
Identification of N-Acyl Hydrazones as New Non-Zinc-Binding MMP-13 Inhibitors by Structure-Based Virtual Screening Studies and Chemical Optimization

 , ,
, ,  ,
,  ,
,  , ,
, ,  ,
, 
Abstract

1. Introduction
2. Results and Discussion
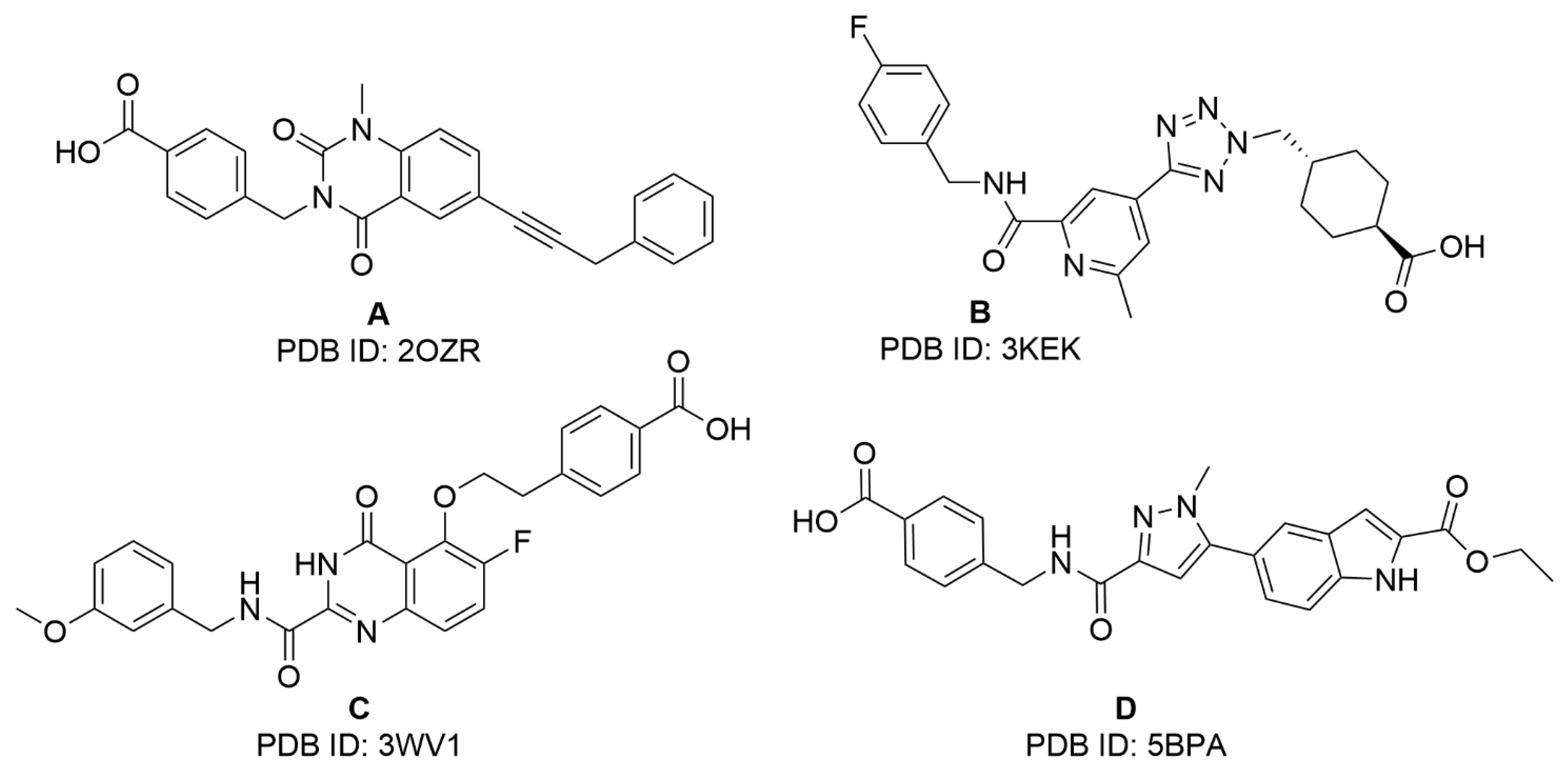
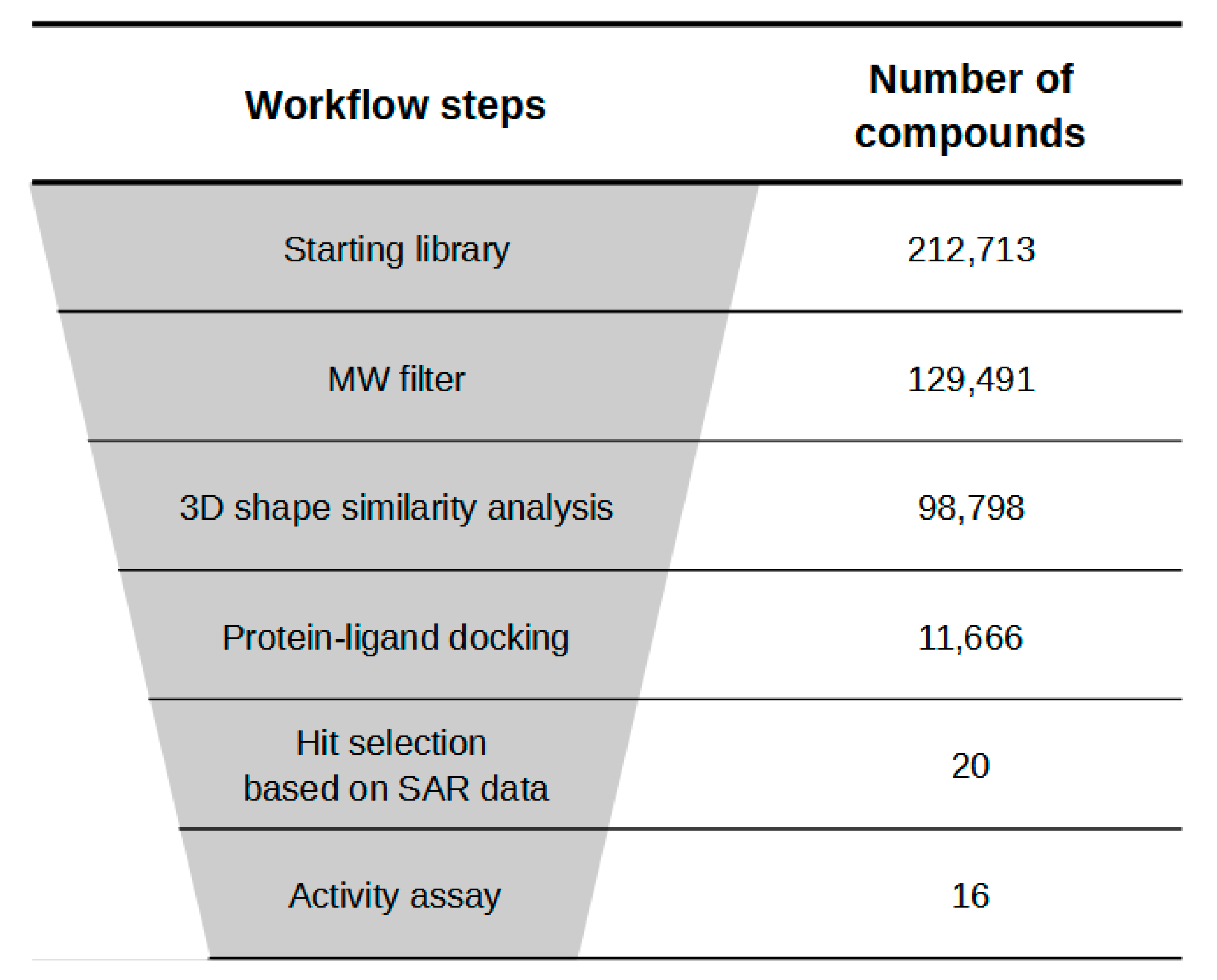
2.1. Virtual Screening Studies
2.2. Enzymatic Assays
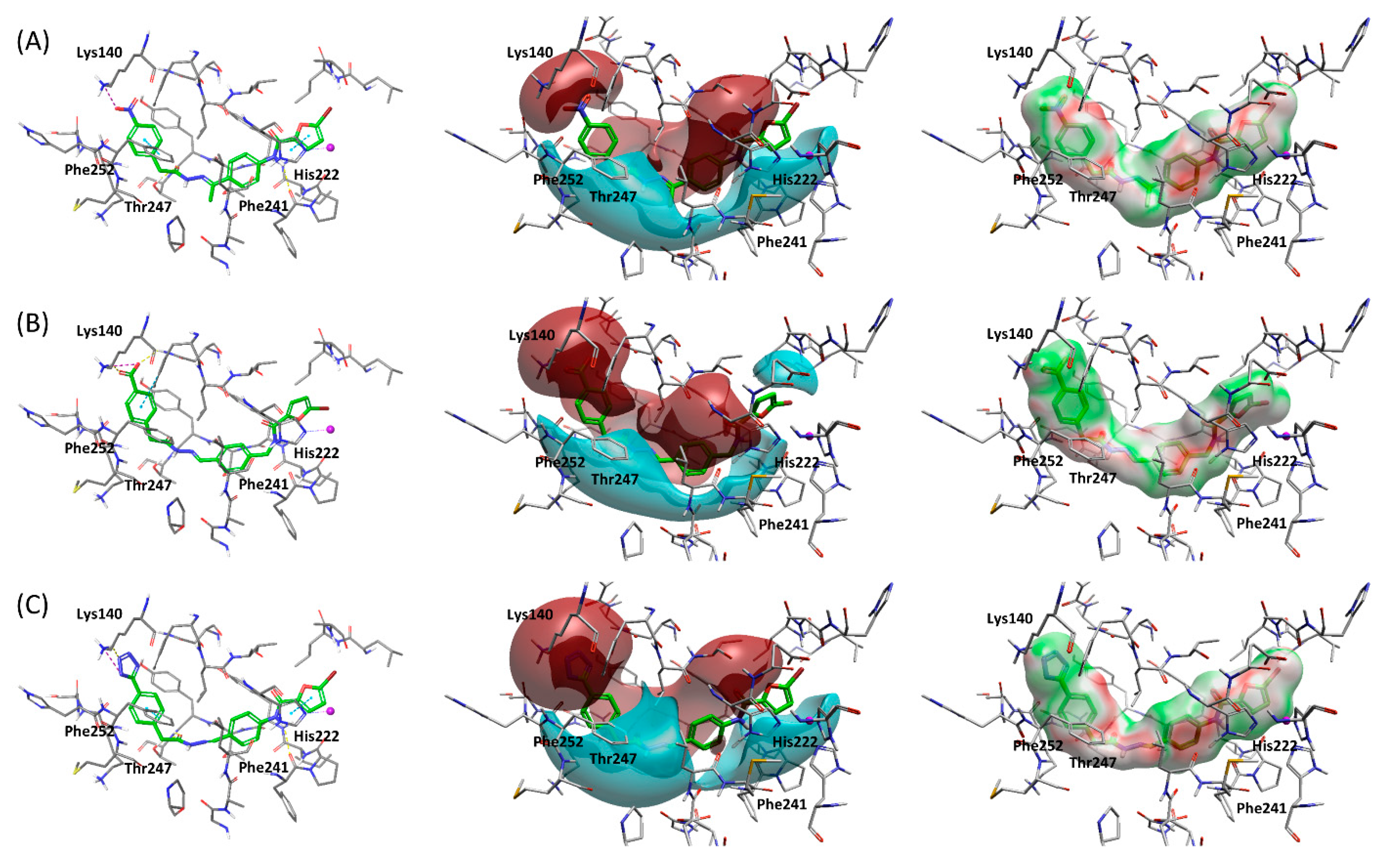
2.3. Structure-Based Hit Optimization
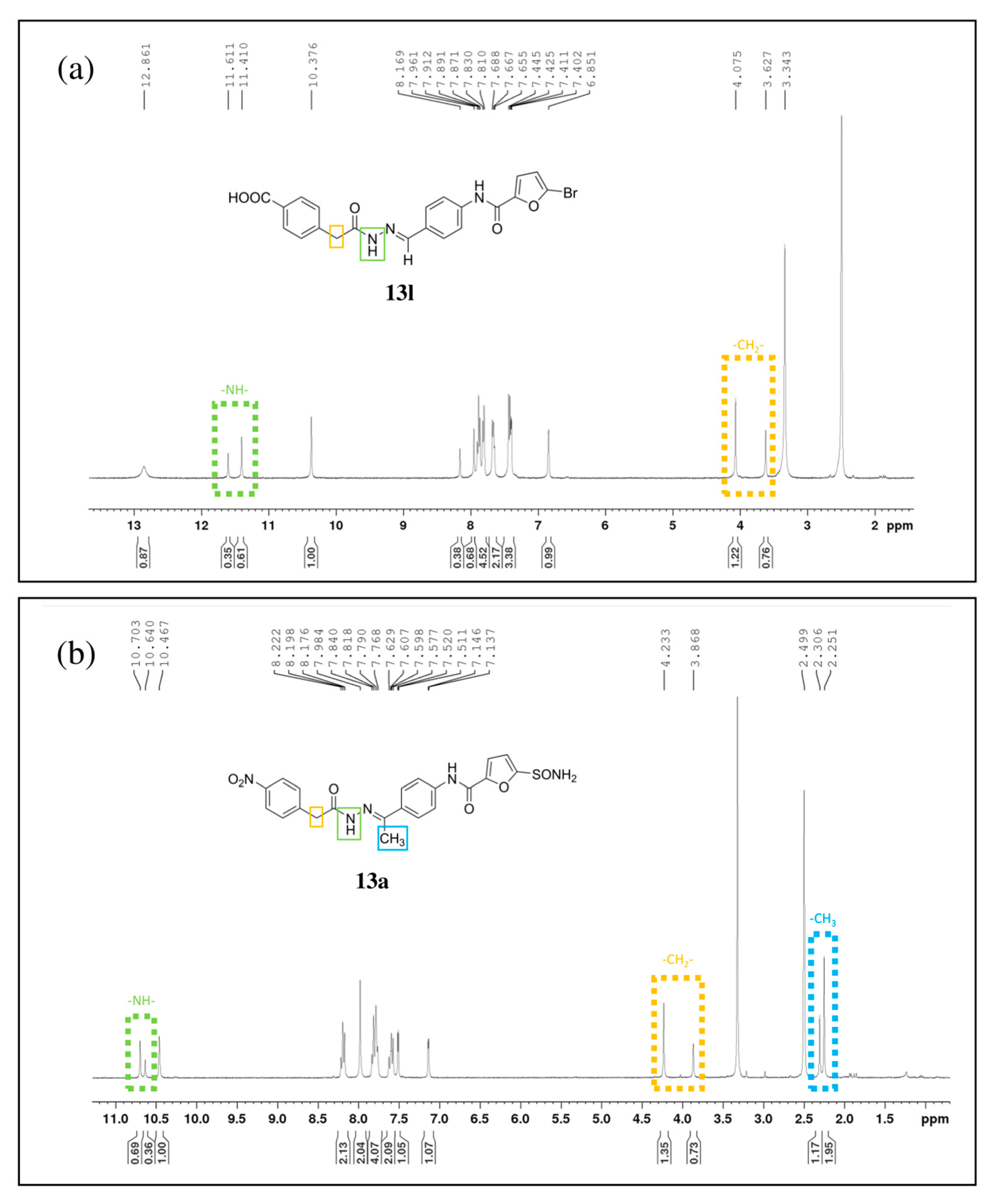
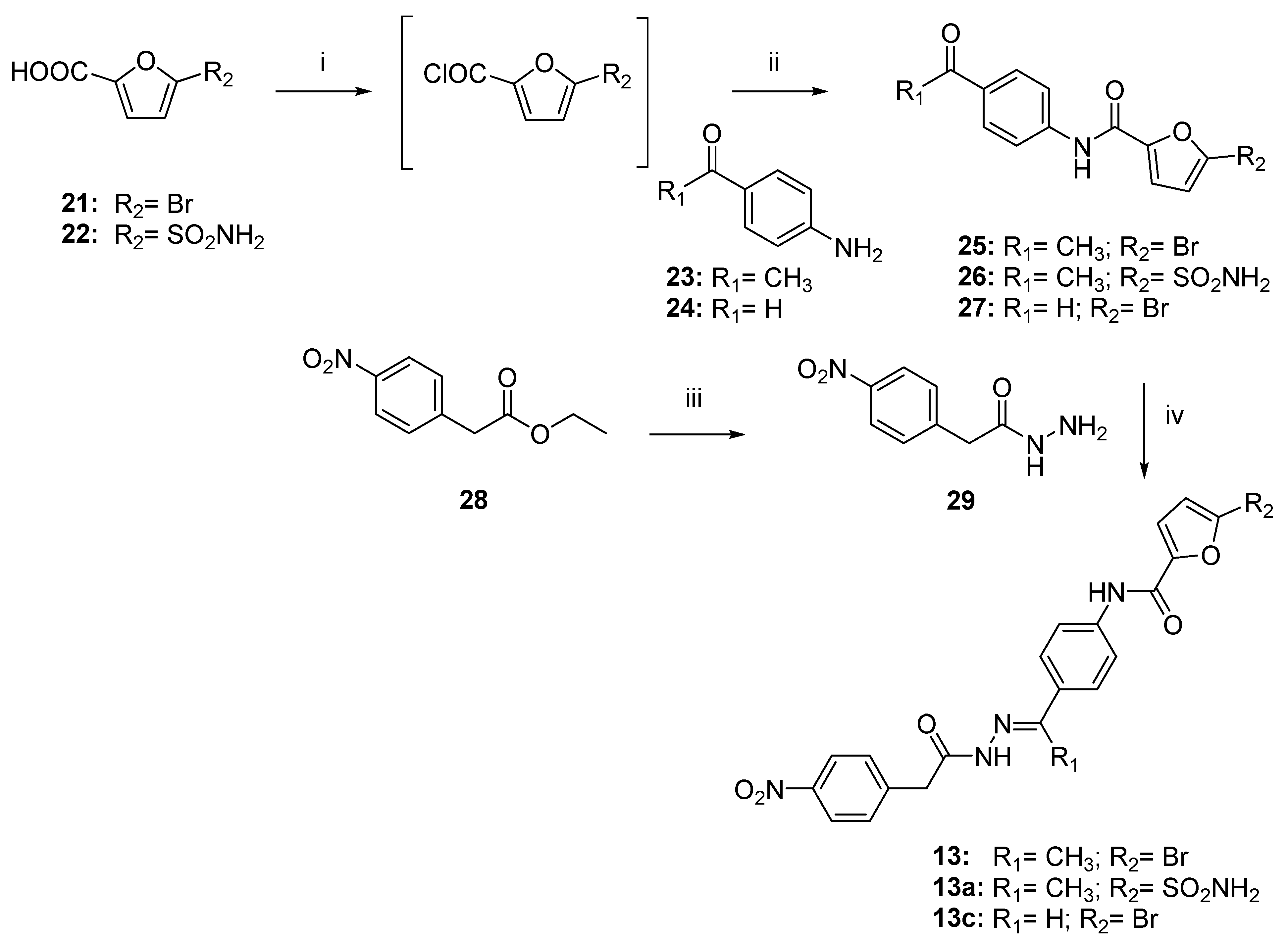
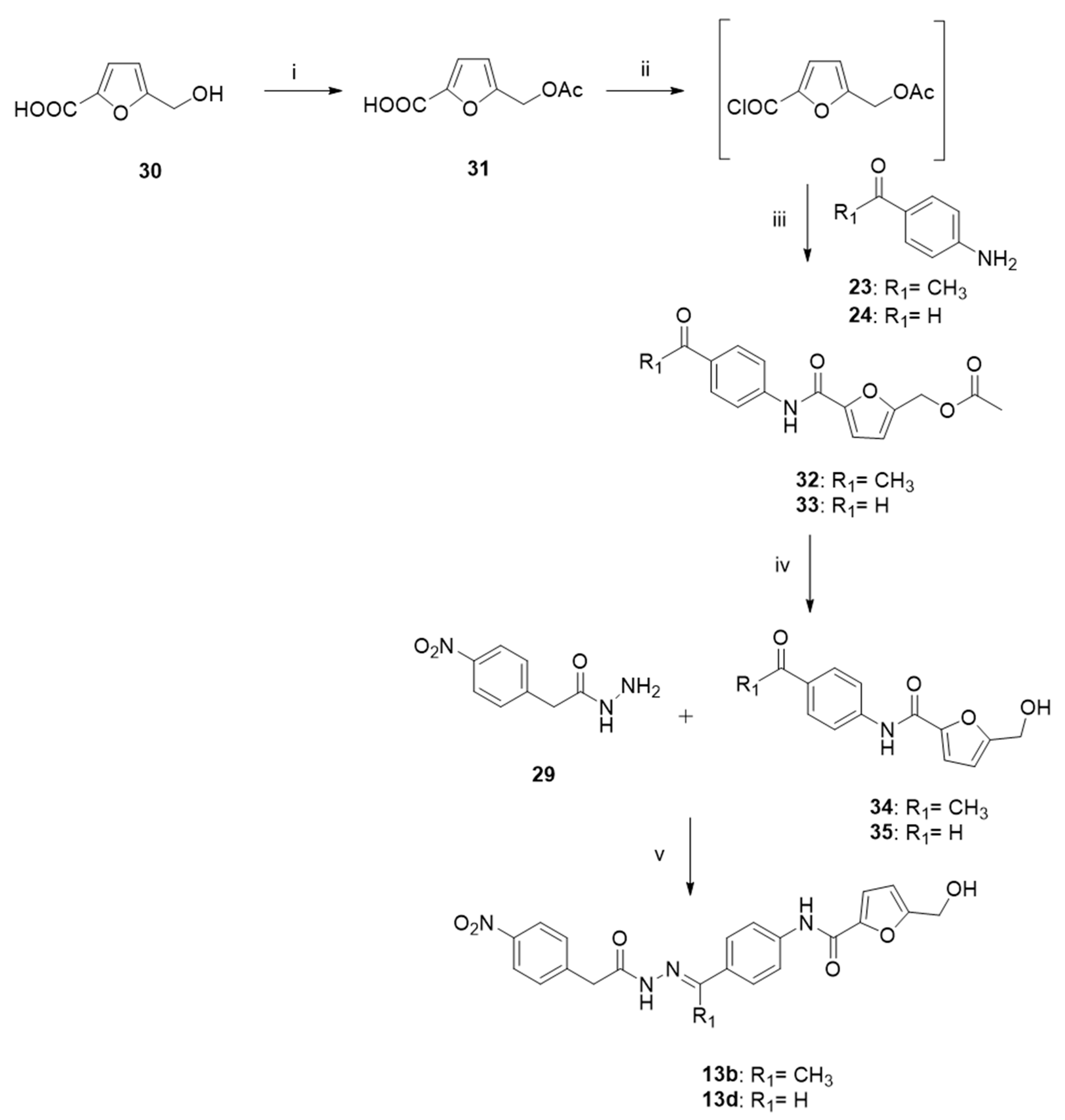
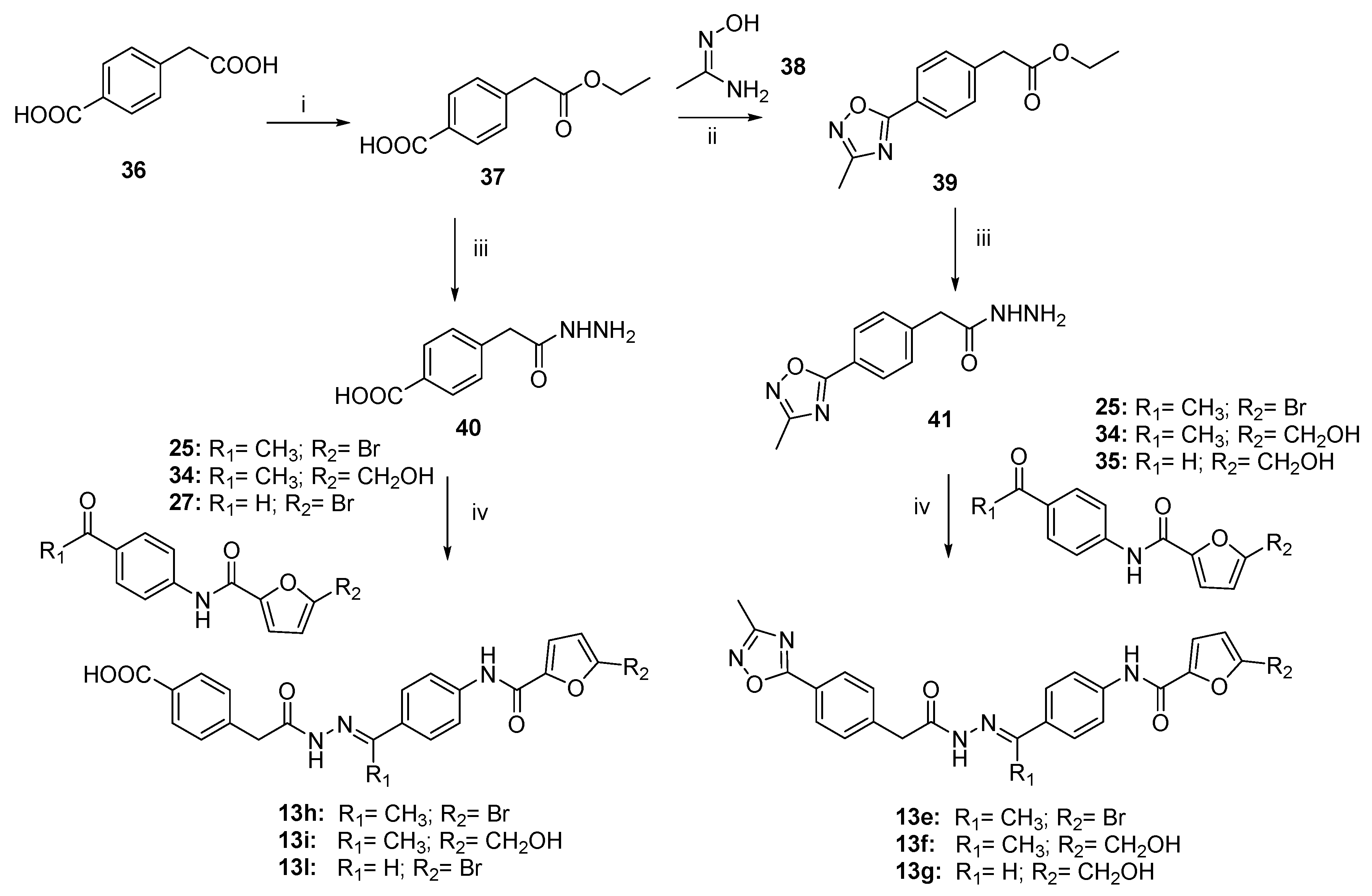
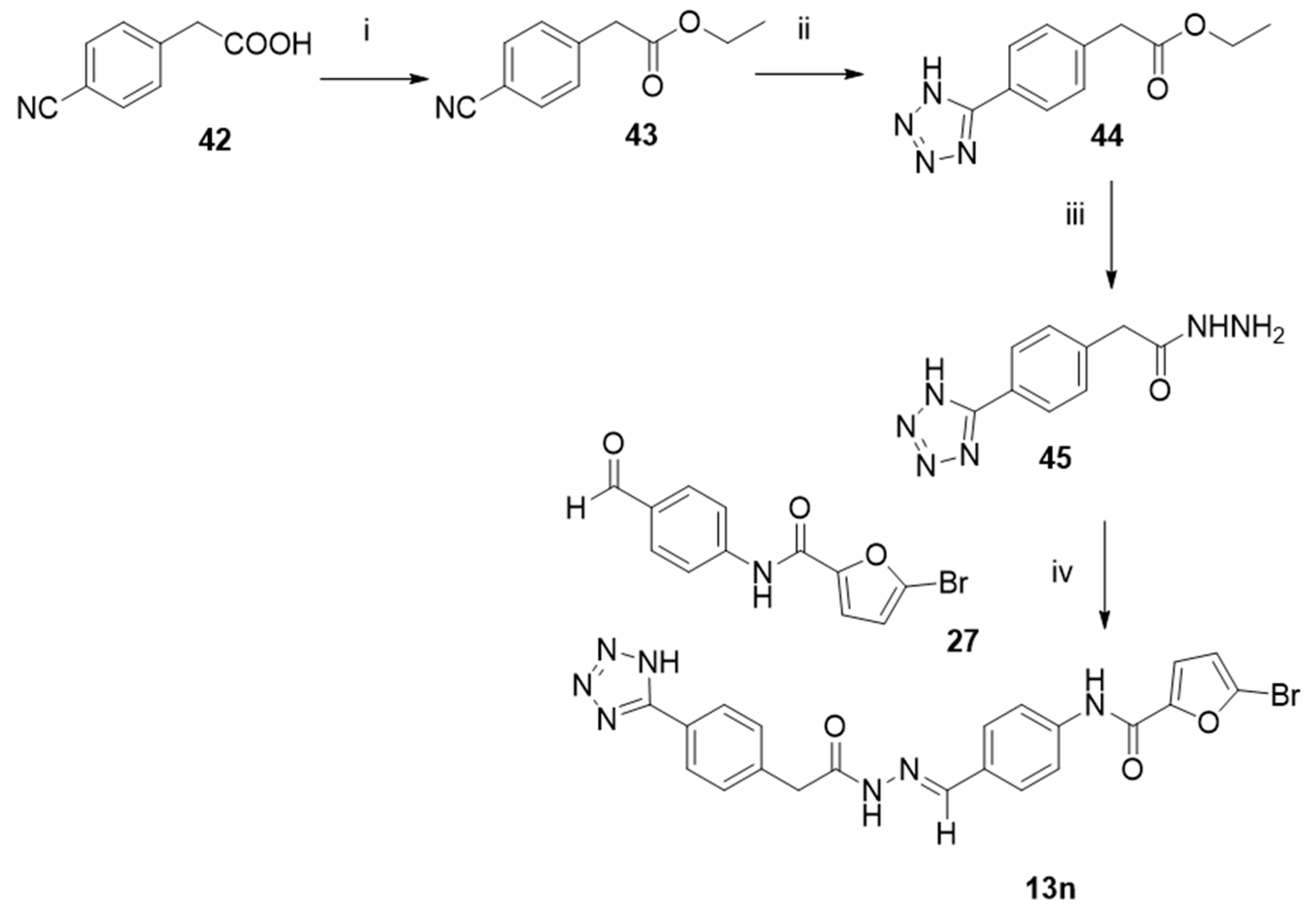
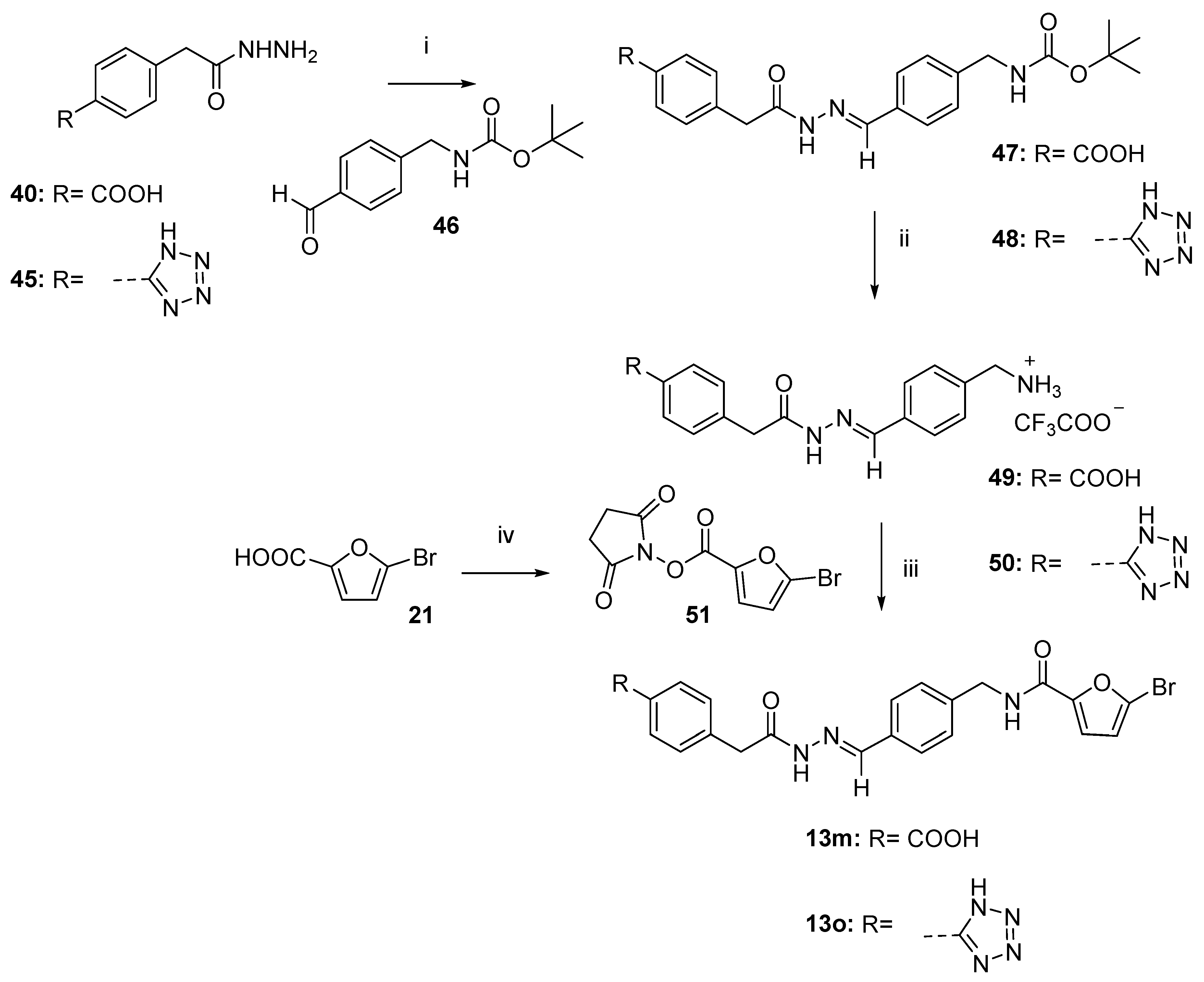
2.4. Chemistry
2.5. SAR Analysis
3. Materials and Methods
3.1. Chemistry
- 5-(acetoxymethyl)furan-2-carboxylic acid (31).
3.1.1. General Procedure to Synthesize Conjugates 25–27 and 32–33
- N-(4-acetylphenyl)-5-bromofuran-2-carboxamide (25).
- N-(4-acetylphenyl)-5-sulfamoylfuran-2-carboxamide (26).
- 5-bromo-N-(4-formylphenyl) furan-2-carboxamide (27).
- (5-((4-acetylphenyl)carbamoyl)furan-2-yl)methyl acetate (32).
- (5-((4-formylphenyl)carbamoyl)furan-2-yl)methyl acetate (33).
- N-(4-acetylphenyl)-5-(hydroxymethyl)furan-2-carboxamide (34).
- N-(4-formylphenyl)-5-(hydroxymethyl)furan-2-carboxamide (35).
- 4-(2-ethoxy-2-oxoethyl)benzoic acid (37).
- (E, Z)-N’-hydroxyacetimidamide (38).
- Ethyl 2-(4-(3-methyl-1,2,4-oxadiazol-5-yl)phenyl)acetate (39).
- Ethyl 2-(4-cyanophenyl)acetate (43).
- Ethyl 2-(4-(1H-tetrazol-5-yl)phenyl)acetate (44).
3.1.2. General Procedure to Synthesize Hydrazides 29, 40–41, and 45
- 2-(4-nitrophenyl) aceto-hydrazide (29).
- 4-(2-hydrazineyl-2-oxoethyl)benzoic acid (40).
- 2-(4-(3-methyl-1,2,4-oxadiazol-5-yl)phenyl)acetohydrazide (41).
- 5-(4-(2-(Hydrazineyloxy)-2-oxoethyl)phenyl)-1H-tetrazole (45).
3.1.3. General Procedure to Synthesize N-Acyl Hydrazones 13, 13a–l, 13n, and 47–48
- (E)-5-bromo-N-(4-(1-(2-(2-(4-nitrophenyl)acetyl)hydrazineylidene)ethyl)phenyl)furan-2-carboxamide (13).
- (E)-N-(4-(1-(2-(2-(4-nitrophenyl)acetyl)hydrazineylidene)ethyl)benzyl)-5-sulfamoylfuran-2-carboxamide (13a).
- (E)-5-(hydroxymethyl)-N-(4-(1-(2-(2-(4nitrophenyl)acetyl)hydrazineylidene)ethyl) phenyl) furan-2-carboxamide (13b).
- (E)-5-bromo-N-(4-((2-(2-(4-nitrophenyl)acetyl)hydrazineylidene)methyl)phenyl)furan-2-carboxamide (13c).
- (E)-5-(hydroxymethyl)-N-(4-((2-(2-(4-nitrophenyl)acetyl)hydrazineylidene)methyl)phenyl) furan-2-carboxamide (13d).
- (E)-5-bromo-N-(4-(1-(2-(2-(4-(3-methyl-1,2,4-oxadiazol-5-yl)phenyl)acetyl)hydrazineylidene)ethyl)phenyl)furan-2-carboxamide (13e).
- (E)-5-(hydroxymethyl)-N-(4-(1-(2-(2-(4-(3-methyl-1,2,4-oxadiazol-5-yl)phenyl)acetyl)hydrazineylidene)ethyl)phenyl)furan-2-carboxamide (13f).
- (E)-5-(hydroxymethyl)-N-(4-((2-(2-(4-(3-methyl-1,2,4-oxadiazol-5-yl)phenyl)acetyl) hydrazono)methyl)phenyl)furan-2-carboxamide (13g).
- (E)-4-(2-(2-(1-(4-(5-bromofuran-2-carboxamido)phenyl)ethylidene)hydrazineyl)-2-oxoethyl)benzoic acid (13h).
- (E)-4-(2-(2-(1-(4-(5-(hydroxymethyl)furan-2-carboxamido)phenyl)ethylidene)hydrazineyl)-2-oxoethyl)benzoic acid (13i).
- (E)-4-(2-(2-(4-(5-bromofuran-2-carboxamido)benzylidene)hydrazineyl)-2-oxoethyl)benzoic acid (13l).
- (E)-N-(4-((2-(2-(4-(1H-tetrazol-5-yl)phenyl)acetyl)hydrazineylidene)methyl)phenyl)-5-bromofuran-2-carboxamide (13n).
- (E)-4-(2-(2-(4-(((tert-butoxycarbonyl)amino)methyl)benzylidene)hydrazineyl)-2-oxoethyl)benzoic acid (47).
- (tert-butyl (E)-(4-((2-(2-(4-(1H-tetrazol-5 yl)phenyl)acetyl)hydrazineylidene)methyl)benzyl) carbamate (48).
- (E)-(4-((2-(2-(4-carboxyphenyl)acetyl)hydrazineylidene)methyl)phenyl)methanaminium 2,2,2-trifluoroacetate (49).
- (E)-(4-((2-(2-(4-(1H-tetrazol-5-yl)phenyl)acetyl)hydrazono)methyl)phenyl) methanaminium 2,2,2-trifluoroacetate (50).
- 2,5-dioxopyrrolidin-1-yl 5-bromofuran-2-carboxylate (51).
3.1.4. General Procedure to Synthesize Final Compounds 13m,o
- (E)-4-(2-(2-(4-((5-bromofuran-2-carboxamido)methyl)benzylidene)hydrazineyl)-2-oxoethyl)benzoic acid (13m).
- (E)-N-(4-((2-(2-(4-(1H-tetrazol-5-yl)phenyl)acetyl)hydrazineylidene)methyl)benzyl)-5-bromofuran-2-carboxamide (13o).
3.2. Biological Assays
3.2.1. Enzyme Activation
3.2.2. Enzyme Inhibition Assays
3.3. Computational Methods
3.3.1. Shape-Based Similarity
3.3.2. Ligand Setup for Docking
3.3.3. Protein Preparation
3.3.4. Grid Preparation
3.3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arabelovic, S.; McAlindon, T.E. Considerations in the Treatment of Early Osteoarthritis. Curr. Rheumatol. Rep. 2005, 7, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, D.; Yuan, Y.; Min, J. New Insights on the MMP-13 Regulatory Network in the Pathogenesis of Early Osteoarthritis. Arthritis Res. Ther. 2017, 19, 248. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, H.; Kimura, T.; Dalal, S.; Okada, Y.; D’Armiento, J. Joint Diseases and Matrix Metalloproteinases: A Role for MMP-13. Curr. Pharm. Biotechnol. 2008, 9, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Troeberg, L.; Nagase, H. Proteases Involved in Cartilage Matrix Degradation in Osteoarthritis. Biochim. Biophys. Acta 2012, 1824, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Ågren, M.S.; Auf dem Keller, U. Matrix Metalloproteinases: How Much Can They Do? Int. J. Mol. Sci. 2020, 21, 2678. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Libert, C. Is There New Hope for Therapeutic Matrix Metalloproteinase Inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Cathcart, J.M.; Cao, J. MMP Inhibitors: Past, Present and Future. Front. Biosci. 2015, 20, 1164–1178. [Google Scholar] [CrossRef]
- Xie, X.-W.; Wan, R.-Z.; Liu, Z.-P. Recent Research Advances in Selective Matrix Metalloproteinase-13 Inhibitors as Anti-Osteoarthritis Agents. ChemMedChem 2017, 12, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Nuti, E.; Cuffaro, D.; Bernardini, E.; Camodeca, C.; Panelli, L.; Chaves, S.; Ciccone, L.; Tepshi, L.; Vera, L.; Orlandini, E.; et al. Development of Thioaryl-Based Matrix Metalloproteinase-12 Inhibitors with Alternative Zinc-Binding Groups: Synthesis, Potentiometric, NMR, and Crystallographic Studies. J. Med. Chem. 2018, 61, 4421–4435. [Google Scholar] [CrossRef]
- Nuti, E.; Rossello, A.; Cuffaro, D.; Camodeca, C.; Van Bael, J.; van der Maat, D.; Martens, E.; Fiten, P.; Pereira, R.V.S.; Ugarte-Berzal, E.; et al. Bivalent Inhibitor with Selectivity for Trimeric MMP-9 Amplifies Neutrophil Chemotaxis and Enables Functional Studies on MMP-9 Proteoforms. Cells 2020, 9, 1634. [Google Scholar] [CrossRef]
- Camodeca, C.; Cuffaro, D.; Nuti, E.; Rossello, A. ADAM Metalloproteinases as Potential Drug Targets. Curr. Med. Chem. 2019, 26, 2661–2689. [Google Scholar] [CrossRef] [PubMed]
- Pece, R.; Tavella, S.; Costa, D.; Varesano, S.; Camodeca, C.; Cuffaro, D.; Nuti, E.; Rossello, A.; Alfano, M.; D’Arrigo, C.; et al. Inhibitors of ADAM10 Reduce Hodgkin Lymphoma Cell Growth in 3D Microenvironments and Enhance Brentuximab-Vedotin Effect. Haematologica 2022, 107, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Cuffaro, D.; Ciccone, L.; Rossello, A.; Nuti, E.; Santamaria, S. Targeting Aggrecanases for Osteoarthritis Therapy: From Zinc Chelation to Exosite Inhibition. J. Med. Chem. 2022, 65, 13505–13532. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Novel Carbonic Anhydrase Inhibitors. Future Med. Chem. 2021, 13, 1935–1937. [Google Scholar] [CrossRef]
- Cuffaro, D.; Nuti, E.; Rossello, A. An Overview of Carbohydrate-Based Carbonic Anhydrase Inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 1906–1922. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Nencetti, S.; Cuffaro, D.; Nuti, E.; Ciccone, L.; Rossello, A.; Fabbi, M.; Ballante, F.; Ortore, G.; Carbotti, G.; Campelli, F.; et al. Identification of Histone Deacetylase Inhibitors with (Arylidene)Aminoxy Scaffold Active in Uveal Melanoma Cell Lines. J. Enzyme Inhib. Med. Chem. 2021, 36, 34–47. [Google Scholar] [CrossRef]
- Fabre, B.; Ramos, A.; de Pascual-Teresa, B. Targeting Matrix Metalloproteinases: Exploring the Dynamics of the S1′ Pocket in the Design of Selective, Small Molecule Inhibitors. J. Med. Chem. 2014, 57, 10205–10219. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.R.; Pavlovsky, A.G.; Ortwine, D.F.; Prior, F.; Man, C.-F.; Bornemeier, D.A.; Banotai, C.A.; Mueller, W.T.; McConnell, P.; Yan, C.; et al. Discovery and Characterization of a Novel Inhibitor of Matrix Metalloprotease-13 That Reduces Cartilage Damage in Vivo without Joint Fibroplasia Side Effects. J. Biol. Chem. 2007, 282, 27781–27791. [Google Scholar] [CrossRef]
- Schnute, M.E.; O’Brien, P.M.; Nahra, J.; Morris, M.; Howard Roark, W.; Hanau, C.E.; Ruminski, P.G.; Scholten, J.A.; Fletcher, T.R.; Hamper, B.C.; et al. Discovery of (Pyridin-4-Yl)-2H-Tetrazole as a Novel Scaffold to Identify Highly Selective Matrix Metalloproteinase-13 Inhibitors for the Treatment of Osteoarthritis. Bioorg. Med. Chem. Lett. 2010, 20, 576–580. [Google Scholar] [CrossRef]
- Nara, H.; Sato, K.; Naito, T.; Mototani, H.; Oki, H.; Yamamoto, Y.; Kuno, H.; Santou, T.; Kanzaki, N.; Terauchi, J.; et al. Discovery of Novel, Highly Potent, and Selective Quinazoline-2-Carboxamide-Based Matrix Metalloproteinase (MMP)-13 Inhibitors without a Zinc Binding Group Using a Structure-Based Design Approach. J. Med. Chem. 2014, 57, 8886–8902. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.J.; Abeywardane, A.; Liang, S.; Muegge, I.; Padyana, A.K.; Xiong, Z.; Hill-Drzewi, M.; Farmer, B.; Li, X.; Collins, B.; et al. Fragment-Based Discovery of Indole Inhibitors of Matrix Metalloproteinase-13. J. Med. Chem. 2011, 54, 8174–8187. [Google Scholar] [CrossRef]
- Specs. Available online: http://www.specs.net (accessed on 15 January 2021).
- Nara, H.; Sato, K.; Naito, T.; Mototani, H.; Oki, H.; Yamamoto, Y.; Kuno, H.; Santou, T.; Kanzaki, N.; Terauchi, J.; et al. Thieno[2,3-d]Pyrimidine-2-Carboxamides Bearing a Carboxybenzene Group at 5-Position: Highly Potent, Selective, and Orally Available MMP-13 Inhibitors Interacting with the S1″ Binding Site. Bioorg. Med. Chem. 2014, 22, 5487–5505. [Google Scholar] [CrossRef] [PubMed]
- Reaxys. Available online: https://www.reaxys.com/ (accessed on 2 February 2019).
- Rossello, A.; Nuti, E.; Orlandini, E.; Carelli, P.; Rapposelli, S.; Macchia, M.; Minutolo, F.; Carbonaro, L.; Albini, A.; Benelli, R.; et al. New N-Arylsulfonyl-N-Alkoxyaminoacetohydroxamic Acids as Selective Inhibitors of Gelatinase A (MMP-2). Bioorg. Med. Chem. 2004, 12, 2441–2450. [Google Scholar] [CrossRef] [PubMed]
- McGovern, S.L.; Helfand, B.T.; Feng, B.; Shoichet, B.K. A Specific Mechanism of Nonspecific Inhibition. J. Med. Chem. 2003, 46, 4265–4272. [Google Scholar] [CrossRef]
- FlareTM, v6.1.0; Cresset®: Litlington, UK; Available online: http://www.cresset-group.com/flare/ (accessed on 14 April 2023).
- Thota, S.; Rodrigues, D.A.; de Sena murteira Pinheiro, P.; Lima, L.M.; Fraga, C.A.M.; Barreiro, E.J. N-Acylhydrazones as Drugs. Bioorg. Med. Chem. Lett. 2018, 28, 2797–2806. [Google Scholar] [CrossRef]
- Socea, L.-I.; Barbuceanu, S.-F.; Pahontu, E.M.; Dumitru, A.-C.; Nitulescu, G.M.; Sfetea, R.C.; Apostol, T.-V. Acylhydrazones and Their Biological Activity: A Review. Molecules 2022, 27, 8719. [Google Scholar] [CrossRef]
- Fortuna, C.G.; Bonaccorso, C.; Bulbarelli, A.; Caltabiano, G.; Rizzi, L.; Goracci, L.; Musumarra, G.; Pace, A.; Palumbo Piccionello, A.; Guarcello, A.; et al. New Linezolid-like 1,2,4-Oxadiazoles Active against Gram-Positive Multiresistant Pathogens. Eur. J. Med. Chem. 2013, 65, 533–545. [Google Scholar] [CrossRef]
- Palla, G.; Predieri, G.; Domiano, P.; Vignali, C.; Turner, W. Conformational Behaviour and E/Z Isomerization of N-Acyl and N-Aroylhydrazones. Tetrahedron 1986, 42, 3649–3654. [Google Scholar] [CrossRef]
- Syakaev, V.V.; Podyachev, S.N.; Buzykin, B.I.; Latypov, S.K.; Habicher, W.D.; Konovalov, A.I. NMR Study of Conformation and Isomerization of Aryl- and Heteroarylaldehyde 4-Tert-Butylphenoxyacetylhydrazones. J. Mol. Struct. 2006, 788, 55–62. [Google Scholar] [CrossRef]
- Wyrzykiewicz, E.; Błaszczyk, A.; Turowska-Tyrk, I. N-(E)-2-Stilbenyloxymethylenecarbonyl Substituted Hydrazones of Ortho, Meta and Para Hydroxybenzaldehydes. Bull. Pol. Acad. Sci. Chem. 2000, 48, 213–229. [Google Scholar]
- Munir, R.; Javid, N.; Zia-Ur-Rehman, M.; Zaheer, M.; Huma, R.; Roohi, A.; Athar, M.M. Synthesis of Novel N-Acylhydrazones and Their C-N/N-N Bond Conformational Characterization by NMR Spectroscopy. Molecules 2021, 26, 4908. [Google Scholar] [CrossRef]
- Tian, B.; He, M.; Tang, S.; Hewlett, I.; Tan, Z.; Li, J.; Jin, Y.; Yang, M. Synthesis and Antiviral Activities of Novel Acylhydrazone Derivatives Targeting HIV-1 Capsid Protein. Bioorg. Med. Chem. Lett. 2009, 19, 2162–2167. [Google Scholar] [CrossRef]
- Kuodis, Z.; Rutavičius, A.; Matijoška, A.; Eicher-Lorka, O. Synthesis and Isomerism of Hydrazones of 2-(5-Thioxo-4,5-Dihydro-1,3,4-Thiadiazol-2-Ylthio)Acetohydrazide. Cent. Eur. J. Chem. 2007, 5, 996–1006. [Google Scholar] [CrossRef]
- Litvinov, I.A.; Kataeva, O.N.; Ermolaeva, L.V.; Vagina, G.A.; Troepol’skaya, T.V.; Naumov, V.A. Crystal and Molecular Structure of Aroyl- and Acetylhydrazones of Acet- and Benzaldehydes. Russ. Chem. Bull. 1991, 40, 62–67. [Google Scholar] [CrossRef]
- Pitasse-Santos, P.; Sueth-Santiago, V.; Lima, M.E.F. 1,2,4- and 1,3,4-Oxadiazoles as Scaffolds in the Development of Antiparasitic Agents. J. Braz. Chem. Soc. 2018, 29, 435–456. [Google Scholar] [CrossRef]
- Bredael, K.; Geurs, S.; Clarisse, D.; De Bosscher, K.; D’hooghe, M. Carboxylic Acid Bioisosteres in Medicinal Chemistry: Synthesis and Properties. J. Chem. 2022, 2022, 2164558. [Google Scholar] [CrossRef]
- Cuffaro, D.; Camodeca, C.; D’Andrea, F.; Piragine, E.; Testai, L.; Calderone, V.; Orlandini, E.; Nuti, E.; Rossello, A. Matrix Metalloproteinase-12 Inhibitors: Synthesis, Structure-Activity Relationships and Intestinal Absorption of Novel Sugar-Based Biphenylsulfonamide Carboxylates. Bioorg. Med. Chem. 2018, 26, 5804–5815. [Google Scholar] [CrossRef]
- Cuffaro, D.; Nuti, E.; Gifford, V.; Ito, N.; Camodeca, C.; Tuccinardi, T.; Nencetti, S.; Orlandini, E.; Itoh, Y.; Rossello, A. Design, Synthesis and Biological Evaluation of Bifunctional Inhibitors of Membrane Type 1 Matrix Metalloproteinase (MT1-MMP). Bioorg. Med. Chem. 2019, 27, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. OMEGA, v.3.0.1.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2018; Available online: http://www.eyesopen.com (accessed on 23 March 2021).
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- ROCS, Version 3.2.2.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2017; Available online: http://www.eyesopen.com (accessed on 6 June 2021).
- Shape Theory—Applications. Available online: https://docs.eyesopen.com/applications/rocs/theory/shape_shape.html (accessed on 23 June 2023).
- Schrödinger Release 2016-3: LigPrep; Schrödinger, LLC: New York, NY, USA, 2016.
- Schrödinger Release 2016-3: Epik; Schrödinger, LLC: New York, NY, USA, 2016.
- Schrödinger Release 2016-3: Schrödinger Suite 2016-3 Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA; Impact, Schrödinger, LLC: New York, NY, USA; Prime, Schrödinger, LLC: New York, NY, USA, 2016.
- Schrödinger Release 2016-3: Maestro; Schrödinger, LLC: New York, NY, USA, 2016.
- Schrödinger Release 2016-3: Glide; Schrödinger, LLC: New York, NY, USA, 2016.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Specs Code | Inhibition % a (100 μM) | clogP b |
|---|---|---|---|
 | AG-205/37007400 | 54.2 | 5.6 |
 | AK-968/13281769 | 48.7 | 6.6 |
 | AK-968/15605222 | 63.1 | 4.3 |
| Compound | MMP-1 | MMP-2 | MMP-9 | MMP-13 | MMP-14 |
|---|---|---|---|---|---|
| 11 | 99 ± 8.0 | 60 ± 4.6 | 66 ± 4.6 | 91 ± 8.5 | 94 ± 5.5 |
| 12 | 75 ± 4 | 47 ± 1.6 | 72 ± 4.8 | 105 ± 5.8 | 67 ± 4.6 |
| 13 | 91 ± 0.8 | 99 ± 6 | 68 ± 3 | 14.6 ± 1.6 | 63 ± 5.4 |
 | |||||
|---|---|---|---|---|---|
| Compound | R | R1 | R2 | n | IC50 (µM) a |
| 13 | NO2 | Me | Br | 0 | 14.6 ± 1.6 |
| 13a | NO2 | Me | -SO2NH2 | 0 | >100 |
| 13b | NO2 | Me | -CH2OH | 0 | >100 |
| 13c | NO2 | H | Br | 0 | 20 ± 1.2 |
| 13d | NO2 | H | -CH2OH | 0 | 40 ± 3.5 |
| 13e |  | Me | Br | 0 | >100 |
| 13f |  | Me | -CH2OH | 0 | 120 ± 7.6 |
| 13g |  | H | -CH2OH | 0 | 33 ± 1.9 |
| 13h | -COOH | Me | Br | 0 | 15 ± 3.6 |
| 13i | -COOH | Me | -CH2OH | 0 | 49 ± 3.6 |
| 13l | -COOH | H | Br | 0 | 11.4 ± 1.0 |
| 13m | -COOH | H | Br | 1 | 1.8 ± 0.5 |
| 13n |  | H | Br | 0 | 2.7 ± 0.2 |
| 13o |  | H | Br | 1 | 163 ± 8.0 |
| Compound | Accomplish Lipinski’s Rule? a | LogP a | Molecular Mass a | Hydrogen Bond Aceptors (HBA) a | Hydrogen Bond Donors (HBD) a | Rotatable Bonds a | BBB Permeability (Cross if > 0.3 and Poorly if <−1) a | CNS Permeability (Penetrate > −2 and Unable <−3) a | CaCo2 Permeability (high > 0.90) a | Intestinal Absorption (Poor < 30%) a | Skin Permeability (low > −2.5) a | HERG I Inibition a | HERG II Inibition a | Hepatotoxicity a | Cardiotoxicity b | Druglikeness (Traded Drugs > 0) c | Mutagenic c | Tumorigenic c | Reproductive Effects c | Irritant c |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 13 | Yes | 4.28550 | 485.294 | 7 | 2 | 7 | −0.648 | −2.187 | 0.630 | 88.652 | −2.745 | No | Yes | Yes | Non-cardiotoxic | −3.0971 | No | No | No | No |
| 13a | Yes | 2.17040 | 485.478 | 8 | 3 | 8 | −1.217 | −3.044 | 0.365 | 71.804 | −2.737 | No | Yes | Yes | Non-cardiotoxic | −5.8165 | No | No | No | No |
| 13b | Yes | 3.01530 | 436.424 | 7 | 3 | 8 | −0.991 | −2.647 | 0.015 | 79.570 | −2.744 | No | Yes | Yes | Non-cardiotoxic | −0.015854 | No | No | No | No |
| 13c | Yes | 3.89540 | 471.267 | 6 | 2 | 7 | −0.670 | −2.279 | 0.631 | 87.820 | −2.746 | No | Yes | Yes | Non-cardiotoxic | −2.2885 | No | No | No | No |
| 13d | Yes | 2.62520 | 422.397 | 7 | 3 | 8 | −0.969 | −2.738 | 0.019 | 78.738 | −2.744 | No | Yes | Yes | Non-cardiotoxic | 0.74757 | No | No | No | No |
| 13e | Yes | 4.73572 | 522.359 | 7 | 2 | 7 | −0.755 | −2.229 | 0.630 | 94.590 | −2.751 | No | Yes | Yes | Non-cardiotoxic | 2.4995 | No | No | No | No |
| 13f | Yes | 3.67972 | 474.497 | 6 | 4 | 8 | −1.410 | −2.724 | 0.143 | 77.682 | −2.736 | No | Yes | Yes | Non-cardiotoxic | 1.4725 | No | No | No | No |
| 13g | Yes | 3.28962 | 460.470 | 6 | 4 | 8 | −1.388 | −2.815 | 0.148 | 76.850 | −2.736 | No | Yes | Yes | Non-cardiotoxic | 2.2082 | No | No | No | No |
| 13h | Yes | 2.74080 | 483.298 | 6 | 2 | 7 | −0.548 | −2.553 | 0.694 | 63.609 | −2.732 | No | No | Yes | Non-cardiotoxic | 2.0347 | No | No | No | No |
| 13i | Yes | 1.47060 | 434.428 | 7 | 3 | 8 | −0.873 | −3.012 | 0.278 | 54.527 | −2.733 | No | No | Yes | Non-cardiotoxic | 5.1125 | No | No | No | No |
| 13l | Yes | 2.35070 | 469.271 | 6 | 2 | 7 | −0.569 | −2.644 | 0.695 | 62.777 | −2.732 | No | No | Yes | Non-cardiotoxic | 2.8355 | No | No | No | No |
| 13m | Yes | 2.02840 | 483.298 | 6 | 2 | 8 | −0.547 | −2.786 | 0.670 | 60.411 | −2.732 | No | No | Yes | Weak or moderate | 3.1821 | No | No | No | No |
| 13n | Yes | 2.79640 | 493.301 | 7 | 2 | 7 | −1.006 | −2.747 | 0.658 | 83.391 | −2.745 | No | Yes | Yes | Weak or moderate | 0.87219 | No | No | No | No |
| 13o | Yes | 2.47410 | 507.328 | 7 | 2 | 8 | −0.98 | −2.888 | 0.632 | 81.025 | −2.745 | No | Yes | Yes | Weak or moderate | 1.2118 | No | No | No | No |
| Compound | MMP-1 | MMP-2 | MMP-9 | MMP-13 | MMP-14 |
|---|---|---|---|---|---|
| 13m | 177 ± 20 | 3.6 ± 0.4 | 50 ± 3.2 | 1.8 ± 0.5 | >200 |
| 13 | 91 ± 0.8 | 99 ± 6 | 68 ± 3 | 14.6 ± 1.6 | 63 ± 5.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuffaro, D.; Gimeno, A.; Bernardoni, B.L.; Di Leo, R.; Pujadas, G.; Garcia-Vallvé, S.; Nencetti, S.; Rossello, A.; Nuti, E. Identification of N-Acyl Hydrazones as New Non-Zinc-Binding MMP-13 Inhibitors by Structure-Based Virtual Screening Studies and Chemical Optimization. Int. J. Mol. Sci. 2023, 24, 11098. https://doi.org/10.3390/ijms241311098
Cuffaro D, Gimeno A, Bernardoni BL, Di Leo R, Pujadas G, Garcia-Vallvé S, Nencetti S, Rossello A, Nuti E. Identification of N-Acyl Hydrazones as New Non-Zinc-Binding MMP-13 Inhibitors by Structure-Based Virtual Screening Studies and Chemical Optimization. International Journal of Molecular Sciences. 2023; 24(13):11098. https://doi.org/10.3390/ijms241311098
Chicago/Turabian StyleCuffaro, Doretta, Aleix Gimeno, Bianca Laura Bernardoni, Riccardo Di Leo, Gerard Pujadas, Santiago Garcia-Vallvé, Susanna Nencetti, Armando Rossello, and Elisa Nuti. 2023. "Identification of N-Acyl Hydrazones as New Non-Zinc-Binding MMP-13 Inhibitors by Structure-Based Virtual Screening Studies and Chemical Optimization" International Journal of Molecular Sciences 24, no. 13: 11098. https://doi.org/10.3390/ijms241311098
APA StyleCuffaro, D., Gimeno, A., Bernardoni, B. L., Di Leo, R., Pujadas, G., Garcia-Vallvé, S., Nencetti, S., Rossello, A., & Nuti, E. (2023). Identification of N-Acyl Hydrazones as New Non-Zinc-Binding MMP-13 Inhibitors by Structure-Based Virtual Screening Studies and Chemical Optimization. International Journal of Molecular Sciences, 24(13), 11098. https://doi.org/10.3390/ijms241311098