
Clinical Potential of Hydrogen Sulfide in Peripheral Arterial Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Current Management of PAD and CLTI
- Fontaine
- Stage I—No symptoms
- Stage II—Intermittent claudication subdivided into:
- Stage IIa—Without pain on resting, but with claudication at a distance of greater than 650 feet (200 m)
- Stage IIb—Without pain on resting, but with a claudication distance of less than 650 feet (200 m)
- Stage III—Nocturnal and/or resting pain
- Stage IV—Necrosis (death of tissue) and/or gangrene in the limb
- Rutherford
- Stage 0—Asymptomatic
- Stage 1—Mild claudication
- Stage 2—Moderate claudication
- Stage 3—Severe claudication
- Stage 4—Rest pain
- Stage 5—Minor tissue loss with ischemic nonhealing ulcer or focal gangrene with diffuse pedal ischemia
- Stage 6—Major tissue loss—Extending above transmetatarsal level, functional foot no longer salvageable
3. Etiology of PAD
3.1. Atherosclerosis
3.2. Vascular Medial Calcification
3.3. Microvascular Dysfunction
3.4. Intimal Hyperplasia: The Unmet Challenge of Post-Operative PAD Management
4. Hydrogen Sulfide
4.1. Endogenous H2S Production
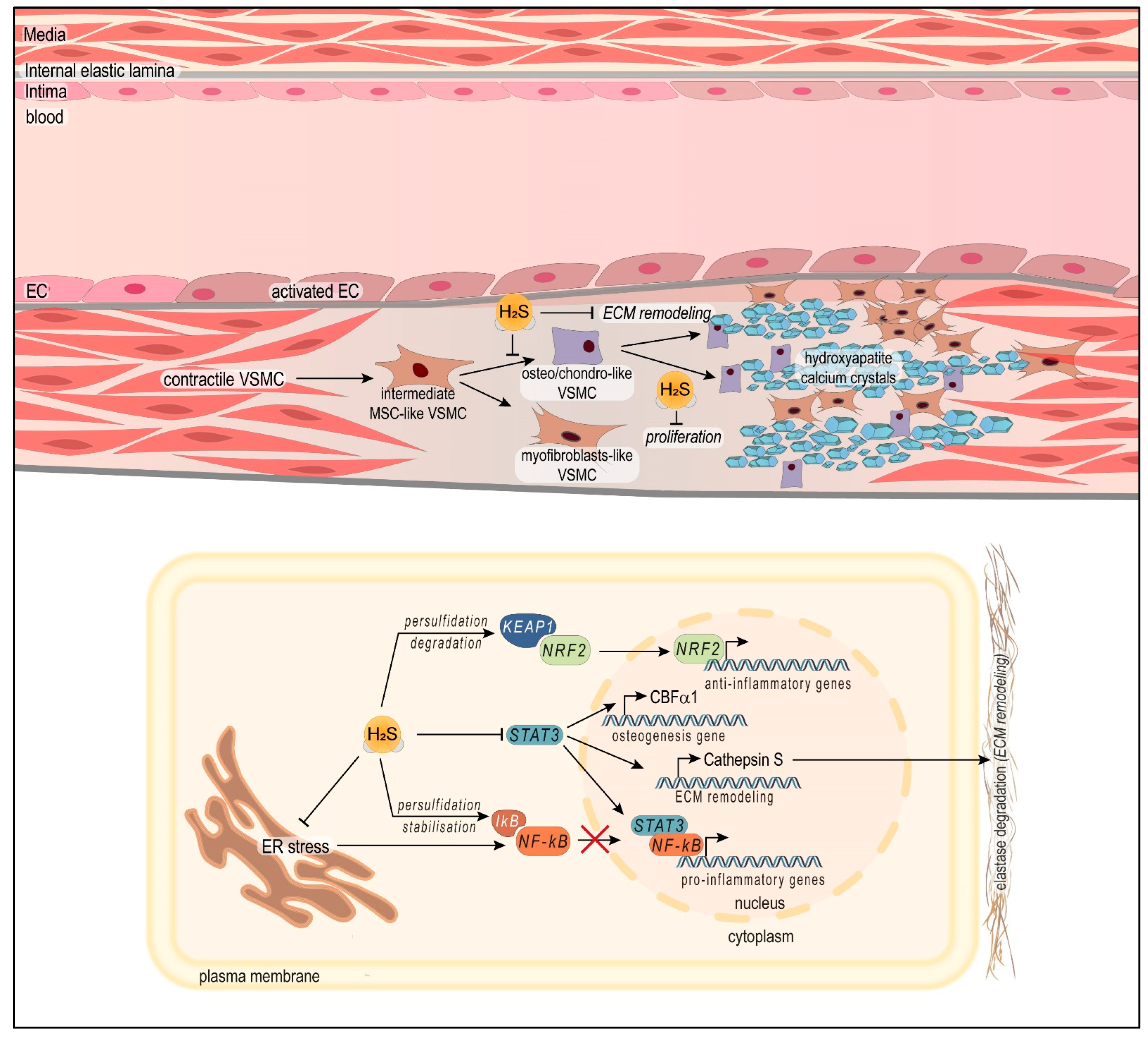
4.2. Vascular Properties of H2S and Benefits in the Context of Peripheral Arterial Disease (PAD and CLTI)
4.2.1. H2S Is a Potent Vasodilator
4.2.2. H2S Protects against Atherosclerosis
4.2.3. H2S Protects against Vascular Medial Calcification
4.2.4. H2S Supports Endothelial Cell Function
4.2.5. H2S Inhibits Intimal Hyperplasia: Post-Operative Management of CLTI Patients
4.3. Further Directions and Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aboyans, V.; Ricco, J.B.; Bartelink, M.E.L.; Bjorck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS): Document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal, upper and lower extremity arteriesEndorsed by: The European Stroke Organization (ESO)The Task Force for the Diagnosis and Treatment of Peripheral Arterial Diseases of the European Society of Cardiology (ESC) and of the European Society for Vascular Surgery (ESVS). Eur. Heart J. 2018, 39, 763–816. [Google Scholar] [CrossRef] [PubMed]
- Aday, A.W.; Matsushita, K. Epidemiology of Peripheral Artery Disease and Polyvascular Disease. Circ. Res. 2021, 128, 1818–1832. [Google Scholar] [CrossRef] [PubMed]
- Hamburg, N.M.; Creager, M.A. Pathophysiology of Intermittent Claudication in Peripheral Artery Disease. Circ. J. 2017, 81, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.L.; Yang, L.S.; Tew, M.; Westcott, M.J.; Spelman, T.D.; Choong, P.F.; Davies, A.H. Quality of Life in Chronic Limb Threatening Ischaemia: Systematic Review and Meta-Analysis. Eur. J. Vasc. Endovasc. Surg. 2022, 64, 666–683. [Google Scholar] [CrossRef]
- Porras, C.P.; Bots, M.L.; Teraa, M.; van Doorn, S.; Vernooij, R.W.M. Differences in Symptom Presentation in Women and Men with Confirmed Lower Limb Peripheral Artery Disease: A Systematic Review and Meta-Analysis. Eur. J. Vasc. Endovasc. Surg. 2022, 63, 602–612. [Google Scholar] [CrossRef]
- Hiatt, W.R.; Armstrong, E.J.; Larson, C.J.; Brass, E.P. Pathogenesis of the limb manifestations and exercise limitations in peripheral artery disease. Circ. Res. 2015, 116, 1527–1539. [Google Scholar] [CrossRef]
- Thukkani, A.K.; Kinlay, S. Endovascular intervention for peripheral artery disease. Circ. Res. 2015, 116, 1599–1613. [Google Scholar] [CrossRef]
- Fowkes, F.G.; Aboyans, V.; Fowkes, F.J.; McDermott, M.M.; Sampson, U.K.; Criqui, M.H. Peripheral artery disease: Epidemiology and global perspectives. Nat. Rev. Cardiol. 2017, 14, 156–170. [Google Scholar] [CrossRef]
- Ying, A.F.; Tang, T.Y.; Jin, A.; Chong, T.T.; Hausenloy, D.J.; Koh, W.P. Diabetes and other vascular risk factors in association with the risk of lower extremity amputation in chronic limb-threatening ischemia: A prospective cohort study. Cardiovasc. Diabetol. 2022, 21, 7. [Google Scholar] [CrossRef]
- Barnes, J.A.; Eid, M.A.; Creager, M.A.; Goodney, P.P. Epidemiology and Risk of Amputation in Patients With Diabetes Mellitus and Peripheral Artery Disease. Atheroscler. Thromb. Vasc. Biol. 2020, 40, 1808–1817. [Google Scholar] [CrossRef]
- Criqui, M.H.; Matsushita, K.; Aboyans, V.; Hess, C.N.; Hicks, C.W.; Kwan, T.W.; McDermott, M.M.; Misra, S.; Ujueta, F.; on behalf of the American Heart Association Council on Epidemiology and Prevention; et al. Lower Extremity Peripheral Artery Disease: Contemporary Epidemiology, Management Gaps, and Future Directions: A Scientific Statement From the American Heart Association. Circulation 2021, 144, e171–e191. [Google Scholar] [CrossRef] [PubMed]
- Abola, M.T.B.; Golledge, J.; Miyata, T.; Rha, S.W.; Yan, B.P.; Dy, T.C.; Ganzon, M.S.V.; Handa, P.K.; Harris, S.; Zhisheng, J.; et al. Asia-Pacific Consensus Statement on the Management of Peripheral Artery Disease: A Report from the Asian Pacific Society of Atherosclerosis and Vascular Disease Asia-Pacific Peripheral Artery Disease Consensus Statement Project Committee. J. Atheroscler. Thromb. 2020, 27, 809–907. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.S.; Bradbury, A.W.; Kolh, P.; White, J.V.; Dick, F.; Fitridge, R.; Mills, J.L.; Ricco, J.B.; Suresh, K.R.; Murad, M.H.; et al. Global vascular guidelines on the management of chronic limb-threatening ischemia. Eur. J. Vasc. Endovasc. Surg. 2019, 58, S1–S109.e33. [Google Scholar] [CrossRef]
- Gerhard-Herman, M.D.; Gornik, H.L.; Barrett, C.; Barshes, N.R.; Corriere, M.A.; Drachman, D.E.; Fleisher, L.A.; Fowkes, F.G.R.; Hamburg, N.M.; Kinlay, S.; et al. 2016 AHA/ACC Guideline on the Management of Patients With Lower Extremity Peripheral Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2017, 69, e71–e126. [Google Scholar] [CrossRef] [PubMed]
- Fowkes, F.G.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Morrison, J.T.; Govsyeyev, N.; Hess, C.N.; Bonaca, M.P. Vorapaxar for Prevention of Major Adverse Cardiovascular and Limb Events in Peripheral Artery Disease. J. Cardiovasc. Pharmacol. Ther. 2022, 27, 10742484211056115. [Google Scholar] [CrossRef]
- Golledge, J. Update on the pathophysiology and medical treatment of peripheral artery disease. Nat. Rev. Cardiol. 2022, 19, 456–474. [Google Scholar] [CrossRef]
- Almasri, J.; Adusumalli, J.; Asi, N.; Lakis, S.; Alsawas, M.; Prokop, L.J.; Bradbury, A.; Kolh, P.; Conte, M.S.; Murad, M.H. A systematic review and meta-analysis of revascularization outcomes of infrainguinal chronic limb-threatening ischemia. J. Vasc. Surg. 2019, 69, 126S–136S. [Google Scholar] [CrossRef]
- Iyer, S.R.; Annex, B.H. Therapeutic Angiogenesis for Peripheral Artery Disease: Lessons Learned in Translational Science. JACC Basic Transl. Sci. 2017, 2, 503–512. [Google Scholar] [CrossRef]
- Bager, L.G.V.; Petersen, J.K.; Havers-Borgersen, E.; Resch, T.; Smolderen, K.G.; Mena-Hurtado, C.; Eiberg, J.; Kober, L.; Fosbol, E.L. The Use of Evidence-Based Medical Therapy in Patients with Critical Limb-Threatening Ischemia. Eur. J. Prev. Cardiol. 2023, zwad022. [Google Scholar] [CrossRef]
- Govsyeyev, N.; Nehler, M.R.; Low Wang, C.C.; Kavanagh, S.; Hiatt, W.R.; Long, C.; Jones, W.S.; Fowkes, F.G.R.; Berger, J.S.; Baumgartner, I.; et al. Etiology and outcomes of amputation in patients with peripheral artery disease in the EUCLID trial. J. Vasc. Surg. 2022, 75, 660–670.e3. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Santulli, G. Update on peripheral artery disease: Epidemiology and evidence-based facts. Atherosclerosis 2018, 275, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.R. Chapter 4—Diseases of Small and Medium-sized Blood Vessels. In Cardiovascular Pathology, 4th ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 125–168. [Google Scholar] [CrossRef]
- Hartmann, F.; Gorski, D.J.; Newman, A.A.C.; Homann, S.; Petz, A.; Owsiany, K.M.; Serbulea, V.; Zhou, Y.Q.; Deaton, R.A.; Bendeck, M.; et al. SMC-Derived Hyaluronan Modulates Vascular SMC Phenotype in Murine Atherosclerosis. Circ. Res. 2021, 129, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef]
- Brandt, K.J.; Burger, F.; Baptista, D.; Roth, A.; Fernandes da Silva, R.; Montecucco, F.; Mach, F.; Miteva, K. Single-Cell Analysis Uncovers Osteoblast Factor Growth Differentiation Factor 10 as Mediator of Vascular Smooth Muscle Cell Phenotypic Modulation Associated with Plaque Rupture in Human Carotid Artery Disease. Int. J. Mol. Sci. 2022, 23, 1796. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cui, Z.Y.; Huang, X.F.; Zhang, D.D.; Guo, R.J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal. Transduct. Target Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M.; et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Gleim, S.; Stitham, J.; Tang, W.H.; Martin, K.A.; Hwa, J. An eicosanoid-centric view of atherothrombotic risk factors. Cell. Mol. Life Sci. 2012, 69, 3361–3380. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Botta, E.; Holinstat, M. Eicosanoids in inflammation in the blood and the vessel. Front. Pharmacol. 2022, 13, 997403. [Google Scholar] [CrossRef] [PubMed]
- Lanzer, P.; Hannan, F.M.; Lanzer, J.D.; Janzen, J.; Raggi, P.; Furniss, D.; Schuchardt, M.; Thakker, R.; Fok, P.W.; Saez-Rodriguez, J.; et al. Medial Arterial Calcification: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1145–1165. [Google Scholar] [CrossRef]
- Kim, T.I.; Guzman, R.J. Medial artery calcification in peripheral artery disease. Front. Cardiovasc. Med. 2023, 10, 1093355. [Google Scholar] [CrossRef] [PubMed]
- Jadidi, M.; Poulson, W.; Aylward, P.; MacTaggart, J.; Sanderfer, C.; Marmie, B.; Pipinos, M.; Kamenskiy, A. Calcification prevalence in different vascular zones and its association with demographics, risk factors, and morphometry. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2313–H2323. [Google Scholar] [CrossRef]
- Sorensen, I.M.H.; Saurbrey, S.A.K.; Hjortkjaer, H.O.; Brainin, P.; Carlson, N.; Ballegaard, E.L.F.; Kamper, A.L.; Christoffersen, C.; Feldt-Rasmussen, B.; Kofoed, K.F.; et al. Regional distribution and severity of arterial calcification in patients with chronic kidney disease stages 1-5: A cross-sectional study of the Copenhagen chronic kidney disease cohort. BMC Nephrol. 2020, 21, 534. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, X.; Wu, H. Arterial Stiffness: A Focus on Vascular Calcification and Its Link to Bone Mineralization. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1078–1093. [Google Scholar] [CrossRef]
- Zununi Vahed, S.; Mostafavi, S.; Hosseiniyan Khatibi, S.M.; Shoja, M.M.; Ardalan, M. Vascular Calcification: An Important Understanding in Nephrology. Vasc. Health Risk Manag. 2020, 16, 167–180. [Google Scholar] [CrossRef]
- Singh, A.; Tandon, S.; Tandon, C. An update on vascular calcification and potential therapeutics. Mol. Biol. Rep. 2021, 48, 887–896. [Google Scholar] [CrossRef]
- Narula, N.; Dannenberg, A.J.; Olin, J.W.; Bhatt, D.L.; Johnson, K.W.; Nadkarni, G.; Min, J.; Torii, S.; Poojary, P.; Anand, S.S.; et al. Pathology of Peripheral Artery Disease in Patients With Critical Limb Ischemia. J. Am. Coll. Cardiol. 2018, 72, 2152–2163. [Google Scholar] [CrossRef]
- Skolnik, J.; Weiss, R.; Meyr, A.J.; Dhanisetty, R.; Choi, E.T.; Cunningham-Hill, M.; Rubin, D.; Oresanya, L. Evaluating the Impact of Medial Arterial Calcification on Outcomes of Infrageniculate Endovascular Interventions for Treatment of Diabetic Foot Ulcers. Vasc. Endovasc. Surg. 2021, 55, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Belur, A.D.; Shah, A.J.; Virani, S.S.; Vorla, M.; Kalra, D.K. Role of Lipid-Lowering Therapy in Peripheral Artery Disease. J. Clin. Med. 2022, 11, 4872. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Camici, P.G.; Bairey Merz, C.N. Coronary microvascular dysfunction: An update. Eur. Heart J. 2014, 35, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Gallinoro, E.; Paolisso, P.; Candreva, A.; Bermpeis, K.; Fabbricatore, D.; Esposito, G.; Bertolone, D.; Fernandez Peregrina, E.; Munhoz, D.; Mileva, N.; et al. Microvascular Dysfunction in Patients With Type II Diabetes Mellitus: Invasive Assessment of Absolute Coronary Blood Flow and Microvascular Resistance Reserve. Front. Cardiovasc. Med. 2021, 8, 765071. [Google Scholar] [CrossRef] [PubMed]
- Kaze, A.D.; Santhanam, P.; Erqou, S.; Ahima, R.S.; Bertoni, A.; Echouffo-Tcheugui, J.B. Microvascular Disease and Incident Heart Failure among Individuals with Type 2 Diabetes Mellitus. J. Am. Heart Assoc. 2021, 10, e018998. [Google Scholar] [CrossRef]
- Owens, C.D.; Mukli, P.; Csipo, T.; Lipecz, A.; Silva-Palacios, F.; Dasari, T.W.; Tarantini, S.; Gardner, A.W.; Montgomery, P.S.; Waldstein, S.R.; et al. Microvascular dysfunction and neurovascular uncoupling are exacerbated in peripheral artery disease, increasing the risk of cognitive decline in older adults. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H924–H935. [Google Scholar] [CrossRef]
- Matsushita, K.; Ballew, S.H.; Coresh, J.; Arima, H.; Arnlov, J.; Cirillo, M.; Ebert, N.; Hiramoto, J.S.; Kimm, H.; Shlipak, M.G.; et al. Measures of chronic kidney disease and risk of incident peripheral artery disease: A collaborative meta-analysis of individual participant data. Lancet Diabetes Endocrinol. 2017, 5, 718–728. [Google Scholar] [CrossRef]
- Yang, C.; Kwak, L.; Ballew, S.H.; Jaar, B.G.; Deal, J.A.; Folsom, A.R.; Heiss, G.; Sharrett, A.R.; Selvin, E.; Sabanayagam, C.; et al. Retinal microvascular findings and risk of incident peripheral artery disease: An analysis from the Atherosclerosis Risk in Communities (ARIC) Study. Atherosclerosis 2020, 294, 62–71. [Google Scholar] [CrossRef]
- De Lorenzo, A.; Moreira, A.S.B.; Muccillo, F.B.; Assad, M.; Tibirica, E.V. Microvascular Function and Endothelial Progenitor Cells in Patients with Severe Hypercholesterolemia and the Familial Hypercholesterolemia Phenotype. Cardiology 2017, 137, 231–236. [Google Scholar] [CrossRef]
- Romero, S.A.; Moralez, G.; Jaffery, M.F.; Huang, M.U.; Engelland, R.E.; Cramer, M.N.; Crandall, C.G. Exercise Training Improves Microvascular Function in Burn Injury Survivors. Med. Sci. Sports Exerc. 2020, 52, 2430–2436. [Google Scholar] [CrossRef]
- Bauer, C.J.; Findlay, M.; Koliamitra, C.; Zimmer, P.; Schick, V.; Ludwig, S.; Gurtner, G.C.; Riedel, B.; Schier, R. Preoperative exercise induces endothelial progenitor cell mobilisation in patients undergoing major surgery—A prospective randomised controlled clinical proof-of-concept trial. Heliyon 2022, 8, e10705. [Google Scholar] [CrossRef]
- Schier, R.; El-Zein, R.; Cortes, A.; Liu, M.; Collins, M.; Rafat, N.; Teschendorf, P.; Wu, H.K.; Heymach, J.; Mehran, R.; et al. Endothelial progenitor cell mobilization by preoperative exercise: A bone marrow response associated with postoperative outcome. Br. J. Anaesth. 2014, 113, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Hurley, D.M.; Williams, E.R.; Cross, J.M.; Riedinger, B.R.; Meyer, R.A.; Abela, G.S.; Slade, J.M. Aerobic Exercise Improves Microvascular Function in Older Adults. Med. Sci. Sports Exerc. 2019, 51, 773–781. [Google Scholar] [CrossRef]
- Dillon, G.A.; Stanhewicz, A.E.; Serviente, C.; Flores, V.A.; Stachenfeld, N.; Alexander, L.M. Seven days of statin treatment improves nitric-oxide mediated endothelial-dependent cutaneous microvascular function in women with endometriosis. Microvasc. Res. 2022, 144, 104421. [Google Scholar] [CrossRef] [PubMed]
- Pajkowski, M.; Dudziak, M.; Chlebus, K.; Hellmann, M. Assessment of microvascular function and pharmacological regulation in genetically confirmed familial hypercholesterolemia. Microvasc. Res. 2021, 138, 104216. [Google Scholar] [CrossRef]
- Tentolouris, A.; Eleftheriadou, I.; Tzeravini, E.; Tsilingiris, D.; Paschou, S.A.; Siasos, G.; Tentolouris, N. Endothelium as a Therapeutic Target in Diabetes Mellitus: From Basic Mechanisms to Clinical Practice. Curr. Med. Chem. 2020, 27, 1089–1131. [Google Scholar] [CrossRef]
- Love, K.M.; Barrett, E.J.; Horton, W.B. Metformin’s Impact on the Microvascular Response to Insulin. Endocrinology 2022, 163, bqac162. [Google Scholar] [CrossRef]
- Liu, J.; Aylor, K.W.; Chai, W.; Barrett, E.J.; Liu, Z. Metformin prevents endothelial oxidative stress and microvascular insulin resistance during obesity development in male rats. Am. J. Physiol. Endocrinol. Metab. 2022, 322, E293–E306. [Google Scholar] [CrossRef]
- Silva, C.; Rodrigues, I.; Andrade, S.; Costa, R.; Soares, R. Metformin Reduces Vascular Assembly in High Glucose-Treated Human Microvascular Endothelial Cells in An AMPK-Independent Manner. Cell J. 2021, 23, 174–183. [Google Scholar] [CrossRef]
- Simpson, E.L.; Kearns, B.; Stevenson, M.D.; Cantrell, A.J.; Littlewood, C.; Michaels, J.A. Enhancements to angioplasty for peripheral arterial occlusive disease: Systematic review, cost-effectiveness assessment and expected value of information analysis. Health Technol. Assess. 2014, 18, 1–252. [Google Scholar] [CrossRef] [PubMed]
- Buccheri, D.; Piraino, D.; Andolina, G.; Cortese, B. Understanding and managing in-stent restenosis: A review of clinical data, from pathogenesis to treatment. J. Thorac. Dis. 2016, 8, E1150–E1162. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Otsuka, F.; Yahagi, K.; Sakakura, K.; Kutys, R.; Ladich, E.R.; Finn, A.V.; Kolodgie, F.D.; Virmani, R. Human autopsy study of drug-eluting stents restenosis: Histomorphological predictors and neointimal characteristics. Eur. Heart J. 2013, 34, 3304–3313. [Google Scholar] [CrossRef]
- Deglise, S.; Bechelli, C.; Allagnat, F. Vascular smooth muscle cells in intimal hyperplasia, an update. Front. Physiol. 2022, 13, 1081881. [Google Scholar] [CrossRef]
- Chakraborty, R.; Chatterjee, P.; Dave, J.M.; Ostriker, A.C.; Greif, D.M.; Rzucidlo, E.M.; Martin, K.A. Targeting smooth muscle cell phenotypic switching in vascular disease. JVS Vasc. Sci. 2021, 2, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yan, K.; Wu, L.; Zheng, Z.; Du, Y.; Liu, Z.; Zhao, L.; Li, W.; Sheng, Y.; Ren, L.; et al. Single-cell RNA-seq reveals cellular heterogeneity of mouse carotid artery under disturbed flow. Cell Death Discov. 2021, 7, 180. [Google Scholar] [CrossRef]
- Yu-Wei, D.; Li, Z.S.; Xiong, S.M.; Huang, G.; Luo, Y.F.; Huo, T.Y.; Zhou, M.H.; Zheng, Y.W. Paclitaxel induces apoptosis through the TAK1-JNK activation pathway. FEBS Open Bio 2020, 10, 1655–1667. [Google Scholar] [CrossRef]
- Cirino, G.; Szabo, C.; Papapetropoulos, A. Physiological roles of hydrogen sulfide in mammalian cells, tissues and organs. Physiol. Rev. 2022, 103, 31–276. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]
- Sen, N. Functional and Molecular Insights of Hydrogen Sulfide Signaling and Protein Sulfhydration. J. Mol. Biol. 2017, 429, 543–561. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Looso, M.; Weigert, A.; Ratiu, C.; Wittig, J.; Drekolia, M.K.; Tombor, L.; Randriamboavonjy, V.; Leisegang, M.S.; et al. Mapping the Endothelial Cell S-Sulfhydrome Highlights the Crucial Role of Integrin Sulfhydration in Vascular Function. Circulation 2021, 143, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Liu, K.; He, J.; Tian, C.; Yu, X.; Yang, J. Direct Proteomic Mapping of Cysteine Persulfidation. Antioxid. Redox Signal. 2020, 33, 1061–1076. [Google Scholar] [CrossRef] [PubMed]
- Bibli, S.I.; Hu, J.; Sigala, F.; Wittig, I.; Heidler, J.; Zukunft, S.; Tsilimigras, D.I.; Randriamboavonjy, V.; Wittig, J.; Kojonazarov, B.; et al. Cystathionine gamma Lyase Sulfhydrates the RNA Binding Protein Human Antigen R to Preserve Endothelial Cell Function and Delay Atherogenesis. Circulation 2019, 139, 101–114. [Google Scholar] [CrossRef]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Antiaging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152–1170.e13. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Agbaga, M.P.; Anderson, R.E. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic. Biol. Med. 2007, 42, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Shackelford, R.E.; Shen, X.; Dominic, P.; Kevil, C.G. Sulfide regulation of cardiovascular function in health and disease. Nat. Rev. Cardiol. 2022, 20, 109–125. [Google Scholar] [CrossRef]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef]
- Longchamp, A.; Mirabella, T.; Arduini, A.; MacArthur, M.R.; Das, A.; Trevino-Villarreal, J.H.; Hine, C.; Ben-Sahra, I.; Knudsen, N.H.; Brace, L.E.; et al. Amino Acid Restriction Triggers Angiogenesis via GCN2/ATF4 Regulation of VEGF and H2S Production. Cell 2018, 173, 117–129.e4. [Google Scholar] [CrossRef]
- Bibli, S.I.; Hu, J.; Leisegang, M.S.; Wittig, J.; Zukunft, S.; Kapasakalidi, A.; Fisslthaler, B.; Tsilimigras, D.; Zografos, G.; Filis, K.; et al. Shear stress regulates cystathionine gamma lyase expression to preserve endothelial redox balance and reduce membrane lipid peroxidation. Redox Biol. 2020, 28, 101379. [Google Scholar] [CrossRef]
- Yuan, S.; Yurdagul, A., Jr.; Peretik, J.M.; Alfaidi, M.; Al Yafeai, Z.; Pardue, S.; Kevil, C.G.; Orr, A.W. Cystathionine gamma-Lyase Modulates Flow-Dependent Vascular Remodeling. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jia, G.; Li, H.; Yan, S.; Qian, J.; Guo, X.; Li, G.; Qin, H.; Zhu, Z.; Wu, Y.; et al. H2O2-Mediated Oxidative Stress Enhances Cystathionine γ-Lyase-Derived H2S Synthesis Via a Sulfenic Acid Intermediate. Antioxidants 2021, 10, 1488. [Google Scholar] [CrossRef]
- Das, M.; Dewan, A.; Shee, S.; Singh, A. The Multifaceted Bacterial Cysteine Desulfurases: From Metabolism to Pathogenesis. Antioxidants 2021, 10, 997. [Google Scholar] [CrossRef] [PubMed]
- Kushkevych, I.; Cejnar, J.; Treml, J.; Dordevic, D.; Kollar, P.; Vitezova, M. Recent Advances in Metabolic Pathways of Sulfate Reduction in Intestinal Bacteria. Cells 2020, 9, 698. [Google Scholar] [CrossRef] [PubMed]
- Figliuolo, V.R.; Coutinho-Silva, R.; Coutinho, C. Contribution of sulfate-reducing bacteria to homeostasis disruption during intestinal inflammation. Life Sci. 2018, 215, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Lin, H.C. Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms 2015, 3, 866–889. [Google Scholar] [CrossRef]
- Rey, F.E.; Gonzalez, M.D.; Cheng, J.; Wu, M.; Ahern, P.P.; Gordon, J.I. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc. Natl. Acad. Sci. USA 2013, 110, 13582–13587. [Google Scholar] [CrossRef]
- Islam, K.N.; Polhemus, D.J.; Donnarumma, E.; Brewster, L.P.; Lefer, D.J. Hydrogen Sulfide Levels and Nuclear Factor-Erythroid 2-Related Factor 2 (NRF2) Activity Are Attenuated in the Setting of Critical Limb Ischemia (CLI). J. Am. Heart Assoc. 2015, 4, e001986. [Google Scholar] [CrossRef]
- Beard, R.S., Jr.; Bearden, S.E. Vascular complications of cystathionine beta-synthase deficiency: Future directions for homocysteine-to-hydrogen sulfide research. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H13–H26. [Google Scholar] [CrossRef]
- Longchamp, A.; MacArthur, M.R.; Trocha, K.; Ganahl, J.; Mann, C.G.; Kip, P.; King, W.W.; Sharma, G.; Tao, M.; Mitchell, S.J.; et al. Plasma Hydrogen Sulfide Is Positively Associated With Post-operative Survival in Patients Undergoing Surgical Revascularization. Front. Cardiovasc. Med. 2021, 8, 750926. [Google Scholar] [CrossRef]
- Wang, R. Roles of Hydrogen Sulfide in Hypertension Development and Its Complications: What, So What, Now What. Hypertension 2023, 80, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Lu, P.C. Early short-term treatment with exogenous hydrogen sulfide postpones the transition from prehypertension to hypertension in spontaneously hypertensive rat. Clin. Exp. Hypertens. 2018, 40, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, L.K.; Zhang, C.Y.; Zeng, X.J.; Yan, H.; Jin, H.F.; Tang, C.S.; Du, J.B. Regulatory effect of hydrogen sulfide on vascular collagen content in spontaneously hypertensive rats. Hypertens. Res. 2008, 31, 1619–1630. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, Y.; Zhang, R.; Chen, Q.; Chen, J.; Zong, Y.; Liu, J.; Feng, S.; Liu, A.D.; Holmberg, L.; et al. Hydrogen sulfide upregulates KATP channel expression in vascular smooth muscle cells of spontaneously hypertensive rats. J. Mol. Med. 2015, 93, 439–455. [Google Scholar] [CrossRef]
- Huang, P.; Chen, S.; Wang, Y.; Liu, J.; Yao, Q.; Huang, Y.; Li, H.; Zhu, M.; Wang, S.; Li, L.; et al. Down-regulated CBS/H2S pathway is involved in high-salt-induced hypertension in Dahl rats. Nitric Oxide 2015, 46, 192–203. [Google Scholar] [CrossRef]
- Liu, X.Y.; Qian, L.L.; Wang, R.X. Hydrogen Sulfide-Induced Vasodilation: The Involvement of Vascular Potassium Channels. Front. Pharmacol. 2022, 13, 911704. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 2011, 109, 1259–1268. [Google Scholar] [CrossRef]
- Naik, J.S.; Osmond, J.M.; Walker, B.R.; Kanagy, N.L. Hydrogen sulfide-induced vasodilation mediated by endothelial TRPV4 channels. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1437–H1444. [Google Scholar] [CrossRef]
- Szabo, C.; Papapetropoulos, A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br. J. Pharmacol. 2011, 164, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Bertoni, G.; Pla, A.F.; Dragoni, S.; Pupo, E.; Merlino, A.; Mancardi, D.; Munaron, L.; Tanzi, F. Hydrogen sulfide regulates intracellular Ca2+ concentration in endothelial cells from excised rat aorta. Curr. Pharm. Biotechnol. 2011, 12, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- d’Emmanuele di Villa Bianca, R.; Sorrentino, R.; Coletta, C.; Mitidieri, E.; Rossi, A.; Vellecco, V.; Pinto, A.; Cirino, G.; Sorrentino, R. Hydrogen sulfide-induced dual vascular effect involves arachidonic acid cascade in rat mesenteric arterial bed. J. Pharmacol. Exp. Ther. 2011, 337, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Orlov, S.N.; Gusakova, S.V.; Smaglii, L.V.; Koltsova, S.V.; Sidorenko, S.V. Vasoconstriction triggered by hydrogen sulfide: Evidence for Na+,K+,2Cl− cotransport and L-type Ca2+ channel-mediated pathway. Biochem. Biophys. Rep. 2017, 12, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Ping, N.N.; Li, S.; Mi, Y.N.; Cao, L.; Cao, Y.X. Hydrogen sulphide induces vasoconstriction of rat coronary artery via activation of Ca2+ influx. Acta Physiol. 2015, 214, 88–96. [Google Scholar] [CrossRef]
- Szijarto, I.A.; Marko, L.; Filipovic, M.R.; Miljkovic, J.L.; Tabeling, C.; Tsvetkov, D.; Wang, N.; Rabelo, L.A.; Witzenrath, M.; Diedrich, A.; et al. Cystathionine gamma-Lyase-Produced Hydrogen Sulfide Controls Endothelial NO Bioavailability and Blood Pressure. Hypertension 2018, 71, 1210–1217. [Google Scholar] [CrossRef]
- Kanagy, N.L.; Szabo, C.; Papapetropoulos, A. Vascular biology of hydrogen sulfide. Am. J. Physiol. Cell Physiol. 2017, 312, C537–C549. [Google Scholar] [CrossRef] [PubMed]
- Greaney, J.L.; Kutz, J.L.; Shank, S.W.; Jandu, S.; Santhanam, L.; Alexander, L.M. Impaired Hydrogen Sulfide-Mediated Vasodilation Contributes to Microvascular Endothelial Dysfunction in Hypertensive Adults. Hypertension 2017, 69, 902–909. [Google Scholar] [CrossRef]
- Aminzadeh, M.A.; Vaziri, N.D. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol. Dial. Transplant. 2012, 27, 498–504. [Google Scholar] [CrossRef]
- Khaledifar, A.; Mobasheri, M.; Kheiri, S.; Zamani, Z. Comparison of N-acetylcysteine and angiotensin converting enzyme inhibitors in blood pressure regulation in hypertensive patients. ARYA Atheroscler. 2015, 11, 5–13. [Google Scholar]
- Dillon, G.A.; Stanhewicz, A.E.; Serviente, C.; Greaney, J.L.; Alexander, L.M. Hydrogen sulfide-dependent microvascular vasodilation is improved following chronic sulfhydryl-donating antihypertensive pharmacotherapy in adults with hypertension. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H728–H734. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, X.; Jin, H.; Wei, H.; Li, W.; Bu, D.; Tang, X.; Ren, Y.; Tang, C.; Du, J. Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 173–179. [Google Scholar] [CrossRef]
- Mani, S.; Li, H.; Untereiner, A.; Wu, L.; Yang, G.; Austin, R.C.; Dickhout, J.G.; Lhotak, S.; Meng, Q.H.; Wang, R. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 2013, 127, 2523–2534. [Google Scholar] [CrossRef]
- Ford, A.; Al-Magableh, M.; Gaspari, T.A.; Hart, J.L. Chronic NaHS Treatment Is Vasoprotective in High-Fat-Fed ApoE−/− Mice. Int. J.Vasc. Med. 2013, 2013, 915983. [Google Scholar] [CrossRef]
- Zhang, H.; Guo, C.; Wu, D.; Zhang, A.; Gu, T.; Wang, L.; Wang, C. Hydrogen sulfide inhibits the development of atherosclerosis with suppressing CX3CR1 and CX3CL1 expression. PLoS ONE 2012, 7, e41147. [Google Scholar] [CrossRef]
- Liu, Z.; Han, Y.; Li, L.; Lu, H.; Meng, G.; Li, X.; Shirhan, M.; Peh, M.T.; Xie, L.; Zhou, S.; et al. The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E−/− mice. Br. J. Pharmacol. 2013, 169, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Guo, C.; Zhang, A.; Fan, Y.; Gu, T.; Wu, D.; Sparatore, A.; Wang, C. Effect of S-aspirin, a novel hydrogen-sulfide-releasing aspirin (ACS14), on atherosclerosis in apoE-deficient mice. Eur. J. Pharmacol. 2012, 697, 106–116. [Google Scholar] [CrossRef]
- Pan, L.L.; Qin, M.; Liu, X.H.; Zhu, Y.Z. The Role of Hydrogen Sulfide on Cardiovascular Homeostasis: An Overview with Update on Immunomodulation. Front. Pharmacol. 2017, 8, 686. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Huang, Y.; Yan, H.; Zhang, Q.; Zhao, M.; Zhu, M.; Liu, J.; Chen, S.X.; Bu, D.; Tang, C.; et al. Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor kappaB (NF-kappaB) pathway. J. Biol. Chem. 2014, 289, 9741–9753. [Google Scholar] [CrossRef]
- Wang, X.H.; Wang, F.; You, S.J.; Cao, Y.J.; Cao, L.D.; Han, Q.; Liu, C.F.; Hu, L.F. Dysregulation of cystathionine gamma-lyase (CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced inflammation in macrophage. Cell Signal. 2013, 25, 2255–2262. [Google Scholar] [CrossRef]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-kappaB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.F.; Liang, C.; Liang, J.M.; Tang, C.S.; Du, J.B. Effects of hydrogen sulfide on vascular inflammation in pulmonary hypertension induced by high pulmonary blood flow: Experiment with rats. Zhonghua Yi Xue Za Zhi 2008, 88, 2235–2239. [Google Scholar] [PubMed]
- Brancaleone, V.; Mitidieri, E.; Flower, R.J.; Cirino, G.; Perretti, M. Annexin A1 mediates hydrogen sulfide properties in the control of inflammation. J. Pharmacol. Exp. Ther. 2014, 351, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Lin, X.; Xu, W.; Zheng, F.; Cai, J.; Yang, J.; Cui, Q.; Tang, C.; Cai, J.; Xu, G.; et al. Sulfhydrated Sirtuin-1 Increasing Its Deacetylation Activity Is an Essential Epigenetics Mechanism of Anti-Atherogenesis by Hydrogen Sulfide. Antioxid. Redox Signal. 2019, 30, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Lu, M.; Hu, L.F.; Wong, P.T.; Webb, G.D.; Bian, J.S. Hydrogen sulfide in the mammalian cardiovascular system. Antioxid. Redox Signal. 2012, 17, 141–185. [Google Scholar] [CrossRef]
- Martelli, A.; Piragine, E.; Gorica, E.; Citi, V.; Testai, L.; Pagnotta, E.; Lazzeri, L.; Pecchioni, N.; Ciccone, V.; Montanaro, R.; et al. The H2S-Donor Erucin Exhibits Protective Effects against Vascular Inflammation in Human Endothelial and Smooth Muscle Cells. Antioxidants 2021, 10, 961. [Google Scholar] [CrossRef]
- Wang, R.; Szabo, C.; Ichinose, F.; Ahmed, A.; Whiteman, M.; Papapetropoulos, A. The role of H2S bioavailability in endothelial dysfunction. Trends Pharmacol. Sci. 2015, 36, 568–578. [Google Scholar] [CrossRef]
- Dufton, N.; Natividad, J.; Verdu, E.F.; Wallace, J.L. Hydrogen sulfide and resolution of acute inflammation: A comparative study utilizing a novel fluorescent probe. Sci. Rep. 2012, 2, 499. [Google Scholar] [CrossRef]
- Miao, L.; Xin, X.; Xin, H.; Shen, X.; Zhu, Y.Z. Hydrogen Sulfide Recruits Macrophage Migration by Integrin beta1-Src-FAK/Pyk2-Rac Pathway in Myocardial Infarction. Sci. Rep. 2016, 6, 22363. [Google Scholar] [CrossRef]
- Zhou, X.; Chu, X.; Xin, D.; Li, T.; Bai, X.; Qiu, J.; Yuan, H.; Liu, D.; Wang, D.; Wang, Z. L-Cysteine-Derived H2S Promotes Microglia M2 Polarization via Activation of the AMPK Pathway in Hypoxia-Ischemic Neonatal Mice. Front. Mol. Neurosci. 2019, 12, 58. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, Y.; Zhu, N.; Zhao, S.; Fan, J.; Liu, E. Hydrogen sulfide inhibits development of atherosclerosis through up-regulating protein S-nitrosylation. Biomed. Pharmacother. 2016, 83, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.F.; Sio, S.W.; Moochhala, S.M.; MacAry, P.A.; Bhatia, M. Hydrogen sulfide upregulates cyclooxygenase-2 and prostaglandin E metabolite in sepsis-evoked acute lung injury via transient receptor potential vanilloid type 1 channel activation. J. Immunol. 2011, 187, 4778–4787. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Pan, L.; Ji, Y. H 2S protects against diabetes-accelerated atherosclerosis by preventing the activation of NLRP3 inflammasome. J. Biomed. Res. 2019, 34, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Daiber, A.; Dopheide, J.F.; Munzel, T.; Espinola-Klein, C. Peripheral artery disease, redox signaling, oxidative stress—Basic and clinical aspects. Redox. Biol. 2017, 12, 787–797. [Google Scholar] [CrossRef]
- Geng, B.; Chang, L.; Pan, C.; Qi, Y.; Zhao, J.; Pang, Y.; Du, J.; Tang, C. Endogenous hydrogen sulfide regulation of myocardial injury induced by isoproterenol. Biochem. Biophys. Res. Commun. 2004, 318, 756–763. [Google Scholar] [CrossRef]
- Muellner, M.K.; Schreier, S.M.; Laggner, H.; Hermann, M.; Esterbauer, H.; Exner, M.; Gmeiner, B.M.; Kapiotis, S. Hydrogen sulfide destroys lipid hydroperoxides in oxidized LDL. Biochem. J. 2009, 420, 277–281. [Google Scholar] [CrossRef]
- Zavaczki, E.; Jeney, V.; Agarwal, A.; Zarjou, A.; Oros, M.; Katko, M.; Varga, Z.; Balla, G.; Balla, J. Hydrogen sulfide inhibits the calcification and osteoblastic differentiation of vascular smooth muscle cells. Kidney Int. 2011, 80, 731–739. [Google Scholar] [CrossRef]
- Du, H.P.; Li, J.; You, S.J.; Wang, Y.L.; Wang, F.; Cao, Y.J.; Hu, L.F.; Liu, C.F. DNA methylation in cystathionine-gamma-lyase (CSE) gene promoter induced by ox-LDL in macrophages and in apoE knockout mice. Biochem. Biophys. Res. Commun. 2016, 469, 776–782. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H.; Kashfi, K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox. Biol. 2021, 38, 101772. [Google Scholar] [CrossRef] [PubMed]
- Corsello, T.; Komaravelli, N.; Casola, A. Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxidants 2018, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G.; et al. Hydrogen Sulfide Induces Keap1 S-sulfhydration and Suppresses Diabetes-Accelerated Atherosclerosis via Nrf2 Activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, J.; Sun, A.; Sun, Y.; Yu, X.; Liu, N.; Dong, S.; Yang, F.; Zhang, L.; Zhong, X.; et al. Hydrogen sulfide decreases high glucose/palmitate-induced autophagy in endothelial cells by the Nrf2-ROS-AMPK signaling pathway. Cell Biosci. 2016, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Shefa, U.; Kim, M.S.; Jeong, N.Y.; Jung, J. Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxid. Med. Cell Longev. 2018, 2018, 1873962. [Google Scholar] [CrossRef]
- Cheung, S.H.; Lau, J.Y.W. Hydrogen sulfide mediates athero-protection against oxidative stress via S-sulfhydration. PLoS ONE 2018, 13, e0194176. [Google Scholar] [CrossRef]
- Xie, Z.Z.; Liu, Y.; Bian, J.S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxid. Med. Cell Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef]
- Mao, Z.; Huang, Y.; Zhang, Z.; Yang, X.; Zhang, X.; Huang, Y.; Sawada, N.; Mitsui, T.; Takeda, M.; Yao, J. Pharmacological levels of hydrogen sulfide inhibit oxidative cell injury through regulating the redox state of thioredoxin. Free Radic. Biol. Med. 2019, 134, 190–199. [Google Scholar] [CrossRef]
- Nicholson, C.K.; Lambert, J.P.; Molkentin, J.D.; Sadoshima, J.; Calvert, J.W. Thioredoxin 1 is essential for sodium sulfide-mediated cardioprotection in the setting of heart failure. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 744–751. [Google Scholar] [CrossRef]
- Tian, D.; Dong, J.; Jin, S.; Teng, X.; Wu, Y. Endogenous hydrogen sulfide-mediated MAPK inhibition preserves endothelial function through TXNIP signaling. Free Radic. Biol. Med. 2017, 110, 291–299. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, N.; Gong, X.; Ni, S.; Xu, L.; Zhang, H. Thioredoxin-1 attenuates atherosclerosis development through inhibiting NLRP3 inflammasome. Endocrine 2020, 70, 65–70. [Google Scholar] [CrossRef] [PubMed]
- El Hadri, K.; Mahmood, D.F.; Couchie, D.; Jguirim-Souissi, I.; Genze, F.; Diderot, V.; Syrovets, T.; Lunov, O.; Simmet, T.; Rouis, M. Thioredoxin-1 promotes anti-inflammatory macrophages of the M2 phenotype and antagonizes atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhao, G.; Chen, B. Protective Mechanism of Thioredoxin-1 against Atherosclerotic Endothelial Injury Induced by Ox-LDL. J. Cardiol. Cardiovasc. Sci. 2018, 2, 13–16. [Google Scholar] [CrossRef]
- Swiatkiewicz, I.; Wroblewski, M.; Nuszkiewicz, J.; Sutkowy, P.; Wroblewska, J.; Wozniak, A. The Role of Oxidative Stress Enhanced by Adiposity in Cardiometabolic Diseases. Int. J. Mol. Sci. 2023, 24, 6382. [Google Scholar] [CrossRef]
- Cheng, C.K.; Ding, H.; Jiang, M.; Yin, H.; Gollasch, M.; Huang, Y. Perivascular adipose tissue: Fine-tuner of vascular redox status and inflammation. Redox. Biol. 2023, 62, 102683. [Google Scholar] [CrossRef]
- Kim, H.W.; Shi, H.; Winkler, M.A.; Lee, R.; Weintraub, N.L. Perivascular Adipose Tissue and Vascular Perturbation/Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2569–2576. [Google Scholar] [CrossRef]
- Liu, L.; Shi, Z.; Ji, X.; Zhang, W.; Luan, J.; Zahr, T.; Qiang, L. Adipokines, adiposity, and atherosclerosis. Cell. Mol. Life Sci. 2022, 79, 272. [Google Scholar] [CrossRef]
- Raman, P.; Khanal, S. Leptin in Atherosclerosis: Focus on Macrophages, Endothelial and Smooth Muscle Cells. Int. J. Mol. Sci. 2021, 22, 5446. [Google Scholar] [CrossRef]
- Maeda, N.; Funahashi, T.; Matsuzawa, Y.; Shimomura, I. Adiponectin, a unique adipocyte-derived factor beyond hormones. Atherosclerosis 2020, 292, 1–9. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, B.; Ma, D.; Wang, L.; Duan, W. Hydrogen Sulfide, Adipose Tissue and Diabetes Mellitus. Diabetes Metab. Syndr. Obes. 2020, 13, 1873–1886. [Google Scholar] [CrossRef]
- Casili, G.; Randi, E.; Panagaki, T.; Zuhra, K.; Petrosino, M.; Szabo, C. Inhibition of the 3-mercaptopyruvate sulfurtransferase-hydrogen sulfide system promotes cellular lipid accumulation. Geroscience 2022, 44, 2271–2289. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Z.; Zhou, H.; Wang, H.Y.; Dai, H.B.; Gao, Q.; Qian, P.; Zhou, Y.B. Hydrogen sulfide prevents arterial medial calcification in rats with diabetic nephropathy. BMC Cardiovasc. Disord. 2021, 21, 495. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signal. 2012, 17, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Hao, D.D.; Sun, J.P.; Li, W.W.; Zhao, M.M.; Li, X.H.; Chen, Y.; Zhu, J.H.; Ding, Y.J.; Liu, J.; et al. Hydrogen sulfide treatment promotes glucose uptake by increasing insulin receptor sensitivity and ameliorates kidney lesions in type 2 diabetes. Antioxid. Redox Signal. 2013, 19, 5–23. [Google Scholar] [CrossRef]
- Zhao, S.; Li, X.; Li, X.; Wei, X.; Wang, H. Hydrogen Sulfide Plays an Important Role in Diabetic Cardiomyopathy. Front. Cell Dev. Biol. 2021, 9, 627336. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, H.; Guo, N. Protective effect of hydrogen sulfide on the kidney (Review). Mol. Med. Rep. 2021, 24, 696. [Google Scholar] [CrossRef]
- Abedin, M.; Tintut, Y.; Demer, L.L. Vascular calcification: Mechanisms and clinical ramifications. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1161–1170. [Google Scholar] [CrossRef]
- Karwowski, W.; Naumnik, B.; Szczepanski, M.; Mysliwiec, M. The mechanism of vascular calcification—A systematic review. Med. Sci. Monit. 2012, 18, RA1–RA11. [Google Scholar] [CrossRef]
- Sage, A.P.; Tintut, Y.; Demer, L.L. Regulatory mechanisms in vascular calcification. Nat. Rev. Cardiol. 2010, 7, 528–536. [Google Scholar] [CrossRef]
- Shao, J.S.; Cheng, S.L.; Sadhu, J.; Towler, D.A. Inflammation and the osteogenic regulation of vascular calcification: A review and perspective. Hypertension 2010, 55, 579–592. [Google Scholar] [CrossRef]
- Wu, S.Y.; Pan, C.S.; Geng, B.; Zhao, J.; Yu, F.; Pang, Y.Z.; Tang, C.S.; Qi, Y.F. Hydrogen sulfide ameliorates vascular calcification induced by vitamin D3 plus nicotine in rats. Acta Pharmacol. Sin. 2006, 27, 299–306. [Google Scholar] [CrossRef]
- Yang, R.; Teng, X.; Li, H.; Xue, H.M.; Guo, Q.; Xiao, L.; Wu, Y.M. Hydrogen Sulfide Improves Vascular Calcification in Rats by Inhibiting Endoplasmic Reticulum Stress. Oxidative Med. Cell. Longev. 2016, 2016, 9095242. [Google Scholar] [CrossRef]
- Aghagolzadeh, P.; Radpour, R.; Bachtler, M.; van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 2017, 265, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.B.; Zhou, H.; Li, L.; Kang, Y.; Cao, X.; Wu, Z.Y.; Ding, L.; Sethi, G.; Bian, J.S. Hydrogen Sulfide Prevents Elastin Loss and Attenuates Calcification Induced by High Glucose in Smooth Muscle Cells through Suppression of Stat3/Cathepsin S Signaling Pathway. Int. J. Mol. Sci. 2019, 20, 4202. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Luciano, M.G.; Ingrosso, D.; Pulzella, P.; Sepe, I.; Lanza, D.; Violetti, E.; Capasso, R.; Lombardi, C.; De Santo, N.G. Hydrogen sulphide-generating pathways in haemodialysis patients: A study on relevant metabolites and transcriptional regulation of genes encoding for key enzymes. Nephrol. Dial. Transplant. 2009, 24, 3756–3763. [Google Scholar] [CrossRef] [PubMed]
- Nasi, S.; Ea, H.K.; Liote, F.; So, A.; Busso, N. Sodium Thiosulfate Prevents Chondrocyte Mineralization and Reduces the Severity of Murine Osteoarthritis. PLoS ONE 2016, 11, e0158196. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Zhuo, L.; Wang, Y.; Jun, M.; Li, G.; Wang, L.; Hong, D. Systematic review of sodium thiosulfate in treating calciphylaxis in chronic kidney disease patients. Nephrology 2018, 23, 669–675. [Google Scholar] [CrossRef]
- Wang, M.J.; Cai, W.J.; Li, N.; Ding, Y.J.; Chen, Y.; Zhu, Y.C. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid. Redox Signal. 2010, 12, 1065–1077. [Google Scholar] [CrossRef]
- Altaany, Z.; Yang, G.; Wang, R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J. Cell Mol. Med. 2013, 17, 879–888. [Google Scholar] [CrossRef]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Modis, K.; Panopoulos, P.; Asimakopoulou, A.; Gero, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Bir, S.C.; Yuan, S.; Shen, X.; Pardue, S.; Wang, R.; Kevil, C.G. Cystathionine gamma-lyase regulates arteriogenesis through NO-dependent monocyte recruitment. Cardiovasc. Res. 2015, 107, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Kiesworo, K.; MacArthur, M.R.; Kip, P.; Agius, T.; Macabrey, D.; Lambelet, M.; Hamard, L.; Ozaki, C.K.; Mitchell, J.R.; Deglise, S.; et al. Cystathionine-gamma-lyase overexpression modulates oxidized nicotinamide adenine dinucleotide biosynthesis and enhances neovascularization. JVS Vasc. Sci. 2023, 4, 100095. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Singh, M.; George, A.K.; Behera, J.; Tyagi, N.; Tyagi, S.C. Hydrogen sulfide improves postischemic neoangiogenesis in the hind limb of cystathionine-beta-synthase mutant mice via PPAR-gamma/VEGF axis. Physiol. Rep. 2018, 6, e13858. [Google Scholar] [CrossRef]
- Xiong, Y.; Chang, L.L.; Tran, B.; Dai, T.; Zhong, R.; Mao, Y.C.; Zhu, Y.Z. ZYZ-803, a novel hydrogen sulfide-nitric oxide conjugated donor, promotes angiogenesis via cross-talk between STAT3 and CaMKII. Acta Pharmacol. Sin. 2020, 41, 218–228. [Google Scholar] [CrossRef]
- Fu, J.; Zou, J.; Chen, C.; Li, H.; Wang, L.; Zhou, Y. Hydrogen molecules (H2) improve perfusion recovery via antioxidant effects in experimental peripheral arterial disease. Mol. Med. Rep. 2018, 18, 5009–5015. [Google Scholar] [CrossRef]
- Cheng, Z.; Garikipati, V.N.; Nickoloff, E.; Wang, C.; Polhemus, D.J.; Zhou, J.; Benedict, C.; Khan, M.; Verma, S.K.; Rabinowitz, J.E.; et al. Restoration of Hydrogen Sulfide Production in Diabetic Mice Improves Reparative Function of Bone Marrow Cells. Circulation 2016, 134, 1467–1483. [Google Scholar] [CrossRef]
- Hayashida, R.; Kondo, K.; Morita, S.; Unno, K.; Shintani, S.; Shimizu, Y.; Calvert, J.W.; Shibata, R.; Murohara, T. Diallyl Trisulfide Augments Ischemia-Induced Angiogenesis via an Endothelial Nitric Oxide Synthase-Dependent Mechanism. Circ. J. 2017, 81, 870–878. [Google Scholar] [CrossRef]
- Pardue, S.; Kolluru, G.K.; Shen, X.; Lewis, S.E.; Saffle, C.B.; Kelley, E.E.; Kevil, C.G. Hydrogen sulfide stimulates xanthine oxidoreductase conversion to nitrite reductase and formation of NO. Redox Biol. 2020, 34, 101447. [Google Scholar] [CrossRef]
- Yang, H.B.; Liu, H.M.; Yan, J.C.; Lu, Z.Y. Effect of Diallyl Trisulfide on Ischemic Tissue Injury and Revascularization in a Diabetic Mouse Model. J. Cardiovasc. Pharmacol. 2018, 71, 367–374. [Google Scholar] [CrossRef]
- Syu, J.N.; Yang, M.D.; Tsai, S.Y.; Chiang, E.I.; Chiu, S.C.; Chao, C.Y.; Rodriguez, R.L.; Tang, F.Y. S-allylcysteine Improves Blood Flow Recovery and Prevents Ischemic Injury by Augmenting Neovasculogenesis. Cell Transplant. 2017, 26, 1636–1647. [Google Scholar] [CrossRef] [PubMed]
- Kan, J.; Guo, W.; Huang, C.; Bao, G.; Zhu, Y.; Zhu, Y.Z. S-propargyl-cysteine, a novel water-soluble modulator of endogenous hydrogen sulfide, promotes angiogenesis through activation of signal transducer and activator of transcription 3. Antioxid. Redox Signal. 2014, 20, 2303–2316. [Google Scholar] [CrossRef]
- Rushing, A.M.; Donnarumma, E.; Polhemus, D.J.; Au, K.R.; Victoria, S.E.; Schumacher, J.D.; Li, Z.; Jenkins, J.S.; Lefer, D.J.; Goodchild, T.T. Effects of a novel hydrogen sulfide prodrug in a porcine model of acute limb ischemia. J. Vasc. Surg. 2019, 69, 1924–1935. [Google Scholar] [CrossRef]
- Macabrey, D.; Joniova, J.; Gasser, Q.; Bechelli, C.; Longchamp, A.; Urfer, S.; Lambelet, M.; Fu, C.Y.; Schwarz, G.; Wagnieres, G.; et al. Sodium thiosulfate, a source of hydrogen sulfide, stimulates endothelial cell proliferation and neovascularization. Front. Cardiovasc. Med. 2022, 9, 965965. [Google Scholar] [CrossRef]
- Sen, U.; Sathnur, P.B.; Kundu, S.; Givvimani, S.; Coley, D.M.; Mishra, P.K.; Qipshidze, N.; Tyagi, N.; Metreveli, N.; Tyagi, S.C. Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am. J. Physiol. Cell Physiol. 2012, 303, C41–C51. [Google Scholar] [CrossRef] [PubMed]
- Qipshidze, N.; Metreveli, N.; Mishra, P.K.; Lominadze, D.; Tyagi, S.C. Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. Int. J. Biol. Sci. 2012, 8, 430–441. [Google Scholar] [CrossRef]
- Tao, B.B.; Liu, S.Y.; Zhang, C.C.; Fu, W.; Cai, W.J.; Wang, Y.; Shen, Q.; Wang, M.J.; Chen, Y.; Zhang, L.J.; et al. VEGFR2 functions as an H2S-targeting receptor protein kinase with its novel Cys1045-Cys1024 disulfide bond serving as a specific molecular switch for hydrogen sulfide actions in vascular endothelial cells. Antioxid. Redox Signal. 2013, 19, 448–464. [Google Scholar] [CrossRef]
- Katsouda, A.; Bibli, S.I.; Pyriochou, A.; Szabo, C.; Papapetropoulos, A. Regulation and role of endogenously produced hydrogen sulfide in angiogenesis. Pharmacol. Res. 2016, 113, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zhu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40. [Google Scholar] [CrossRef]
- Jang, H.; Oh, M.Y.; Kim, Y.J.; Choi, I.Y.; Yang, H.S.; Ryu, W.S.; Lee, S.H.; Yoon, B.W. Hydrogen sulfide treatment induces angiogenesis after cerebral ischemia. J. Neurosci. Res. 2014, 92, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: Mechanisms and implications. Am. J. Physiol. Cell Physiol. 2017, 312, C3–C15. [Google Scholar] [CrossRef] [PubMed]
- Zecchin, A.; Kalucka, J.; Dubois, C.; Carmeliet, P. How Endothelial Cells Adapt Their Metabolism to Form Vessels in Tumors. Front. Immunol. 2017, 8, 1750. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [PubMed]
- Macabrey, D.; Longchamp, A.; MacArthur, M.R.; Lambelet, M.; Urfer, S.; Deglise, S.; Allagnat, F. Sodium thiosulfate acts as a hydrogen sulfide mimetic to prevent intimal hyperplasia via inhibition of tubulin polymerisation. EBioMedicine 2022, 78, 103954. [Google Scholar] [CrossRef]
- Yang, G.; Li, H.; Tang, G.; Wu, L.; Zhao, K.; Cao, Q.; Xu, C.; Wang, R. Increased neointimal formation in cystathionine gamma-lyase deficient mice: Role of hydrogen sulfide in alpha5beta1-integrin and matrix metalloproteinase-2 expression in smooth muscle cells. J. Mol. Cell Cardiol. 2012, 52, 677–688. [Google Scholar] [CrossRef]
- Trocha, K.M.; Kip, P.; Tao, M.; MacArthur, M.R.; Trevino-Villarreal, J.H.; Longchamp, A.; Toussaint, W.; Lambrecht, B.N.; de Vries, M.R.; Quax, P.H.A.; et al. Short-term preoperative protein restriction attenuates vein graft disease via induction of cystathionine gamma-lyase. Cardiovasc. Res. 2020, 116, 416–428. [Google Scholar] [CrossRef]
- Meng, Q.H.; Yang, G.; Yang, W.; Jiang, B.; Wu, L.; Wang, R. Protective effect of hydrogen sulfide on balloon injury-induced neointima hyperplasia in rat carotid arteries. Am. J. Pathol. 2007, 170, 1406–1414. [Google Scholar] [CrossRef]
- Ma, B.; Liang, G.; Zhang, F.; Chen, Y.; Zhang, H. Effect of hydrogen sulfide on restenosis of peripheral arteries after angioplasty. Mol. Med. Rep. 2012, 5, 1497–1502. [Google Scholar] [CrossRef]
- Macabrey, D.; Deslarzes-Dubuis, C.; Longchamp, A.; Lambelet, M.; Ozaki, C.K.; Corpataux, J.M.; Allagnat, F.; Deglise, S. Hydrogen Sulphide Release via the Angiotensin Converting Enzyme Inhibitor Zofenopril Prevents Intimal Hyperplasia in Human Vein Segments and in a Mouse Model of Carotid Artery Stenosis. Eur. J. Vasc. Endovasc. Surg. 2022, 63, 336–346. [Google Scholar] [CrossRef]
- Longchamp, A.; Kaur, K.; Macabrey, D.; Dubuis, C.; Corpataux, J.M.; Deglise, S.; Matson, J.B.; Allagnat, F. Hydrogen sulfide-releasing peptide hydrogel limits the development of intimal hyperplasia in human vein segments. Acta Biomater. 2019, 97, 374–384. [Google Scholar] [CrossRef]
- Kip, P.; Tao, M.; Trocha, K.M.; MacArthur, M.R.; Peters, H.A.B.; Mitchell, S.J.; Mann, C.G.; Sluiter, T.J.; Jung, J.; Patterson, S.; et al. Periprocedural Hydrogen Sulfide Therapy Improves Vascular Remodeling and Attenuates Vein Graft Disease. J. Am. Heart Assoc. 2020, 9, e016391. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Wang, R. Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 2006, 20, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Bryan, S.; Khaper, N.; Mani, S.; Wang, R. Cystathionine gamma-lyase deficiency and overproliferation of smooth muscle cells. Cardiovasc. Res. 2010, 86, 487–495. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Liang, X.; Wu, J.; Dong, S.; Li, H.; Jin, M.; Sun, D.; Zhang, W.; Zhong, X. Inhibition of hydrogen sulfide on the proliferation of vascular smooth muscle cells involved in the modulation of calcium sensing receptor in high homocysteine. Exp. Cell Res. 2016, 347, 184–191. [Google Scholar] [CrossRef]
- Zhong, X.; Wang, Y.; Wu, J.; Sun, A.; Yang, F.; Zheng, D.; Li, T.; Dong, S.; Zhao, Y.; Yang, G.; et al. Calcium sensing receptor regulating smooth muscle cells proliferation through initiating cystathionine-gamma-lyase/hydrogen sulfide pathway in diabetic rat. Cell Physiol. Biochem. 2015, 35, 1582–1598. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Werlin, E.C.; Chen, M.; Mottola, G.; Chatterjee, A.; Lance, K.D.; Bernards, D.A.; Sansbury, B.E.; Spite, M.; Desai, T.A.; et al. Perivascular delivery of resolvin D1 inhibits neointimal hyperplasia in a rabbit vein graft model. J. Vasc. Surg. 2018, 68, 188S–200S.e4. [Google Scholar] [CrossRef]
- Razavi, M.K.; Donohoe, D.; D’Agostino, R.B., Jr.; Jaff, M.R.; Adams, G.; Investigators, D. Adventitial Drug Delivery of Dexamethasone to Improve Primary Patency in the Treatment of Superficial Femoral and Popliteal Artery Disease: 12-Month Results From the DANCE Clinical Trial. JACC Cardiovasc. Interv. 2018, 11, 921–931. [Google Scholar] [CrossRef]
- Ling, K.; Xu, A.; Chen, Y.; Chen, X.; Li, Y.; Wang, W. Protective effect of a hydrogen sulfide donor on balloon injury-induced restenosis via the Nrf2/HIF-1alpha signaling pathway. Int. J. Mol. Med. 2019, 43, 1299–1310. [Google Scholar] [CrossRef]
- Borghi, C.; Bacchelli, S.; Esposti, D.D.; Bignamini, A.; Magnani, B.; Ambrosioni, E. Effects of the administration of an angiotensin-converting enzyme inhibitor during the acute phase of myocardial infarction in patients with arterial hypertension. SMILE Study Investigators. Survival of Myocardial Infarction Long-term Evaluation. Am. J. Hypertens. 1999, 12, 665–672. [Google Scholar] [CrossRef]
- Borghi, C.; Omboni, S.; Novo, S.; Vinereanu, D.; Ambrosio, G.; Ambrosioni, E. Efficacy and Safety of Zofenopril Versus Ramipril in the Treatment of Myocardial Infarction and Heart Failure: A Review of the Published and Unpublished Data of the Randomized Double-Blind SMILE-4 Study. Adv. Ther. 2018, 35, 604–618. [Google Scholar] [CrossRef]
- Ambrosioni, E.; Borghi, C.; Magnani, B. The effect of the angiotensin-converting-enzyme inhibitor zofenopril on mortality and morbidity after anterior myocardial infarction. The Survival of Myocardial Infarction Long-Term Evaluation (SMILE) Study Investigators. N. Engl. J. Med. 1995, 332, 80–85. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bechelli, C.; Macabrey, D.; Deglise, S.; Allagnat, F. Clinical Potential of Hydrogen Sulfide in Peripheral Arterial Disease. Int. J. Mol. Sci. 2023, 24, 9955. https://doi.org/10.3390/ijms24129955
Bechelli C, Macabrey D, Deglise S, Allagnat F. Clinical Potential of Hydrogen Sulfide in Peripheral Arterial Disease. International Journal of Molecular Sciences. 2023; 24(12):9955. https://doi.org/10.3390/ijms24129955
Chicago/Turabian StyleBechelli, Clémence, Diane Macabrey, Sebastien Deglise, and Florent Allagnat. 2023. "Clinical Potential of Hydrogen Sulfide in Peripheral Arterial Disease" International Journal of Molecular Sciences 24, no. 12: 9955. https://doi.org/10.3390/ijms24129955
APA StyleBechelli, C., Macabrey, D., Deglise, S., & Allagnat, F. (2023). Clinical Potential of Hydrogen Sulfide in Peripheral Arterial Disease. International Journal of Molecular Sciences, 24(12), 9955. https://doi.org/10.3390/ijms24129955