
Vitamin E and Its Molecular Effects in Experimental Models of Neurodegenerative Diseases

 , ,
, ,
Abstract

1. Introduction
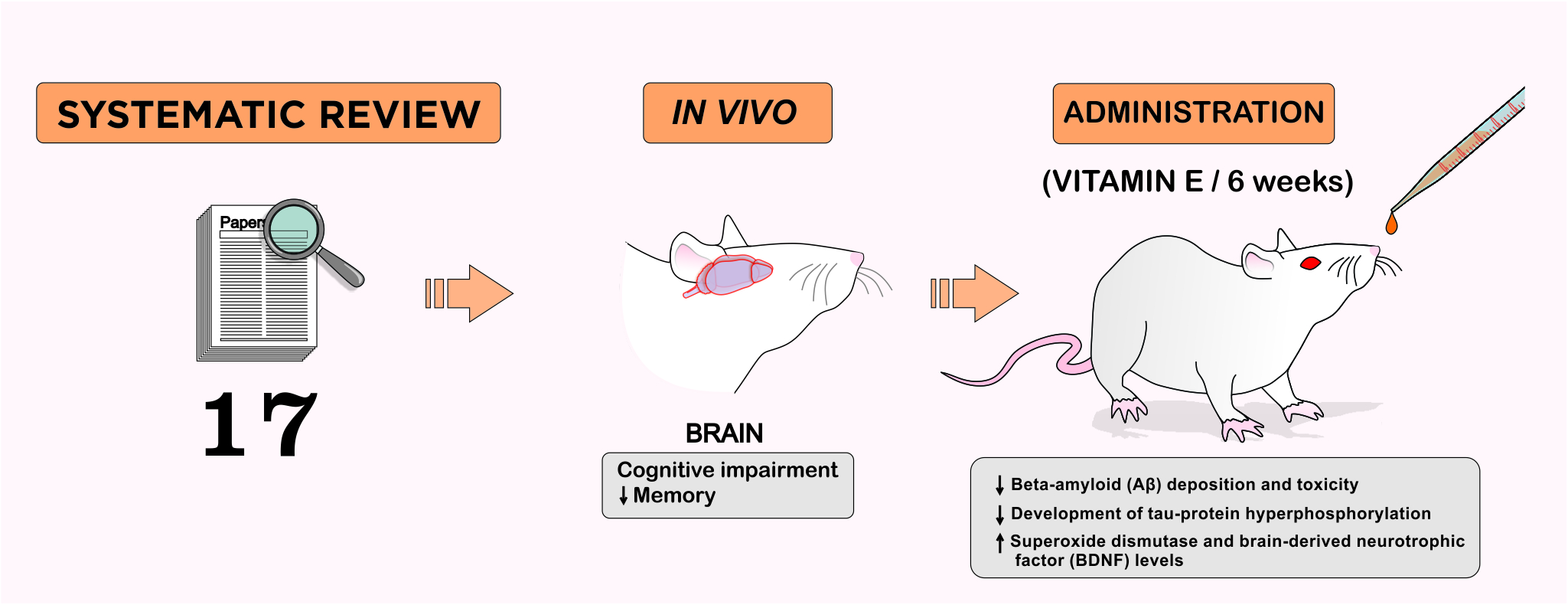
2. Methodology
3. Results
3.1. Neuroprotective Mechanisms of Vitamin E in Neurodegenerative Diseases
3.1.1. Memory and Learning
3.1.2. Cognitive
3.1.3. Motor Coordination
3.1.4. Oxidative Stress and Neurodegenerative Diseases
3.1.5. Neuroprotection and Neuroregeneration
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Heemels, M.T. Neurodegenerative diseases. Nature 2016, 539, 179. [Google Scholar] [CrossRef]
- Guo, L.; Lee, V.M.Y. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. A Unifying Role for Prions in Neurodegenerative Diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [PubMed]
- Chekani, F.; Bali, V.; Aparasu, R.R. Quality of life of patients with Parkinson’s disease and neurodegenerative dementia: A nationally representative study. Res. Soc. Adm. Pharm. 2016, 12, 604–613. [Google Scholar] [CrossRef]
- Dadhania, V.P.; Trivedi, P.P.; Vikram, A.; Tripathi, D.N. Nutraceuticals against Neurodegeneration: A Mechanistic Insight. Curr. Neuropharmacol. 2016, 14, 627–640. [Google Scholar] [CrossRef]
- Rasool, M.; Malik, A.; Qureshi, M.S.; Manan, A.; Pushparaj, P.N.; Asif, M.; Qazi, M.H.; Qazi, A.M.; Kamal, M.A.; Gan, S.H.; et al. Recent Updates in the Treatment of Neurodegenerative Disorders Using Natural Compounds. Evid. Based Complement. Alternat. Med. 2014, 2014, 979730. [Google Scholar] [CrossRef]
- Leonoudakis, D.; Rane, A.; Angeli, S.; Lithgow, G.J.; Andersen, J.K.; Chinta, S.J. Anti-Inflammatory and Neuroprotective Role of Natural Product Securinine in Activated Glial Cells: Implications for Parkinson’s Disease. Mediat. Inflamm. 2017, 2017, 8302636. [Google Scholar] [CrossRef]
- Bagli, E.; Goussia, A.; Moschos, M.M.; Agnantis, N.; Kitsos, G. Natural Compounds and Neuroprotection: Mechanisms of Action and Novel Delivery Systems. In Vivo 2016, 30, 535–547. [Google Scholar]
- Starkov, A.A.; Beal, F.M. Portal to Alzheimer’s disease. Nat. Med. 2008, 14, 1020–1021. [Google Scholar] [CrossRef]
- Venkatesan, R.; Ji, E.; Kim, S.Y. Phytochemicals That Regulate Neurodegenerative Disease by Targeting Neurotrophins: A Comprehensive Review. BioMed Res. Int. 2015, 2015, 814068. [Google Scholar] [CrossRef]
- Deshpande, P.; Gogia, N.; Singh, A. Exploring the efficacy of natural products in alleviating Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1321. [Google Scholar] [CrossRef]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Ulatowski, L.; Ghelfi, M.; West, R.; Atkinson, J.; Finno, C.J.; Manor, D. The tocopherol transfer protein mediates vitamin E trafficking between cerebellar astrocytes and neurons. J. Biol. Chem. 2022, 298, 101–712. [Google Scholar] [CrossRef]
- Lee, P.; Ulatowski, L.M. Vitamin E: Mechanism of transport and regulation in the CNS. IUBMB Life 2019, 71, 424–429. [Google Scholar] [CrossRef]
- Kohlschütter, A.; Finckh, B.; Nickel, M.; Bley, A.; Hübner, C. First Recognized Patient with Genetic Vitamin E Deficiency Stable after 36 Years of Controlled Supplement Therapy. Neurodegener. Dis. 2020, 20, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Head, B.; Traber, M.G. Expanding role of vitamin E in protection against metabolic dysregulation: Insights gained from model systems, especially the developing nervous system of zebrafish embryos. Free Radic. Biol. Med. 2021, 176, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Traber, M. Vitamin E: Necessary nutrient for neural development and cognitive function. Proc. Nutr. Soc. 2021, 80, 319–326. [Google Scholar] [CrossRef]
- Jiang, Q.; Im, S.; Wagner, J.G.; Hernandez, M.L.; Peden, D.B. Gamma-tocopherol, a major form of vitamin E in diets: Insights into antioxidant and anti-inflammatory effects, mechanisms, and roles in disease management. Free Radic. Biol. Med. 2022, 178, 347–359. [Google Scholar] [CrossRef]
- Shibata, H.; Katsuki, H.; Okawara, M.; Kume, T.; Akaike, A. C-Jun N-terminal kinase inhibition and α-tocopherol protect midbrain dopaminergic neurons from interferon-γ/lipopolysaccharide-induced injury without affecting nitric oxide production. J. Neurosci. Res. 2006, 83, 102–109. [Google Scholar] [CrossRef]
- Godbout, J. α-Tocopherol reduces lipopolysaccharide-induced peroxide radical formation and interleukin-6 secretion in primary murine microglia and in brain. J. Neuroimmunol. 2004, 149, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Hernangomez, M.; Carrillo-Salinas, F.; Mecha, M.; Correa, F.; Mestre, L.; Loria, F.; Feliu, A.; Docagne, F.; Guaza, C. Brain Innate Immunity in the Regulation of Neuroinflammation: Therapeutic Strategies by Modulating CD200-CD200R Interaction Involve the Cannabinoid System. Curr. Pharm. Des. 2014, 20, 4707–4722. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Davies, K.J.A. Is vitamin E an antioxidant, a regulator of signal transduction and gene expression, or a ‘junk’ food? Comments on the two accompanying papers: “Molecular mechanism of α-tocopherol action” by A. Azzi and “Vitamin E, antioxidant and nothing more” by M. Traber and J. Atkinson. Free Rad. Biol. Med. 2007, 43, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Rad. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef]
- Ambrogini, P.; Minelli, A.; Galati, C.; Betti, M.; Lattanzi, D.; Ciffolilli, S.; Piroddi, M.; Galli, F.; Cuppini, R. Post-Seizure α-Tocopherol Treatment Decreases Neuroinflammation and Neuronal Degeneration Induced by Status Epilepticus in Rat Hippocampus. Mol. Neurobiol. 2014, 50, 246–256. [Google Scholar] [CrossRef]
- Icer, M.A.; Arslan, N.; Gezmen-Karadag, M. Effects of vitamin E on neurodegenerative diseases: An update. Acta Neurobiol. Exp. 2021, 81, 21–33. [Google Scholar] [CrossRef]
- Regner-Nelke, L.; Nelke, C.; Schroeter, C.B.; Dziewas, R.; Warnecke, T.; Ruck, T.; Meuth, S.G. Enjoy Carefully: The Multifaceted Role of Vitamin E in Neuro-Nutrition. Int. J. Mol. Sci. 2021, 22, 10087. [Google Scholar] [CrossRef]
- Sung, S.; Yao, Y.; Uryu, K.; Yang, H.; Lee, V.M.Y.; Trojanowski, J.Q.; Praticò, D. Early Vitamin E supplementation in young but not aged mice reduces Aβ levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J. 2004, 18, 323–325. [Google Scholar] [CrossRef]
- Conte, V.; Uryu, K.; Fujimoto, S.; Yao, Y.; Rokach, J.; Longhi, L.; Trojanowski, J.Q.; Lee, V.M.Y.; McIntosh, T.K.; Pratico, D. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J. Neurochem. 2004, 90, 758–764. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Dodwell, S.A.; Meyer-Luehmann, M.; Hyman, B.T.; Bacskai, B.J. Plaque-Derived Oxidative Stress Mediates Distorted Neurite Trajectories in the Alzheimer Mouse Model. J. Neuropathol. Exp. Neurol. 2006, 65, 1082–1089. [Google Scholar] [CrossRef]
- Annaházi, A.; Mracskó, É.; Süle, Z.; Karg, E.; Penke, B.; Bari, F.; Farkas, E. Pre-treatment and post-treatment with α-tocopherol attenuates hippocampal neuronal damage in experimental cerebral hypoperfusion. Eur. J. Pharmacol. 2007, 571, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Pasbakhsh, P.; Omidi, N.; Mehrannia, K.; Sobhani, A.G.; Kashani, I.R.; Abbasi, M.; Valeshabad, A.K. The protective effect of vitamin E on locus coeruleus in early model of Parkinson’s disease in rat: Immunoreactivity evidence. Iran. Biomed. J. 2008, 12, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Kuhad, A.; Bishnoi, M.; Chopra, K. Chronic treatment with tocotrienol, an isoform of vitamin E, prevents intracerebroventricular streptozotocin-induced cognitive impairment and oxidative–nitrosative stress in rats. Pharmacol. Biochem. Behav. 2009, 93, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Bostanci, M.Ö.; Bas, O.; Bagirici, F. Alpha-Tocopherol Decreases Iron-Induced Hippocampal and Nigral Neuron Loss. Cell. Mol. Neurobiol. 2010, 30, 389–394. [Google Scholar] [CrossRef]
- Alzoubi, K.H.; Khabour, O.F.; Rashid, B.A.; Damaj, I.M.; Salah, H.A. The neuroprotective effect of vitamin E on chronic sleep deprivation-induced memory impairment: The role of oxidative stress. Behav. Brain Res. 2012, 226, 205–210. [Google Scholar] [CrossRef]
- Desrumaux, C.; Pisoni, A.; Meunier, J.; Deckert, V.; Athias, A.; Perrier, V.; Villard, V.; Lagrost, L.; Verdier, J.M.; Maurice, T. Increased Amyloid-β Peptide-Induced Memory Deficits in Phospholipid Transfer Protein (PLTP) Gene Knockout Mice. Neuropsychopharmacology 2013, 38, 817–825. [Google Scholar] [CrossRef]
- Ishihara, Y.; Itoh, K.; Mitsuda, Y.; Shimada, T.; Kubota, T.; Kato, C.; Song, S.Y.; Kobayashi, Y.; Yasumoto, S.M.; Sekita, S.; et al. Involvement of brain oxidation in the cognitive impairment in a triple transgenic mouse model of Alzheimer’s disease: Noninvasive measurement of the brain redox state by magnetic resonance imaging. Free Radic. Res. 2013, 47, 731–739. [Google Scholar] [CrossRef]
- An, H.M.; Tan, Y.L.; Shi, J.; Wang, Z.; Lv, M.H.; Soares, J.C.; Zhou, D.; Yang, F.; Zhang, X.Y. Ginkgo biloba leaf extract and alpha-tocopherol attenuate haloperidol-induced orofacial dyskinesia in rats: Possible implication of antiapoptotic mechanisms by preventing Bcl-2 decrease and Bax elevation. Phytomedicine 2016, 23, 1653–1660. [Google Scholar] [CrossRef]
- Wang, S.; Yang, S.; Liu, W.; Zhang, Y.; Xu, P.; Wang, T.; Ling, T.; Liu, R. Alpha-tocopherol quinine ameliorates spatial memory deficits by reducing beta-amyloid oligomers, neuroinflammation and oxidative stress in transgenic mice with Alzheimer’s disease. Behav. Brain Res. 2016, 296, 109–117. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Yang, X. PM 2.5 induced neurodegenerative-like changes in mice and the antagonistic effects of vitamin E. Toxicol. Res. 2019, 8, 172–179. [Google Scholar] [CrossRef]
- Jahanshahi, M.; Nikmahzar, E.; Sayyahi, A. Vitamin E therapy prevents the accumulation of congophilic amyloid plaques and neurofibrillary tangles in the hippocampus in a rat model of Alzheimer’s disease. Iran. J. Basic Med. Sci. 2020, 23, 86–92. [Google Scholar] [CrossRef]
- Rana, A.; Singh, S.; Deshmukh, R.; Kumar, A. Pharmacological potential of tocopherol and doxycycline against traumatic brain injury-induced cognitive/motor impairment in rats. Brain Inj. 2020, 34, 1039–1050. [Google Scholar] [CrossRef]
- Nesari, A.; Mansouri, M.T.; Khodayar, M.J.; Rezaei, M. Preadministration of high-dose alpha-tocopherol improved memory impairment and mitochondrial dysfunction induced by proteasome inhibition in rat hippocampus. Nutr. Neurosci. 2021, 24, 119–129. [Google Scholar] [CrossRef]
- Shahidi, S.; Ghahremanitamadon, F.; Asl, S.S.; Komaki, A.; Afshar, S.; Hashemi-Firouzi, N. Electrophysiological, Behavioral and Molecular Study of Vitamin E and Ginkgo biloba in a Rat Model of Alzheimer’s Disease. Res. J. Pharmacogn. (RJP) 2021, 8, 39–51. [Google Scholar]
- Singh, S.; Chauhan, K. Pharmacological Approach using Doxycycline and Tocopherol in Rotenone induced Oxidative Stress, Neuroinflammation and Parkinson’s like symptoms. Int. J. Neurosci. 2022, 28, 192–205. [Google Scholar] [CrossRef]
- Iqbal, A.; Anwar, F.; Saleem, U.; Khan, S.S.; Karim, A.; Ahmad, B.; Gul, M.; Iqbal, Z.; Ismall, T. Inhibition of Oxidative Stress and the NF-κB Pathway by a Vitamin E Derivative: Pharmacological Approach against Parkinson’s Disease. OCS Omega 2022, 7, 45088–45095. [Google Scholar] [CrossRef] [PubMed]
- Zappe, K.; Pointner, A.; Switzeny, O.J.; Magnet, U.; Tomeva, E.; Heller, J.; Mare, G.; Wagner, K.-H.; Knasmueller, S.; Haslberger, A.G. Counteraction of Oxidative Stress by Vitamin E Affects Epigenetic Regulation by Increasing Global Methylation and Gene Expression of MLH1 and DNMT1 Dose Dependently in Caco-2 Cells. Oxid. Med. Cell. Longev. 2018, 2018, 3734250. [Google Scholar] [CrossRef] [PubMed]
- Takatsu, H.; Owada, K.; Abe, K.; Nakano, M.; Urano, S. Effect of Vitamin E on Learning and Memory Deficit in Aged Rats. J. Nutr. Sci. Vitaminol. 2009, 55, 389–393. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Vitamin E Protects Against Oxidative Damage and Learning Disability After Mild Traumatic Brain Injury in Rats. Neurorehabil. Neural Repair. 2010, 24, 290–298. [Google Scholar] [CrossRef]
- Mangialasche, F.; Xu, W.; Kivipelto, M.; Costanzi, E.; Ercolani, S.; Pigliautile, M.; Cecchetti, R.; Baglioni, M.; Simmons, A.; Soininen, H.; et al. Tocopherols and tocotrienols plasma levels are associated with cognitive impairment. Neurobiol. Aging 2012, 33, 2282–2290. [Google Scholar] [CrossRef]
- Mangialasche, F.; Solomon, A.; Kåreholt, I.; Hooshmand, B.; Cecchetti, R.; Fratiglioni, L.; Soininen, H.; Laatikainen, T.; Mecocci, P.; Kivipelto, M. Serum levels of vitamin E forms and risk of cognitive impairment in a Finnish cohort of older adults. Exp. Gerontol. 2013, 48, 1428–1435. [Google Scholar] [CrossRef]
- Bowman, G.L.; Silbert, L.C.; Howieson, D.; Dodge, H.H.; Traber, M.G.; Frei, B.; Kaye, J.A.; Shannon, J.; Quinn, J.F. Nutrient biomarker patterns, cognitive function, and MRI measures of brain aging. Neurology 2012, 78, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Deng, F.; Chen, L. Parkinsonism Caused by Viral Encephalitis Affecting the Bilateral Substantia Nigra. Clin. Neuroradiol. 2019, 29, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Ulatowski, L.; Parker, R.; Warrier, G.; Sultana, R.; Butterfield, D.A.; Manor, D. Vitamin E is essential for Purkinje neuron integrity. Neuroscience 2014, 260, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Igarashi, K.; Uchihara, T.; Jishage, K.; Tomita, H.; Inaba, A.; Li, Y.; Arita, M.; Suzuki, H.; Mizusawa, H.; et al. Delayed-onset ataxia in mice lacking α-tocopherol transfer protein: Model for neuronal degeneration caused by chronic oxidative stress. Proc. Natl. Acad. Sci. USA 2001, 98, 15185–15190. [Google Scholar] [CrossRef]
- El-shaer, N.; El Attar, A. A Study on the Effect of Vitamin E on Histomorphological and Immunohistochemical Changes Induced By Electromagnetic Field. Egypt Acad. J. Biol. Sci. B Zool. 2019, 11, 85–99. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Kitada, T.; Pisani, A.; Porter, D.R.; Yamaguchi, H.; Tscherter, A.; Martella, G.; Bonsi, P.; Zhang, C.; Pothos, E.N.; Shen, J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1 -deficient mice. Proc. Natl. Acad. Sci. USA 2007, 104, 11441–11446. [Google Scholar] [CrossRef]
- Madeo, G.; Schirinzi, T.; Martella, G.; Latagliata, E.C.; Puglisi, F.; Shen, J.; Valente, E.M.; Federici, M.; Mercuri, N.B.; Puglisi-Allegra, S.; et al. PINK1 heterozygous mutations induce subtle alterations in dopamine-dependent synaptic plasticity. Mov. Disordem. 2014, 29, 41–53. [Google Scholar] [CrossRef]
- Martella, G.; Madeo, G.; Maltese, M.; Vanni, V.; Puglisi, F.; Ferraro, E.; Schirinzi, T.; Valente, E.M.; Bonanni, L.; Shen, J.; et al. Exposure to low-dose rotenone precipitates synaptic plasticity alterations in PINK1 heterozygous knockout mice. Neurobiol. Dis. 2016, 91, 21–36. [Google Scholar] [CrossRef]
- Rahman, H.; Qureshi, M.; Khan, R. Influence of Dietary Zinc on Semen Traits and Seminal Plasma Antioxidant Enzymes and Trace Minerals of Beetal Bucks. Reprod. Dom. Anim. 2014, 49, 1004–1007. [Google Scholar] [CrossRef]
- Boujbiha, M.A.M.; Hamden, K.; Guermazi, F.; Bouslama, A.; Omezzine, A.; El Feki, A. Impairment of Spermatogenesis in Rats by Mercuric Chloride: Involvement of Low 17β-Estradiol Level in Induction of Acute Oxidative Stress. Biol. Trace Elem. Res. 2011, 142, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Idris, M.; Khalique, M.A.; Zia-Ur-Rahman; Alhidary, I.A.; Abdelrahman, M.M.; Khan, R.U.; Chand, N.; Farooq, U.; Ahmad, S. The activity and use of zinc in poultry diets. World’s Poult. Sci. Journal. 2016, 72, 159–167. [Google Scholar] [CrossRef]
- Ward, R.J. Ageing neuroinflammation and neurodegeneration. Front./Biosci. 2015, 7, 433. [Google Scholar] [CrossRef] [PubMed]
- Chon, H.; Choi, B.; Jeong, G.; Lee, E.; Lee, S. Suppression of proinflammatory cytokine production by specific metabolites of Lactobacillus plantarum 10hk2 via inhibiting NF-κB and p38 MAPK expressions. Comp. Immunol. Microbiol. Infect. Dis. 2010, 33, e41–e49. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Sugimoto, N.; Ohta, K.; Shimizu, T.; Ohtani, K.; Nakayama, Y.; Nakamura, T.; Hitomi, Y.; Nakamura, H.; Koizumi, S.; et al. Phosphodiesterase Inhibitors Suppress Lactobacillus casei Cell-Wall-Induced NF-κ B and MAPK Activations and Cell Proliferation through Protein Kinase A—Or Exchange Protein Activated by cAMP-Dependent Signal Pathway. Sci. World J. 2012, 2012, 748572. [Google Scholar] [CrossRef]
- Celikoglu, E.; Aslanturk, A.; Kalender, Y. Vitamin E and Sodium Selenite Against Mercuric Chloride-Induced Lung Toxicity in the Rats. Braz. Arch. Biol. Technol. 2015, 58, 587–594. [Google Scholar] [CrossRef]
- Chaudhary, G.; Sinha, K.; Gupta, Y.K. Protective effect of exogenous administration of α -tocopherol in middle cerebral artery occlusion model of cerebral ischemia in rats. Fundam Clin. Pharmacol. 2003, 17, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Tovmasyan, A.; Sheng, H.; Weitner, T.; Arulpragasam, A.; Lu, M.; Warner, D.S.; Vujaskovic, Z.; Spasojevic, I.; Batinic-Haberle, I. Design, Mechanism of Action, Bioavailability and Therapeutic Effects of Mn Porphyrin-Based Redox Modulators. Med. Princ. Pract. 2013, 22, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Guidotti, G.; Racagni, G.; Riva, M.A. Reduced neuroplasticity in aged rats: A role for the neurotrophin brain-derived neurotrophic factor. Neurobiol. Aging 2013, 34, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.I.; Prakash, R.S.; Voss, M.W.; Chaddock, L.; Heo, S.; McLaren, M.; Pence, B.D.; Martin, S.A.; Vieira, V.J.; Woods, A.; et al. Brain-Derived Neurotrophic Factor Is Associated with Age-Related Decline in Hippocampal Volume. J. Neurosci. 2010, 30, 5368–5375. [Google Scholar] [CrossRef]
- von Bohlen und Halbach, O. Involvement of BDNF in age-dependent alterations in the hippocampus. Front. Aging Neurosci. 2010, 2, 36. [Google Scholar] [CrossRef]
- Murer, M.G.; Yan, Q.; Raisman-Vozari, R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog. Neurobiol. 2001, 63, 71–124. [Google Scholar] [CrossRef]
- Guimarães, M.R.M.; Murad, L.B.; Paganelli, A.; de Oliveira, C.A.B.; Vianna, L.M.A. Effects of alpha-tocopherol associated with lovastatin on brain tissue and memory function in SHRSPs. Physiol. Behav. 2015, 149, 303–309. [Google Scholar] [CrossRef]
- Murad, L.B.; Guimarães, M.R.M.; Paganelli, A.; de Oliveira, C.A.B.; Vianna, L.M. Alpha-tocopherol in the brain tissue preservation of stroke-prone spontaneously hypertensive rats. J. Physiol. Biochem. 2014, 70, 49–60. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Reference | Species/Strain | Gender | Control Group | Induction Method | Experimental Model | Type of Subst. | Duration | Dose | Admin. Route | Data Collection Time | Outcome Measurement | Main Findings |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sung 2003 [28] | Tg 2576 mice | M/F | Placebo | Transgenic rats | AD a | Vitamin E | 6 to 8 months | 2 mg/g | Oral | 8 months | Neuroprotection | Reduced lipid peroxidation, soluble Aβ b and amyloid plaque deposition |
| Conte et al., 2004 [29] | Tg 2576 mice | F | Normal chow | RCBI c | AD a | Vitamin E | 4 weeks before lesion and 8 weeks after | 2 mg/g | Injection into the left parieto-temporal region. | 8 weeks | Neuroprotection | Reduced BLP (Brain Lipid Peroxidation) Alleviated the learning deficit Improved behavioral commitment |
| Garcia-Alloza 2006 [30] | APPswe/PS1d9 mice | * | 2 received distilled water 2 received cremophor 25% in distilled water | Transgenic rats | AD a | Trolox (Vitamin E) | 15 days | 1 day before sugery 210 mg/kg, and after for 15 days | Gavage | 15 days | Neuroregeneration | Slowed the progress of AD a Significantly reduced oxidative stress Changed the structures of the neurites |
| Annaházi, 2007 [31] | Wistar rats | M | Operated group Nothing applied | Chronic brain hypoperfusion | Brain injury in bilateral common carotid arteries | α-tocopherol | 5 days before surgery and 5 days after | 100 mg/kg | Intraperitoneal | 17 days | Neuroprotection | Improved the learning process Prevented loss of stained pyramidal cells in the CA1 hippocampus Preserved dendritic arborizations Attenuated microglial activation |
| Pasbakhsh, 2009 [32] | Sprague-Dawley rats | M | Sham group operated + Sham group treated with vehicle + sham group treated with vitamin E | 6-OHDA d | PD e initial model | Vitamin E | 8 weeks | D-a-tocopheryl succinate (16 mg/kg, i.m, Bioglan, UK) and 0.8 mL/kg of propylene glycol | Intramuscular | 2 weeks after surgery | Neuroprotection | Delayed functional decline |
| Tiwari et al., 2009 [33] | Wistar rats | M | Injected citrate buffer | ST’Z f | AD a Cognitive deficit | α-tocopherol | 21 days after lesion | 100 mg/kg | Oral | 21 days | Neuroprotection | Prevention of cognitive impairment Prevented a reduction in the levels of GSH g and catalase Reduced MDA h, nitrite and cholinesterase activity |
| Bostanci, 2010 [34] | Wistar rats | M | Saline solution | Iron | Neurotoxicity Oxidative stress | α-tocopherol | 10 days | 100 mg/kg/day | Intraperitoneal | 10 days | Neuroprotection |
Attenuated the loss of neurons
Decreased cell loss in the hippocampus and substantia nigra Protective effect on pyramidal cells of the hippocampus |
| Alzoubi, 2012 [35] | Wistar rats | M | Vehicle | Sleep deprivation | Memory deficit Learning impairment | Vitamin E | 6 weeks | 100 mg/kg | Oral by gavage | 6 weeks | Neuroprotection | Prevented memory impairment It normalized the reduction in oxidative stress markers (GSH g/GSSG i), SOD j and GPx k catalase activity |
| Desrumaux, 2013 [36] | PLTO-KO mice | M | 3 μL of vehicle | Aβ b 25–35 | AD a Memory impairment | Vitamin E | 10 days | 800 mg/kg | Oral | 10 days | Neuroprotection | Reduced short-term memory impairment Prevented PLTP-KO l compromise |
| Ishihara, 2013 [37] | 3 Tg—AD a mice | * | Normal diet | Transgenic rats | AD a pathogenesis Cognitive deficit | α-tocopherol | 4 months | 1.342 mg/g and normal diet 0.076 mg/g | Oral | 4 months and 4 days | Neuroprotection | Prevented cognitive impairment Attenuated the reduction in GSH g levels and the increase in GSSG i and TBARS m Decreased the levels of reactive radicals in the brain |
| An, 2016 [38] | Sprague Dawley rats | M | Saline solution | HAL n | Orofacial dyskinesia VCM o | α-tocopherol | 5 weeks | 20 mg/kg/day | Oral | 5 weeks from the last administration | Neuroprotection | Reduced stereotypical behavior Decreased the expression of anti-apoptotic protein Bcl-2 p Increased the expression of pro-apoptotic Bax q protein Decreased Bax q/Bcl-2 p ratio in prefrontal cortex, striatum, substantia nigra, and globus pallidus |
| Wang, 2016 [39] | APPswe/PS1dE9 mice | M | Saline solution | Transgenic rats | AD a | α-tocopherol | 4 weeks | 100 mg/kg | Oral gavage | 4 weeks | Neuroprotection Neuroregeneration | Improved memory impairment Improved cognitive dysfunction Counteracted oxidative stress Decreased the levels of Aβ b oligomer |
| Liu, 2019 [40] | C57BL/6J mice | M | 0 mg kg –1 day –1 of PM 2.5 r | PM 2.5 r | Cognitive deficit Oxidative stress | Vitamin E | 7 days | 50 mg/kg–1 day–1 | Intragastric | 7 days | Neuroprotection Neuroregeneration | Improved cognitive function Reduced cellular damage Increased the number of cells Decreased the expression of Aβ b 1–42 Reduced oxidative stress |
| Jahanshahi, 2020 [41] | Wistar rats | M | No medication | Scopolamin | AD a | Vitamin E | 14 days after induction | 25, 50, and 100 mg/kg/day | Intraperitoneal | 16 days | Neuroprotection | Amyloid plaque reduction Prevented an increase in neurofibrillary tangles in hippocampal subregions |
| Rana et al., 2020 [42] | Wistar rats | M | Vehicle | TBI s | Cognitive impairment Motor damage | α-tocopherol | 28 days after induction | 5 mg/kg, po. 10 mg/kg, po. | Weight drop model | 28 days | Neuroprotection Neuromodulation | Attenuated locomotor performance Reduced cognitive impairment Reduced neuroinflammatory markers Restored neurotransmitter levels Balanced oxidative stress |
| Nesari, 2021 [43] | Wistar rats | M | DMSO t + Saline solution | Lactacystin | Proteasome inhibition Oxidative stress Memory impairment | α-tocopherol | 5 days before induction | 60 and 200 mg/kg, i.p. | Bilateral hippocampal injection | 7 days | Neuroprotection | High doses of α-tocopherol exhibited remarkable mitochondrial protection Improved memory impairment Increased the levels of glutathione |
| Shahidi, 2021 [44] | Wistar rats | M | Saline solution + Non operated | Aβ b 25–35 | AD a | Vitamin E | 10 days | 200 mg/kg | Oral by gavage | 2 weeks 10 days | Neuroprotection | Improved passive avoidance of memory impairment The amplitude of the PS u was increased Alleviated LTP v deficiency Reverted the increase in Bcl-2 p and Bax q ratio in the hippocampus |
| Singh, 2022 [45] | Wistar rats | M | Ropinirol | Rotenona | Sintomas semelhantes a PD e | Tocopherol | 40 days | 5 and 10 mg/kg | Intraperitoneal | 41 days | Neuroprotection and neuroinflammation |
Attenuated behavioral changes
Enhanced the expression of neurotransmitters Reduced the levels of inflammatory markers |
| Iqbal, 2022 [46] | Albino swiss mice | M | CMC w | HAL n | PD e | Tocopherol | 23 days | 5, 10, 20 and 40 mg/kg | Oral route | 23 days | Neuroprotection | Increased the levels of antioxidant enzymes and neurotransmitters Decreased the levels of inflammatory cytokines and the expression of α-synuclein mRNA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Cunha Germano, B.C.; de Morais, L.C.C.; Idalina Neta, F.; Fernandes, A.C.L.; Pinheiro, F.I.; do Rego, A.C.M.; Araújo Filho, I.; de Azevedo, E.P.; de Paiva Cavalcanti, J.R.L.; Guzen, F.P.; et al. Vitamin E and Its Molecular Effects in Experimental Models of Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 11191. https://doi.org/10.3390/ijms241311191
da Cunha Germano BC, de Morais LCC, Idalina Neta F, Fernandes ACL, Pinheiro FI, do Rego ACM, Araújo Filho I, de Azevedo EP, de Paiva Cavalcanti JRL, Guzen FP, et al. Vitamin E and Its Molecular Effects in Experimental Models of Neurodegenerative Diseases. International Journal of Molecular Sciences. 2023; 24(13):11191. https://doi.org/10.3390/ijms241311191
Chicago/Turabian Styleda Cunha Germano, Bianca Caroline, Lara Cristina Carlos de Morais, Francisca Idalina Neta, Amélia Carolina Lopes Fernandes, Francisco Irochima Pinheiro, Amália Cinthia Meneses do Rego, Irami Araújo Filho, Eduardo Pereira de Azevedo, José Rodolfo Lopes de Paiva Cavalcanti, Fausto Pierdona Guzen, and et al. 2023. "Vitamin E and Its Molecular Effects in Experimental Models of Neurodegenerative Diseases" International Journal of Molecular Sciences 24, no. 13: 11191. https://doi.org/10.3390/ijms241311191
APA Styleda Cunha Germano, B. C., de Morais, L. C. C., Idalina Neta, F., Fernandes, A. C. L., Pinheiro, F. I., do Rego, A. C. M., Araújo Filho, I., de Azevedo, E. P., de Paiva Cavalcanti, J. R. L., Guzen, F. P., & Cobucci, R. N. (2023). Vitamin E and Its Molecular Effects in Experimental Models of Neurodegenerative Diseases. International Journal of Molecular Sciences, 24(13), 11191. https://doi.org/10.3390/ijms241311191