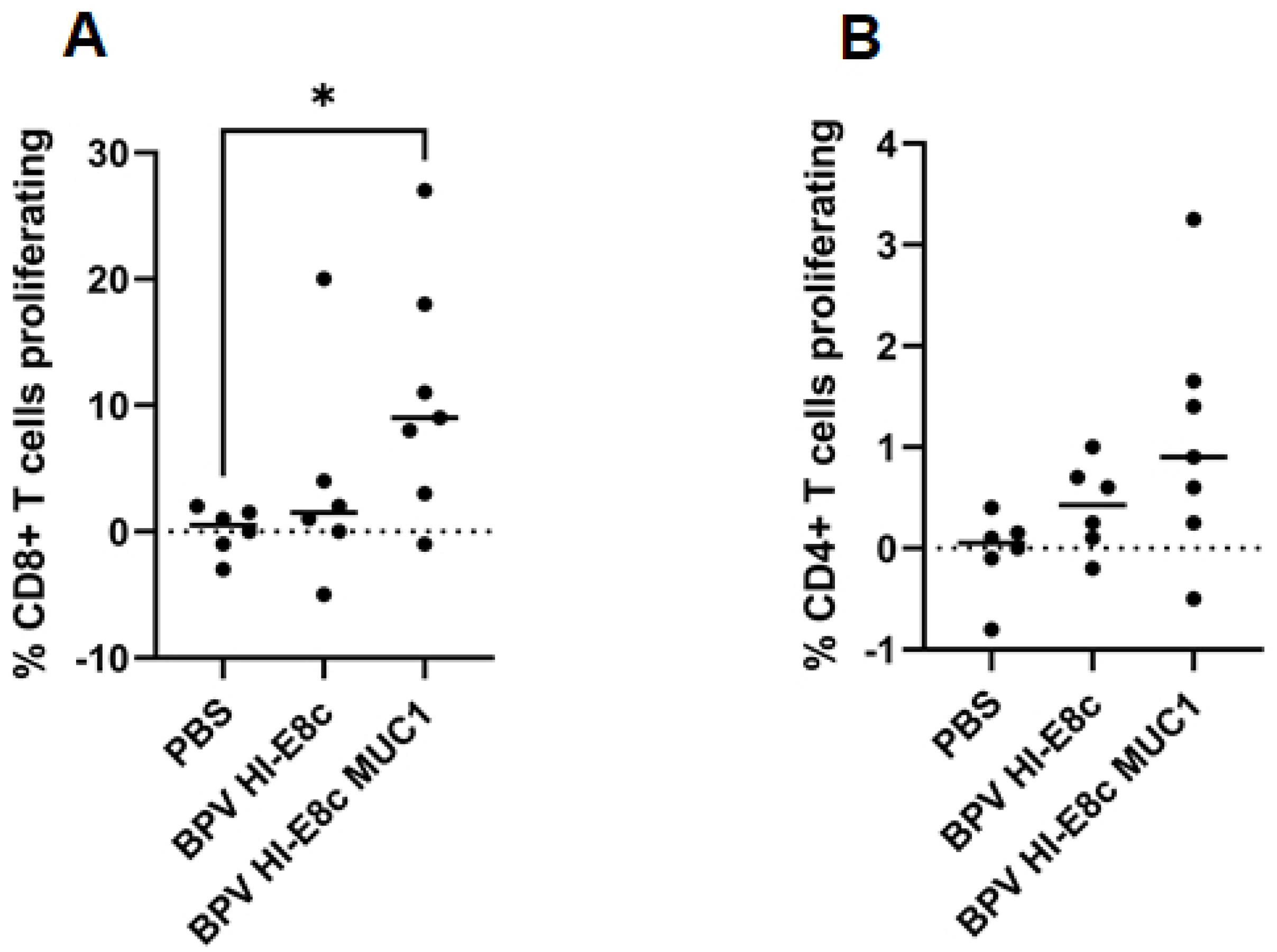
6.2. Immunogenicity of Polyionic VLPs Formulated with Microbial Antigens
7. Comparison of Immunogenicity of Polyionic VLPs and Other Vaccine Platforms
7.1. Prostate Tumor Antigens
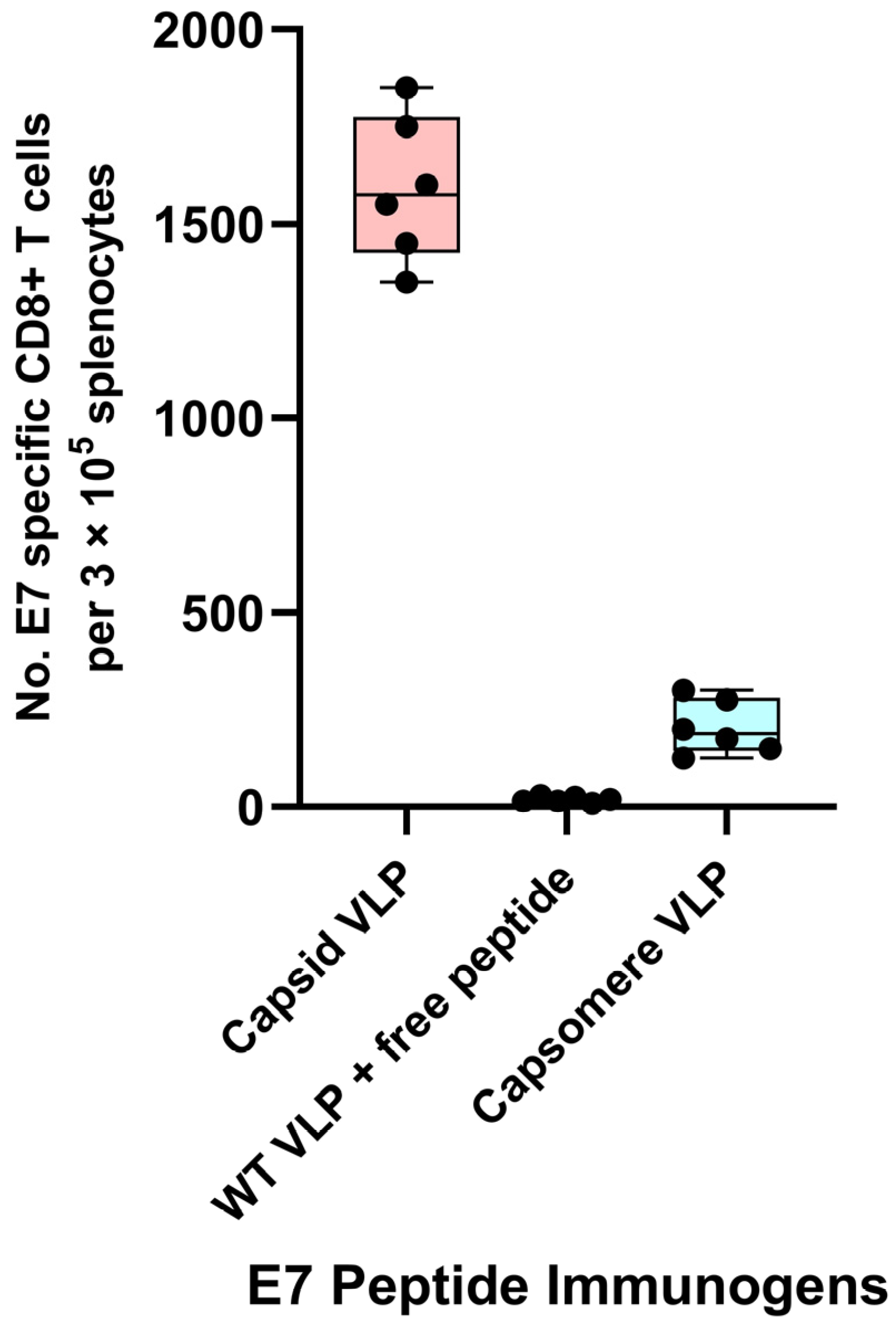
7.2. HPV 16 E7 Antigens
8. Application of Other VLP Platforms Technologies against Tumor Antigens
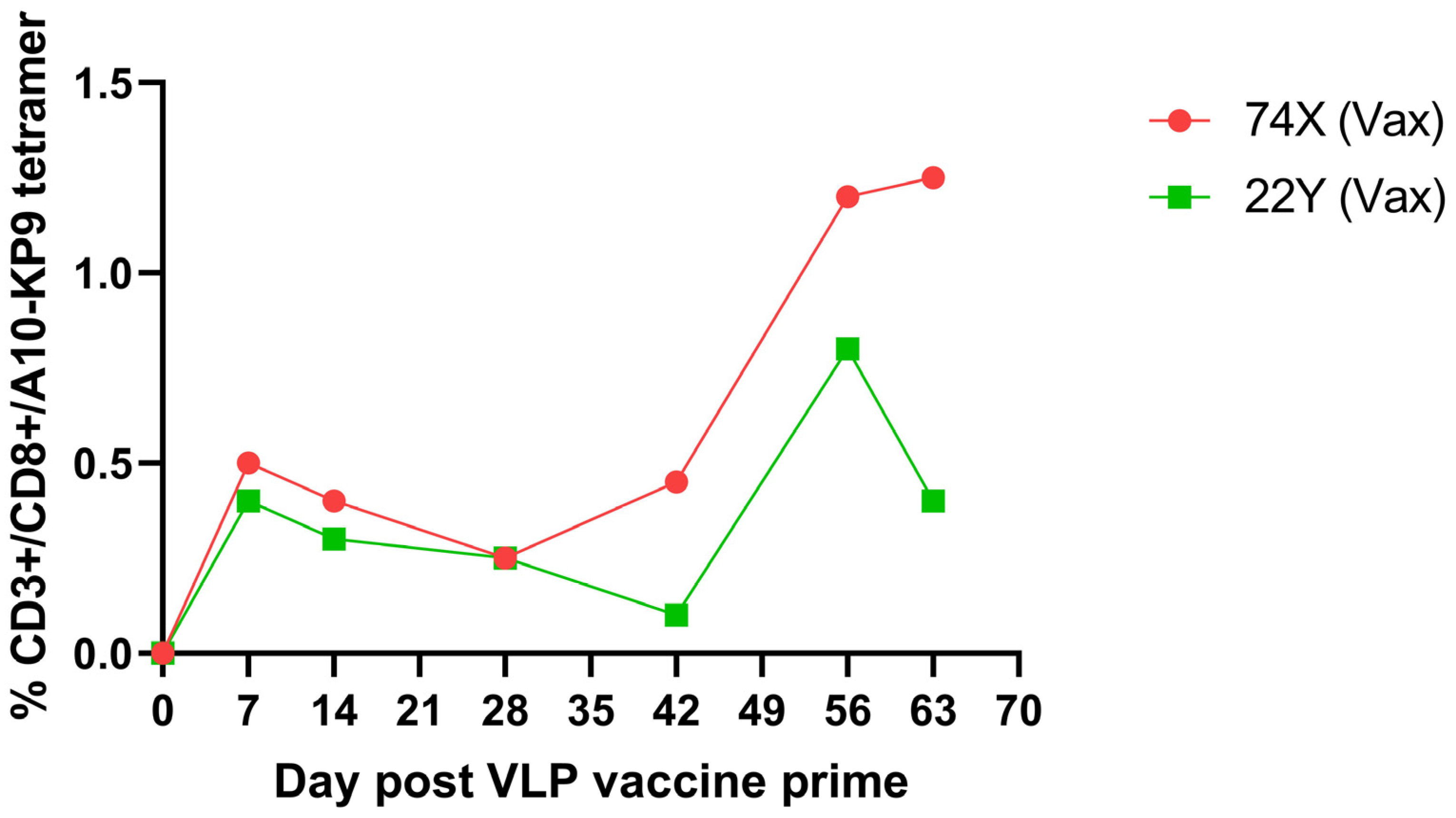
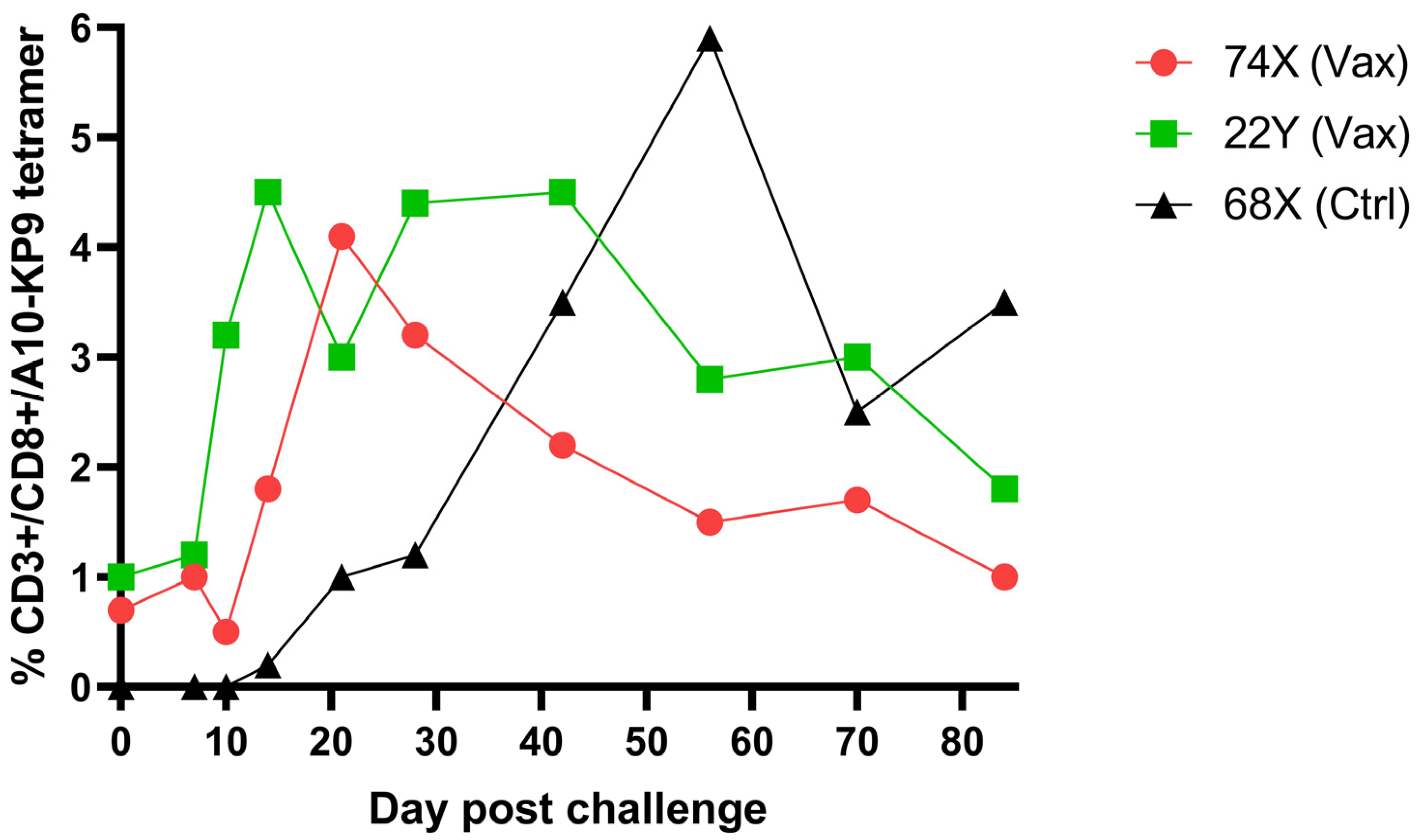
9. Immunogenicity of Polyionic VLP Vaccines in Non-Human Primates
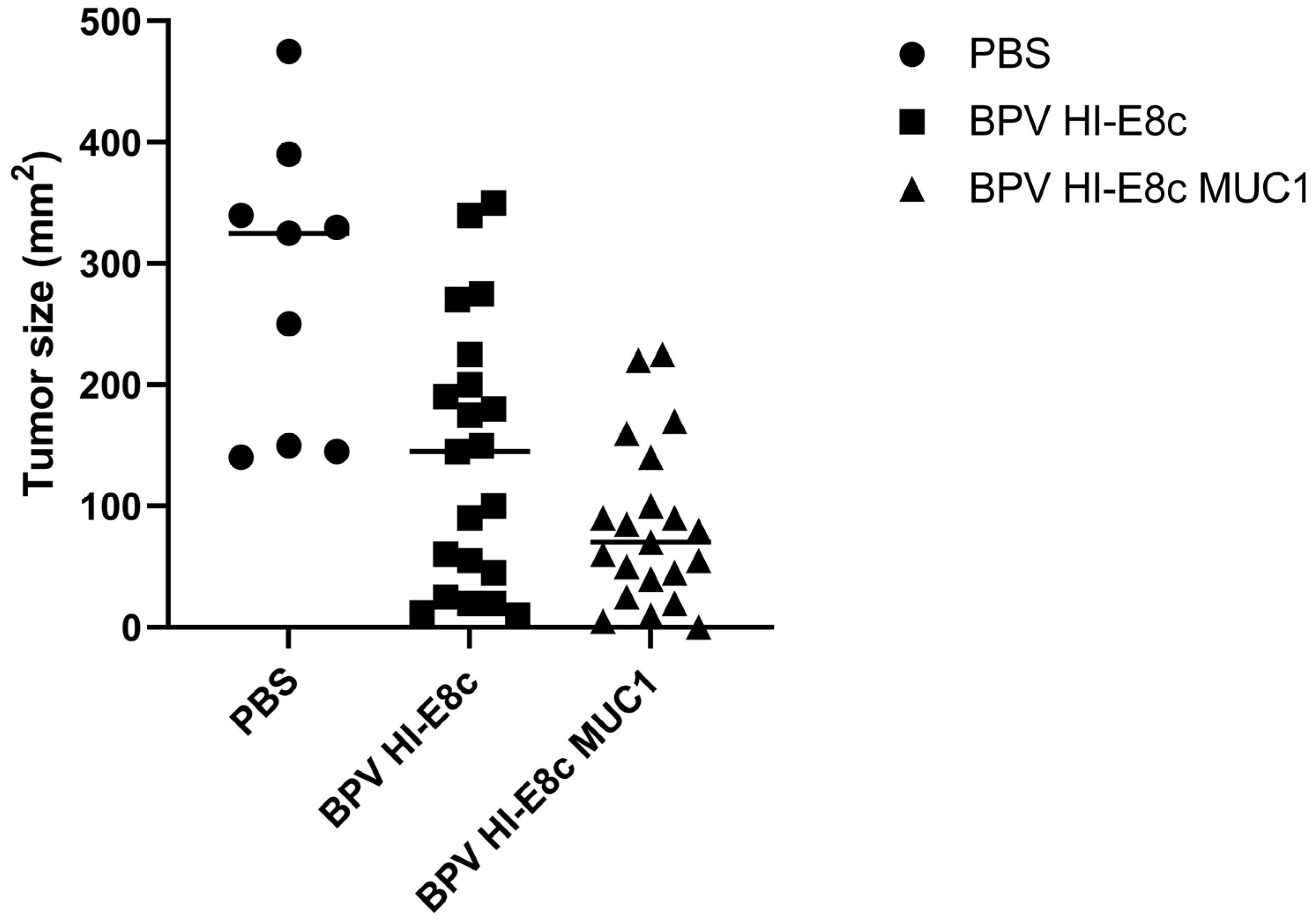
10. Efficacy of MUC1 Polyionic VLP Vaccine in a Murine Cancer Model
11. Efficacy of Prostate Cancer Polyionic VLP Vaccine in a Physiologically Relevant Murine Prostate Cancer Model
11.1. The TRAMP Mouse Model of Prostate Cancer
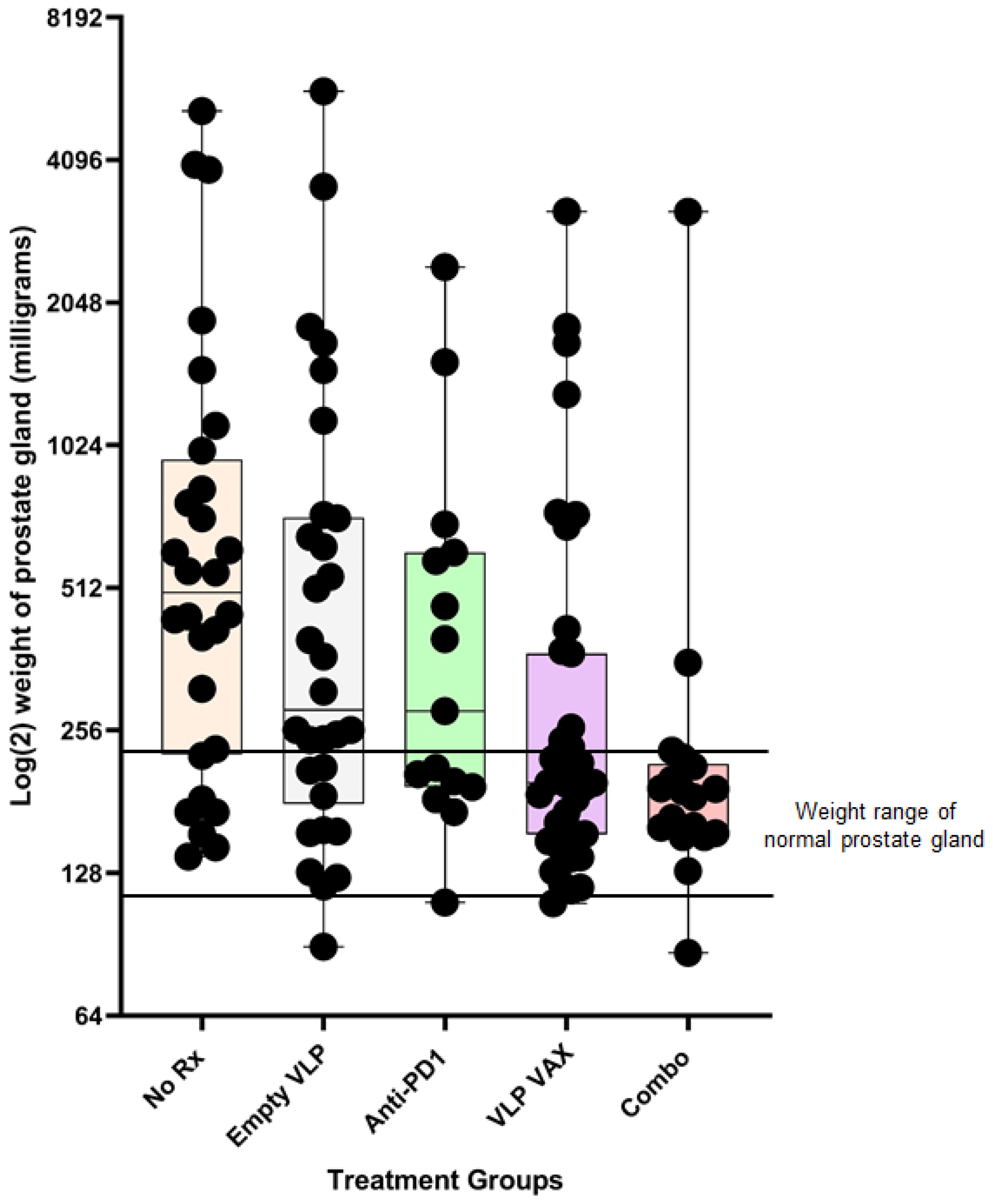
11.2. Efficacy of Polyionic VLP Vaccination of Advanced Stage Cancer in TRAMP Mice
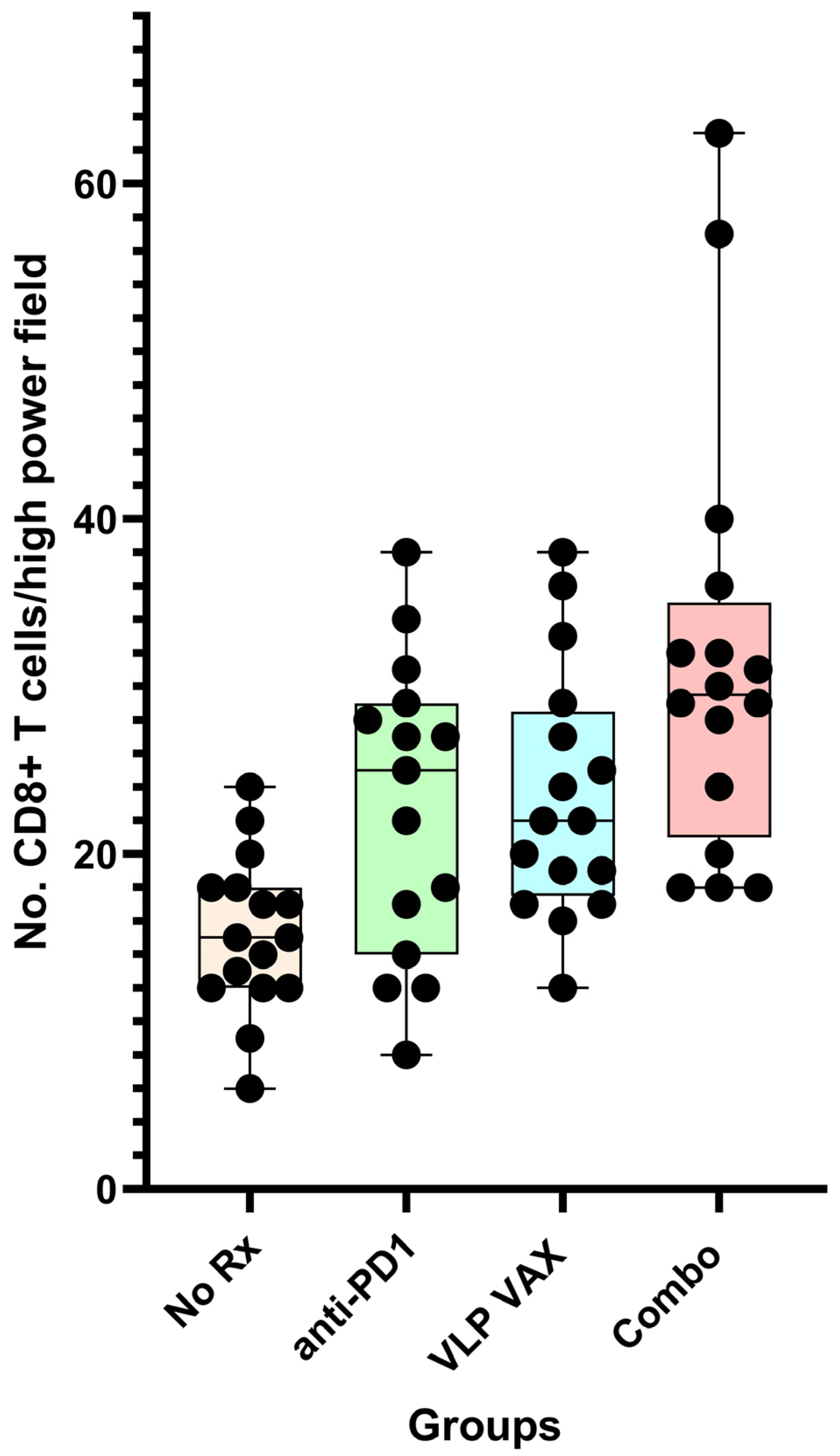
11.3. Generation of Tissue Infiltrating CD8+ T Cells in the TRAMP Model
11.4. Comparative Studies of Efficacy of Vaccine Platforms in TRAMP Mice
12. Mechanisms of Polyionic VLP Vaccine-Induced Immunogenicity
12.1. Adjuvant Effect
12.2. Particle Size and Immunogenicity
12.3. Reversible Linkage of Antigens to VLPs and Immunogenicity
12.4. Immunogenicity and Cell Penetrating Amino Acids
12.5. Transcriptional Profile of Papillomavirus VLP-Treated Dendritic Cells
12.6. Papillomavirus VLP Treatment of Dendritic Cell Subsets
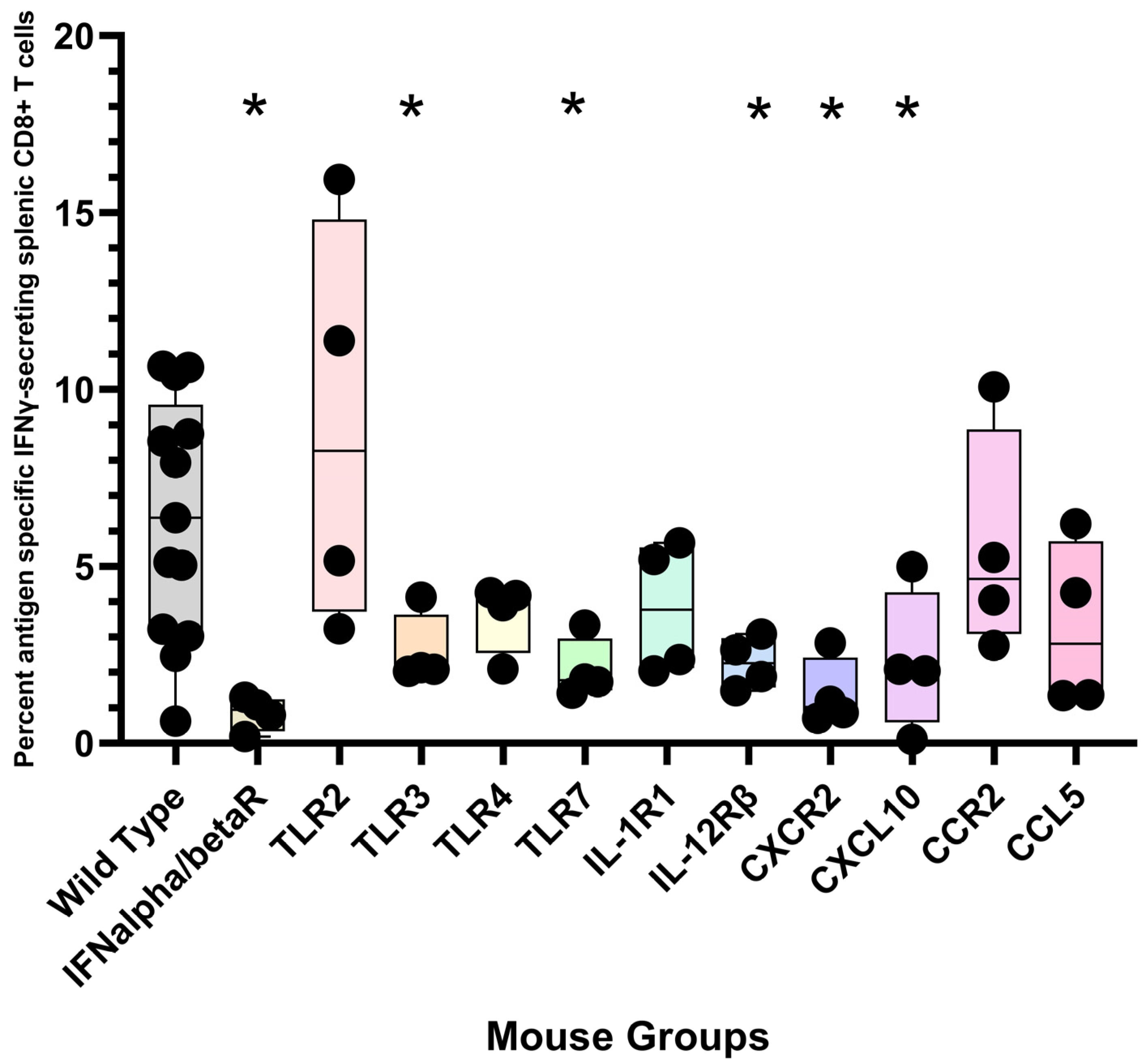
12.7. CD8+ T Cell Response to Polyionic VLPs in Gene-Deficient Mice
13. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hobernik, D.; Bros, M. DNA Vaccines-How Far From Clinical Use? Int. J. Mol. Sci. 2018, 19, 3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myhr, A.I. DNA Vaccines: Regulatory Considerations and Safety Aspects. Curr. Issues Mol. Biol. 2017, 22, 79–88. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors: Humoral, cellular, and innate response, what’s important? Hum. Vaccin. Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, N.H.; Sørvoll, I.H.; Michelsen, A.E.; Munthe, L.A.; Lund-Johansen, F.; Ahlen, M.T.; Wiedmann, M.; Aamodt, A.-H.; Skattør, T.H.; Tjønnfjord, G.E.; et al. Thrombosis and Thrombocytopenia after ChAdOx1 nCoV-19 Vaccination. N. Engl. J. Med. 2021, 384, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Embgenbroich, M.; Burgdorf, S. Current Concepts of Antigen Cross-Presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef] [Green Version]
- Bevan, M.J. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 1976, 143, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Iborra, S.; Martínez-López, M.; Khouili, S.C.; Enamorado, M.; Cueto, F.J.; Conde-Garrosa, R.; del Fresno, C.; Sancho, D. Optimal Generation of Tissue-Resident but Not Circulating Memory T Cells during Viral Infection Requires Crosspriming by DNGR-1 + Dendritic Cells. Immunity 2016, 45, 847–860. [Google Scholar] [CrossRef] [Green Version]
- Okła, K.; Farber, D.L.; Zou, W. Tissue-resident memory T cells in tumor immunity and immunotherapy. J. Exp. Med. 2021, 218, e20201605. [Google Scholar] [CrossRef]
- Rosato, P.C.; Beura, L.K.; Masopust, D. Tissue resident memory T cells and viral immunity. Curr. Opin. Virol. 2017, 22, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pejawar-Gaddy, S.; Rajawat, Y.; Hilioti, Z.; Xue, J.; Gaddy, D.F.; Finn, O.J.; Viscidi, R.P.; Bossis, I. Generation of a tumor vaccine candidate based on conjugation of a MUC1 peptide to polyionic papillomavirus virus-like particles. Cancer Immunol. Immunother. 2010, 59, 1685–1696. [Google Scholar] [CrossRef] [Green Version]
- Bishop, B.; Dasgupta, J.; Klein, M.; Garcea, R.L.; Christensen, N.D.; Zhao, R.; Chen, X.S. Crystal structures of four types of human papillomavirus L1 capsid proteins: Understanding the specificity of neutralizing monoclonal antibodies. J. Biol. Chem. 2007, 282, 31803–31811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, P.; Day, P.M.; Pang, Y.-Y.S.; Frye, S.A.; Jensen, P.N.; Lowy, D.R.; Schiller, J.T. Papillomavirus-like particles induce acute activation of dendritic cells. J. Immunol. 2001, 166, 5346–5355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, P.; Thompson, C.D.; Day, P.M.; Bacot, S.M.; Lowy, D.R.; Schiller, J.T. Interaction of papillomavirus virus-like particles with human myeloid antigen-presenting cells. Clin. Immunol. 2003, 106, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, M.P.; Fausch, S.C.; Da Silva, D.M.; Kast, W.M. Human dendritic cells are activated by chimeric human papillomavirus type-16 virus-like particles and induce epitope-specific human T cell responses in vitro. J. Immunol. 2001, 166, 5917–5924. [Google Scholar] [CrossRef] [Green Version]
- Fausch, S.C.; Da Silva, D.M.; Kast, W.M. Heterologous papillomavirus virus-like particles and human papillomavirus virus-like particle immune complexes activate human Langerhans cells. Vaccine 2005, 23, 1720–1729. [Google Scholar] [CrossRef]
- Lenz, P.; Lowy, D.R.; Schiller, J.T. Papillomavirus virus-like particles induce cytokines characteristic of innate immune responses in plasmacytoid dendritic cells. Eur. J. Immunol. 2005, 35, 1548–1556. [Google Scholar] [CrossRef]
- Peng, S.; Frazer, I.H.; Fernando, G.J.; Zhou, J. Papillomavirus virus-like particles can deliver defined CTL epitopes to the MHC class I pathway. Virology 1998, 240, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Peng, J.; Jabbar, I.A.; Liu, X.; Filgueira, L.; Frazer, I.H.; Thomas, R. Despite differences between dendritic cells and Langerhans cells in the mechanism of papillomavirus-like particle antigen uptake, both cells cross-prime T cells. Virology 2004, 324, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Simons, B.W.; Cannella, F.; Rowley, D.T.; Viscidi, R.P. Bovine papillomavirus prostate cancer antigen virus-like particle vaccines are efficacious in advanced cancers in the TRAMP mouse spontaneous prostate cancer model. Cancer Immunol. Immunother. 2020, 69, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Cappuccini, F.; Stribbling, S.; Pollock, E.; Hill, A.V.S.; Redchenko, I. Immunogenicity and efficacy of the novel cancer vaccine based on simian adenovirus and MVA vectors alone and in combination with PD-1 mAb in a mouse model of prostate cancer. Cancer Immunol. Immunother. 2016, 65, 701–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spies, E.; Reichardt, W.; Alvarez, G.; Groettrup, M.; Öhlschläger, P. An Artificial PAP Gene Breaks Self-tolerance and Promotes Tumor Regression in the TRAMP Model for Prostate Carcinoma. Mol. Ther. 2012, 20, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Hernandez, M.L.; Gray, A.; Hubby, B.; Klinger, O.J.; Kast, W.M. Prostate stem cell antigen vaccination induces a long-term protective immune response against prostate cancer in the absence of autoimmunity. Cancer Res. 2008, 68, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Hung, C.-F.; Juang, J.; He, L.; Kim, T.W.; Armstrong, D.K.; Pai, S.I.; Chen, P.-J.; Lin, C.-T.; Boyd, D.A.; et al. Comparison of HPV DNA vaccines employing intracellular targeting strategies. Gene Ther. 2004, 11, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- da Silva, J.R.; Bitencourt Rodrigues, K.; Formoso Pelegrin, G.; Silva Sales, N.; Muramatsu, H.; de Oliveira Silva, M.; Porchia, B.F.M.M.; Moreno, A.C.R.; Aps, L.R.M.M.; Venceslau-Carvalho, A.A.; et al. Single immunizations of self-amplifying or non-replicating mRNA-LNP vaccines control HPV-associated tumors in mice. Sci. Transl. Med. 2023, 15, eabn3464. [Google Scholar] [CrossRef]
- Rahimian, S.; Fransen, M.F.; Kleinovink, J.W.; Christensen, J.R.; Amidi, M.; Hennink, W.E.; Ossendorp, F. Polymeric nanoparticles for co-delivery of synthetic long peptide antigen and poly IC as therapeutic cancer vaccine formulation. J. Control. Release 2015, 203, 16–22. [Google Scholar] [CrossRef]
- Galliverti, G.; Tichet, M.; Domingos-Pereira, S.; Hauert, S.; Nardelli-Haefliger, D.; Swartz, M.A.; Hanahan, D.; Wullschleger, S. Nanoparticle Conjugation of Human Papillomavirus 16 E7-long Peptides Enhances Therapeutic Vaccine Efficacy against Solid Tumors in Mice. Cancer Immunol. Res. 2018, 6, 1301–1313. [Google Scholar] [CrossRef]
- Palladini, A.; Thrane, S.; Janitzek, C.M.; Pihl, J.; Clemmensen, S.B.; De Jongh, W.A.; Clausen, T.M.; Nicoletti, G.; Landuzzi, L.; Penichet, M.L.; et al. Virus-like particle display of HER2 induces potent anti-cancer responses. Oncoimmunology 2018, 7, e1408749. [Google Scholar] [CrossRef] [Green Version]
- Rolih, V.; Caldeira, J.; Bolli, E.; Salameh, A.; Conti, L.; Barutello, G.; Riccardo, F.; Magri, J.; Lamolinara, A.; Parra, K.; et al. Development of a VLP-Based Vaccine Displaying an xCT Extracellular Domain for the Treatment of Metastatic Breast Cancer. Cancers 2020, 12, 1492. [Google Scholar] [CrossRef]
- Bolli, E.; O’Rourke, J.P.; Conti, L.; Lanzardo, S.; Rolih, V.; Christen, J.M.; Barutello, G.; Forni, M.; Pericle, F.; Cavallo, F. A Virus-Like-Particle immunotherapy targeting Epitope-Specific anti-xCT expressed on cancer stem cell inhibits the progression of metastatic cancer in vivo. Oncoimmunology 2018, 7, e1408746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yong, L.-K.; Li, D.; Cubas, R.; Chen, C.; Yao, Q. Mesothelin virus-like particle immunization controls pancreatic cancer Growth through CD8+ T cell induction and reduction in the frequency of CD4+foxp3+ICOS− regulatory T Cells. PLoS ONE 2013, 8, e68303. [Google Scholar] [CrossRef] [PubMed]
- Cubas, R.; Zhang, S.; Li, M.; Chen, C.; Yao, Q. Chimeric Trop2 virus-like particles: A potential immunotherapeutic approach against pancreatic cancer. J. Immunother. 2011, 34, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Lizotte, P.H.; Wen, A.M.; Sheen, M.R.; Fields, J.; Rojanasopondist, P.; Steinmetz, N.F.; Fiering, S. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat. Nanotechnol. 2015, 11, 295–303. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Vogel, M.; Riether, C.; Muller, J.; Salatino, S.; Ternette, N.; Gomes, A.C.; Cabral-Miranda, G.; El-Turabi, A.; Ruedl, C.; et al. Targeting Mutated Plus Germline Epitopes Confers Pre-clinical Efficacy of an Instantly Formulated Cancer Nano-Vaccine. Front. Immunol. 2019, 10, 1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, S.E.; Queen, S.E.; Viscidi, R.; Johnson, D.; Kent, S.J.; Adams, R.J.; Tarwater, P.M.; Mankowski, J.L. Central nervous system-specific consequences of simian immunodeficiency virus Gag escape from major histocompatibility complex class I-mediated control. J. Neurovirol. 2016, 22, 498–507. [Google Scholar] [CrossRef] [Green Version]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, N.M.; DeMayo, F.; Finegold, M.J.; Medina, D.; Tilley, W.D.; Aspinall, J.O.; Cunha, G.R.; Donjacour, A.A.; Matusik, R.J.; Rosen, J.M. Prostate cancer in a transgenic mouse. Proc. Natl. Acad. Sci. USA 1995, 92, 3439–3443. [Google Scholar] [CrossRef] [Green Version]
- Gingrich, J.R.; Barrios, R.J.; Foster, B.A.; Greenberg, N.M. Pathologic progression of autochthonous prostate cancer in the TRAMP model. Prostate Cancer Prostatic Dis. 1999, 2, 70–75. [Google Scholar] [CrossRef]
- Kido, L.A.; Lamas, C.D.A.; Maróstica, M.R., Jr.; Cagnon, V.H.A. Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) model: A good alternative to study PCa progression and chemoprevention approaches. Life Sci. 2018, 217, 141–147. [Google Scholar] [CrossRef]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, A.A.; Foster, B.A.; Kwon, E.D.; Truong, T.; Choi, E.M.; Greenberg, N.M.; Burg, M.B.; Allison, J. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 2000, 60, 2444–2448. [Google Scholar] [PubMed]
- Kines, R.C.; Cerio, R.J.; Roberts, J.N.; Thompson, C.D.; de Los Pinos, E.; Lowy, D.R.; Schiller, J.T. Human papillomavirus capsids preferentially bind and infect tumor cells. Int. J. Cancer 2015, 138, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hojeij, R.; Domingos-Pereira, S.; Nkosi, M.; Gharbi, D.; Derré, L.; Schiller, J.T.; Jichlinski, P.; Nardelli-Haefliger, D. Immunogenic Human Papillomavirus Pseudovirus-Mediated Suicide-Gene Therapy for Bladder Cancer. Int. J. Mol. Sci. 2016, 17, 1125. [Google Scholar] [CrossRef] [Green Version]
- Kines, R.C.; Schiller, J.T. Harnessing Human Papillomavirus’ Natural Tropism to Target Tumors. Viruses 2022, 14, 1656. [Google Scholar] [CrossRef]
- Goldstein, D.; Laszlo, J. The role of interferon in cancer therapy: A current perspective. CA Cancer J. Clin. 1988, 38, 258–277. [Google Scholar] [CrossRef]
- Milner, J.J.; Toma, C.; He, Z.; Kurd, N.S.; Nguyen, Q.P.; McDonald, B.; Quezada, L.; Widjaja, C.E.; Witherden, D.A.; Crowl, J.T.; et al. Heterogenous Populations of Tissue-Resident CD8+ T Cells Are Generated in Response to Infection and Malignancy. Immunity 2020, 52, 808–824.e7. [Google Scholar] [CrossRef]
- Mikyskova, R.; Indrova, M.; Stepanek, I.; Kanchev, I.; Bieblova, J.; Vosahlikova, S.; Moserova, I.; Truxova, I.; Fucikova, J.; Bartunkova, J.; et al. Dendritic cells pulsed with tumor cells killed by high hydrostatic pressure inhibit prostate tumor growth in TRAMP mice. Oncoimmunology 2017, 6, e1362528. [Google Scholar] [CrossRef]
- Krupa, M.; Canamero, M.; Gomez, C.E.; Najera, J.L.; Gil, J.; Esteban, M. Immunization with recombinant DNA and modified vaccinia virus Ankara (MVA) vectors delivering PSCA and STEAP1 antigens inhibits prostate cancer progression. Vaccine 2011, 29, 1504–1513. [Google Scholar] [CrossRef]
- Mueller, M.; Reichardt, W.; Koerner, J.; Groettrup, M. Coencapsulation of tumor lysate and CpG-ODN in PLGA-microspheres enables successful immunotherapy of prostate carcinoma in TRAMP mice. J. Control. Release 2012, 162, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Suresh, M. Vaccine adjuvants to engage the cross-presentation pathway. Front. Immunol. 2022, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Brewitz, A.; Eickhoff, S.; Dähling, S.; Quast, T.; Bedoui, S.; Kroczek, R.A.; Kurts, C.; Garbi, N.; Barchet, W.; Iannacone, M.; et al. CD8(+) T Cells Orchestrate pDC-XCR1(+) Dendritic Cell Spatial and Functional Cooperativity to Optimize Priming. Immunity 2017, 46, 205–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foged, C. Subunit vaccines of the future: The need for safe, customized and optimized particulate delivery systems. Ther. Deliv. 2011, 2, 1057–1077. [Google Scholar] [CrossRef]
- Irvine, D.J.; Swartz, M.A.; Szeto, G.L. Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 2013, 12, 978–990. [Google Scholar] [CrossRef] [Green Version]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Cover Picture: Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef]
- Mottram, P.L.; Leong, D.; Crimeen-Irwin, B.; Gloster, S.; Xiang, S.D.; Meanger, J.; Ghildyal, R.; Vardaxis, N.; Plebanski, M. Type 1 and 2 immunity following vaccination is influenced by nanoparticle size: Formulation of a model vaccine for respiratory syncytial Virus. Mol. Pharm. 2006, 4, 73–84. [Google Scholar] [CrossRef]
- Hirai, T.; Yoshioka, Y.; Takahashi, H.; Ichihashi, K.-I.; Yoshida, T.; Tochigi, S.; Nagano, K.; Abe, Y.; Kamada, H.; Tsunoda, S.-I.; et al. Amorphous silica nanoparticles enhance cross-presentation in murine dendritic cells. Biochem. Biophys. Res. Commun. 2012, 427, 553–556. [Google Scholar] [CrossRef]
- Sharma, G.; Valenta, D.T.; Altman, Y.; Harvey, S.; Xie, H.; Mitragotri, S.; Smith, J.W. Polymer particle shape independently influences binding and internalization by macrophages. J. Control. Release 2010, 147, 408–412. [Google Scholar] [CrossRef] [Green Version]
- Howland, S.W.; Wittrup, K.D. Antigen release kinetics in the phagosome are critical to cross-presentation efficiency. J. Immunol. 2008, 180, 1576–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirosue, S.; Kourtis, I.C.; van der Vlies, A.J.; Hubbell, J.A.; Swartz, M.A. Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation. Vaccine 2010, 28, 7897–7906. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, K.; Gleiter, S.; Brinkmann, U.; Rudolph, R.; Lilie, H. Conjugation of an antibody Fv fragment to a virus coat protein: Cell-specific targeting of recombinant polyoma-virus-like particles. Biochem. J. 2001, 356 Pt 3, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Rosalia, R.A.; Quakkelaar, E.D.; Redeker, A.; Khan, S.; Camps, M.; Drijfhout, J.W.; Silva, A.L.; Jiskoot, W.; van Hall, T.; van Veelen, P.A.; et al. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. Eur. J. Immunol. 2013, 43, 2554–2565. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Cheong, H.; Park, Y.S.; Kim, H.; Zhang, M.; Moon, C.; Huang, Y. Cell-penetrating peptide-mediated topical delivery of biomacromolecular drugs. Curr. Pharm. Biotechnol. 2014, 15, 231–239. [Google Scholar] [CrossRef]
- Futaki, S.; Nakase, I.; Tadokoro, A.; Takeuchi, T.; Jones, A.T. Arginine-rich peptides and their internalization mechanisms. Biochem. Soc. Trans. 2007, 35, 784–787. [Google Scholar] [CrossRef]
- Behr, J.-P. The Proton Sponge: A Trick to Enter Cells the Viruses Did Not Exploit. Chimia 1997, 51, 34–36. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Benjaminsen, R.V.; Mattebjerg, M.A.; Henriksen, J.R.; Moghimi, S.M.; Andresen, T.L. The possible “proton sponge” effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol. Ther. 2013, 21, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, F.; Huang, Y. Smart Cell-Penetrating Peptide-Based Techniques for Intracellular Delivery of Therapeutic Macromolecules. Adv. Protein Chem. Struct. Biol. 2018, 112, 183–220. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Murillo, F.M.; Cui, H.; Blosser, R.; Uematsu, S.; Takeda, K.; Akira, S.; Viscidi, R.P.; Roden, R.B.S. Papillomavirus-like particles stimulate murine bone marrow-derived dendritic cells to produce alpha interferon and Th1 immune responses via MyD88. J. Virol. 2004, 78, 11152–11160. [Google Scholar] [CrossRef] [Green Version]
- Kroczek, R.A.; Henn, V. The Role of XCR1 and its Ligand XCL1 in Antigen Cross-Presentation by Murine and Human Dendritic Cells. Front. Immunol. 2012, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.-L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8α+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef] [Green Version]
- Bachem, A.; Güttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, K.P.; Munster, D.J.; Clark, G.J.; Dzionek, A.; Schmitz, J.; Hart, D.N.J. Characterization of human blood dendritic cell subsets. Blood 2002, 100, 4512–4520. [Google Scholar] [CrossRef]
- Pooley, J.L.; Heath, W.R.; Shortman, K. Cutting edge: Intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 2001, 166, 5327–5330. [Google Scholar] [CrossRef] [Green Version]
- Haniffa, M.; Shin, A.; Bigley, V.; McGovern, N.; Teo, P.; See, P.; Wasan, P.S.; Wang, X.-N.; Malinarich, F.; Malleret, B.; et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 2012, 37, 60–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnorrer, P.; Behrens, G.M.N.; Wilson, N.S.; Pooley, J.L.; Smith, C.M.; El-Sukkari, D.; Davey, G.; Kupresanin, F.; Li, M.; Maraskovsky, E.; et al. The dominant role of CD8+dendritic cells in cross-presentation is not dictated by antigen capture. Proc. Natl. Acad. Sci. USA 2006, 103, 10729–10734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belz, G.T.; Smith, C.M.; Eichner, D.; Shortman, K.; Karupiah, G.; Carbone, F.R.; Heath, W.R. Cutting edge: Conventional CD8α+ dendritic cells are generally involved in priming CTL immunity to viruses. J. Immunol. 2004, 172, 1996–2000. [Google Scholar] [CrossRef] [Green Version]
- Belz, G.T.; Shortman, K.; Bevan, M.J.; Heath, W.R. CD8α+ dendritic cells selectively present MHC class I-restricted noncytolytic viral and intracellular bacterial antigens in vivo. J. Immunol. 2005, 175, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Clark, R.A.; Liu, L.; Wagers, A.J.; Fuhlbrigge, R.C.; Kupper, T.S. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 2012, 483, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amsen, D.; van Gisbergen, K.; Hombrink, P.; van Lier, R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef]
- Yang, R.; Murillo, F.M.; Lin, K.-Y.; Yutzy, W.H.; Uematsu, S.; Takeda, K.; Akira, S.; Viscidi, R.P.; Roden, R.B.S. Human papillomavirus type-16 virus-like particles activate complementary defense responses in key dendritic cell subpopulations. J. Immunol. 2004, 173, 2624–2631. [Google Scholar] [CrossRef] [Green Version]
- Förster, R.; Schubel, A.; Breitfeld, D.; Kremmer, E.; Renner-Müller, I.; Wolf, E.; Lipp, M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 1999, 99, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Kapp, E.; Gupta, N.; Wong, J.; Lim, J.; Ji, H.; Heath, W.R.; Simpson, R.; Villadangos, J.A. Differential expression of pathogen-recognition molecules between dendritic cell subsets revealed by plasma membrane proteomic analysis. Mol. Immunol. 2010, 47, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Kaisho, T. Pathogen sensors and chemokine receptors in dendritic cell subsets. Vaccine 2012, 30, 7652–7657. [Google Scholar] [CrossRef] [PubMed]
- Davey, G.M.; Wojtasiak, M.; Proietto, A.I.; Carbone, F.R.; Heath, W.R.; Bedoui, S. Cutting edge: Priming of CD8 T cell immunity to herpes simplex virus type 1 requires cognate TLR3 expression in vivo. J. Immunol. 2010, 184, 2243–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasilakos, J.P.; Tomai, M.A. The use of Toll-like receptor 7/8 agonists as vaccine adjuvants. Expert Rev. Vaccines 2013, 12, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Z.; Kurche, J.S.; Burchill, M.A.; Kedl, R.M. TLR7 enables cross-presentation by multiple dendritic cell subsets through a type I IFN-dependent pathway. Blood 2011, 118, 3028–3038. [Google Scholar] [CrossRef] [Green Version]
- Schulz, O.; Diebold, S.S.; Chen, M.; Näslund, T.I.; Nolte, M.A.; Alexopoulou, L.; Azuma, Y.-T.; Flavell, R.A.; Liljeström, P.; e Sousa, C.R. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar] [CrossRef]
- Edwards, A.D.; Diebold, S.S.; Slack, E.M.C.; Tomizawa, H.; Hemmi, H.; Kaisho, T.; Akira, S.; Reis e Sousa, C. Toll-like receptor expression in murine DC subsets: Lack of TLR7 expression by CD8α+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 2003, 33, 827–833. [Google Scholar] [CrossRef]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Gately, M.K.; Wolitzky, A.G.; Quinn, P.M.; Chizzonite, R. Regulation of human cytolytic lymphocyte responses by interleukin-12. Cell. Immunol. 1992, 143, 127–142. [Google Scholar] [CrossRef]
- Mehrotra, P.T.; Wu, D.; Crim, J.A.; Mostowski, H.S.; Siegel, J.P. Effects of IL-12 on the generation of cytotoxic activity in human CD8+ T lymphocytes. J. Immunol. 1993, 151, 2444–2452. [Google Scholar] [CrossRef]
- Starbeck-Miller, G.R.; Harty, J.T. The Role of Il-12 and Type I Interferon in Governing the Magnitude of CD8 T Cell Responses. Adv. Exp. Med. Biol. 2015, 850, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Swadling, L.; Diniz, M.O.; Schmidt, N.M.; Amin, O.E.; Chandran, A.; Shaw, E.; Pade, C.; Gibbons, J.M.; Le Bert, N.; Tan, A.T.; et al. Pre-existing polymerase-specific T cells expand in abortive seronegative SARS-CoV-2. Nature 2022, 601, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Kundu, R.; Narean, J.S.; Wang, L.; Fenn, J.; Pillay, T.; Fernandez, N.D.; Conibear, E.; Koycheva, A.; Davies, M.; Tolosa-Wright, M.; et al. Cross-reactive memory T cells associate with protection against SARS-CoV-2 infection in COVID-19 contacts. Nat. Commun. 2022, 13, 80. [Google Scholar] [CrossRef]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef]
- Masopust, D.; Choo, D.; Vezys, V.; Wherry, E.J.; Duraiswamy, J.; Akondy, R.; Wang, J.; Casey, K.A.; Barber, D.L.; Kawamura, K.S.; et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 2010, 207, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Enamorado, M.; Iborra, S.; Priego, E.; Cueto, F.J.; Quintana, J.A.; Martínez-Cano, S.; Mejías-Pérez, E.; Esteban, M.; Melero, I.; Hidalgo, A.; et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat. Commun. 2017, 8, 16073. [Google Scholar] [CrossRef]
- Zens, K.D.; Chen, J.K.; Farber, D.L. Vaccine-generated lung tissue–resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 2016, 1, e85832. [Google Scholar] [CrossRef]
- Mikhak, Z.; Strassner, J.P.; Luster, A.D. Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. J. Exp. Med. 2013, 210, 1855–1869. [Google Scholar] [CrossRef] [Green Version]
- Mora, J.R.; Bono, M.R.; Manjunath, N.; Weninger, W.; Cavanagh, L.L.; Rosemblatt, M.; von Andrian, U.H. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 2003, 424, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Mason, D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today 1998, 19, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Gras, S.; Kedzierski, L.; Valkenburg, S.A.; Laurie, K.; Liu, Y.C.; Denholm, J.T.; Richards, M.J.; Rimmelzwaan, G.F.; Kelso, A.; Doherty, P.C.; et al. Cross-reactive CD8+ T-cell immunity between the pandemic H1N1-2009 and H1N1-1918 influenza A viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 12599–12604. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-K.; Cornberg, M.; Wang, X.Z.; Chen, H.D.; Selin, L.K.; Welsh, R.M. Private specificities of CD8 T cell responses control patterns of heterologous immunity. J. Exp. Med. 2005, 201, 523–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souquette, A.; Thomas, P.G. Past Life and Future Effects—How Heterologous Infections Alter Immunity to Influenza Viruses. Front. Immunol. 2018, 9, 1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, R.M.; Selin, L.K. No one is naive: The significance of heterologous T-cell immunity. Nat. Rev. Immunol. 2002, 2, 417–426. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vaccine | Age at Administration | Measure of Tumor Burden | Efficacy | Comment | Reference |
|---|---|---|---|---|---|
| Dendritic cells pulsed with tumor cells | 8 wks | Genitourinary tract weight to whole body weight ratio | 33% | [49] | |
| STEAP Simian adenovirus prime and vaccinia boost | 6–8 wks prime 7–11 wks boost | Genitourinary tract weight to whole body weight ratio | 20% | [22] | |
| Tumor cell lysate + anti-CTLA4 | 14 wks | Prostate weight | 0% | Incidence of tumors was reduced by 26% | [43] |
| PSCA and STEAP DNA prime and vaccinia boost | 7 wks prime 11 wks boost | Genitourinary tract weight | 40% | [50] | |
| Tumor cell lysate + CpG adjuvant in microspheres | Repeated at 10, 12, 14, and 16 wks | Prostate tumor volume by magnetic resonance imaging | 80% | Adjuvanted microspheres alone had 60% efficacy | [51] |
| PSCA DNA prime and VEE self-replicating mRNA boost | 8–10 wks prime 10–12 wks boost | Survival at 200 days | 20% | 80% efficacy at 1 year | [23] |
| PSCA, SPAS-1 and PAP Polyionic VLP vaccine | 19–20 wks | Prostate weight | 43% | [21] | |
| PSCA, SPAS-1 and PAP Polyionic VLPs + anti-PD1 | 19–20 wks | Prostate weight | 63% | [21] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viscidi, R.P.; Rowley, T.; Bossis, I. Bioengineered Bovine Papillomavirus L1 Protein Virus-like Particle (VLP) Vaccines for Enhanced Induction of CD8 T Cell Responses through Cross-Priming. Int. J. Mol. Sci. 2023, 24, 9851. https://doi.org/10.3390/ijms24129851
Viscidi RP, Rowley T, Bossis I. Bioengineered Bovine Papillomavirus L1 Protein Virus-like Particle (VLP) Vaccines for Enhanced Induction of CD8 T Cell Responses through Cross-Priming. International Journal of Molecular Sciences. 2023; 24(12):9851. https://doi.org/10.3390/ijms24129851
Chicago/Turabian StyleViscidi, Raphael P., Treva Rowley, and Ioannis Bossis. 2023. "Bioengineered Bovine Papillomavirus L1 Protein Virus-like Particle (VLP) Vaccines for Enhanced Induction of CD8 T Cell Responses through Cross-Priming" International Journal of Molecular Sciences 24, no. 12: 9851. https://doi.org/10.3390/ijms24129851
APA StyleViscidi, R. P., Rowley, T., & Bossis, I. (2023). Bioengineered Bovine Papillomavirus L1 Protein Virus-like Particle (VLP) Vaccines for Enhanced Induction of CD8 T Cell Responses through Cross-Priming. International Journal of Molecular Sciences, 24(12), 9851. https://doi.org/10.3390/ijms24129851