

The Role of SOX9 in IGF-II-Mediated Pulmonary Fibrosis

 , and
, and
Abstract
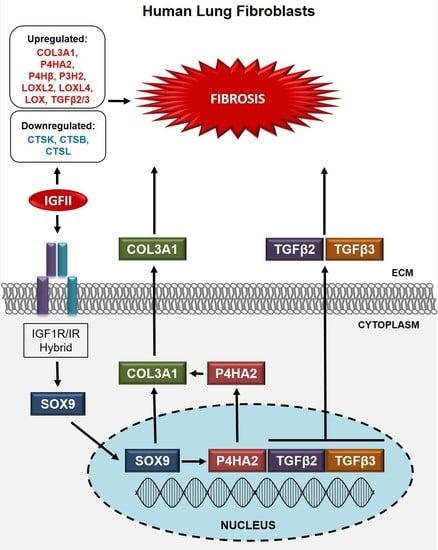
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
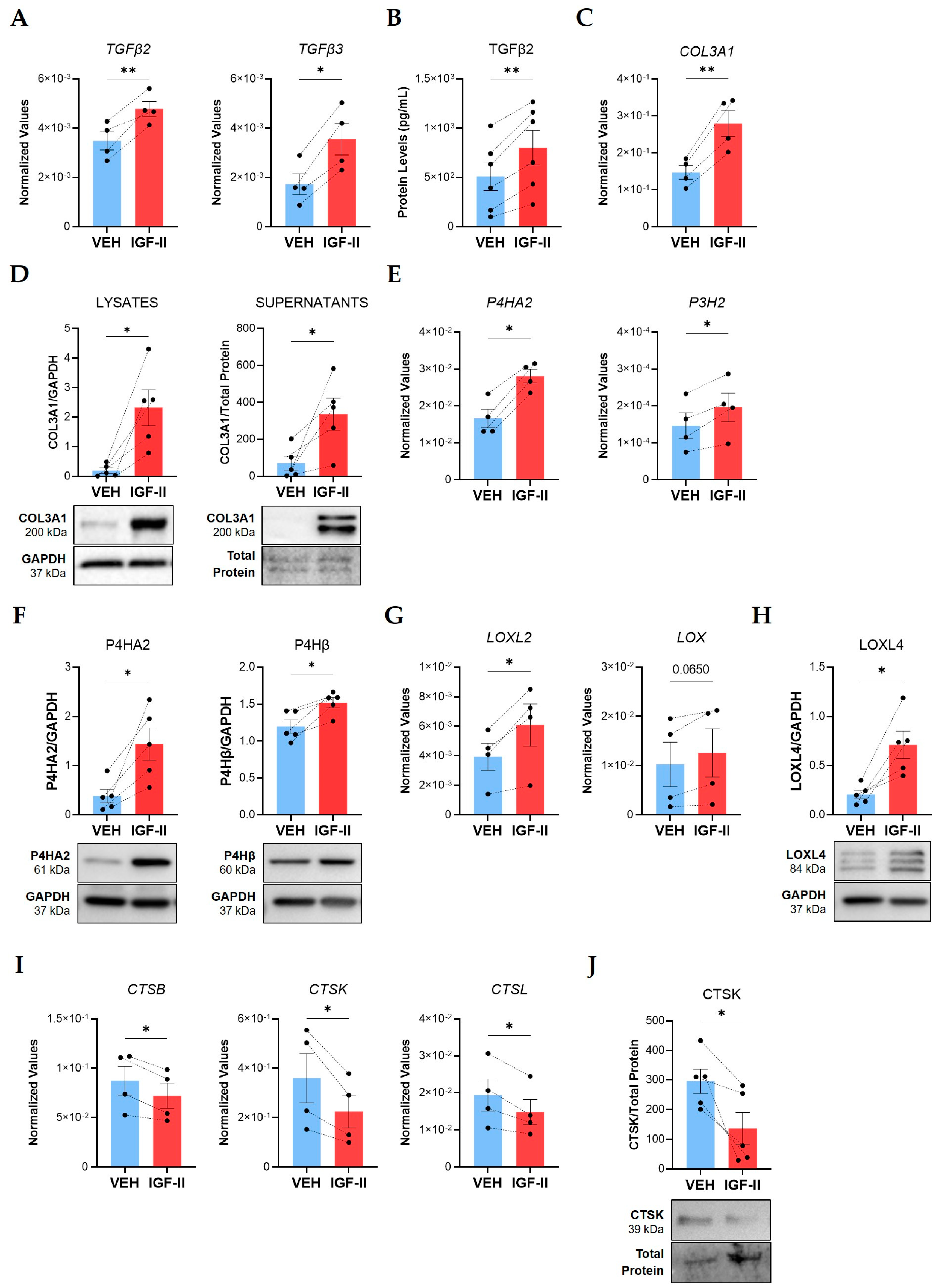
2.1. IGF-II Promotes Fibrosis in Normal Human Lung Fibroblasts and Tissues from Different Donors
2.2. SOX9 Is Overexpressed in SSc Lung Tissues
2.3. SOX9 Is an Immediate Early Gene Activated by IGF-II through the IGF-1R/IR Hybrid Receptor in Human Lung Fibroblasts
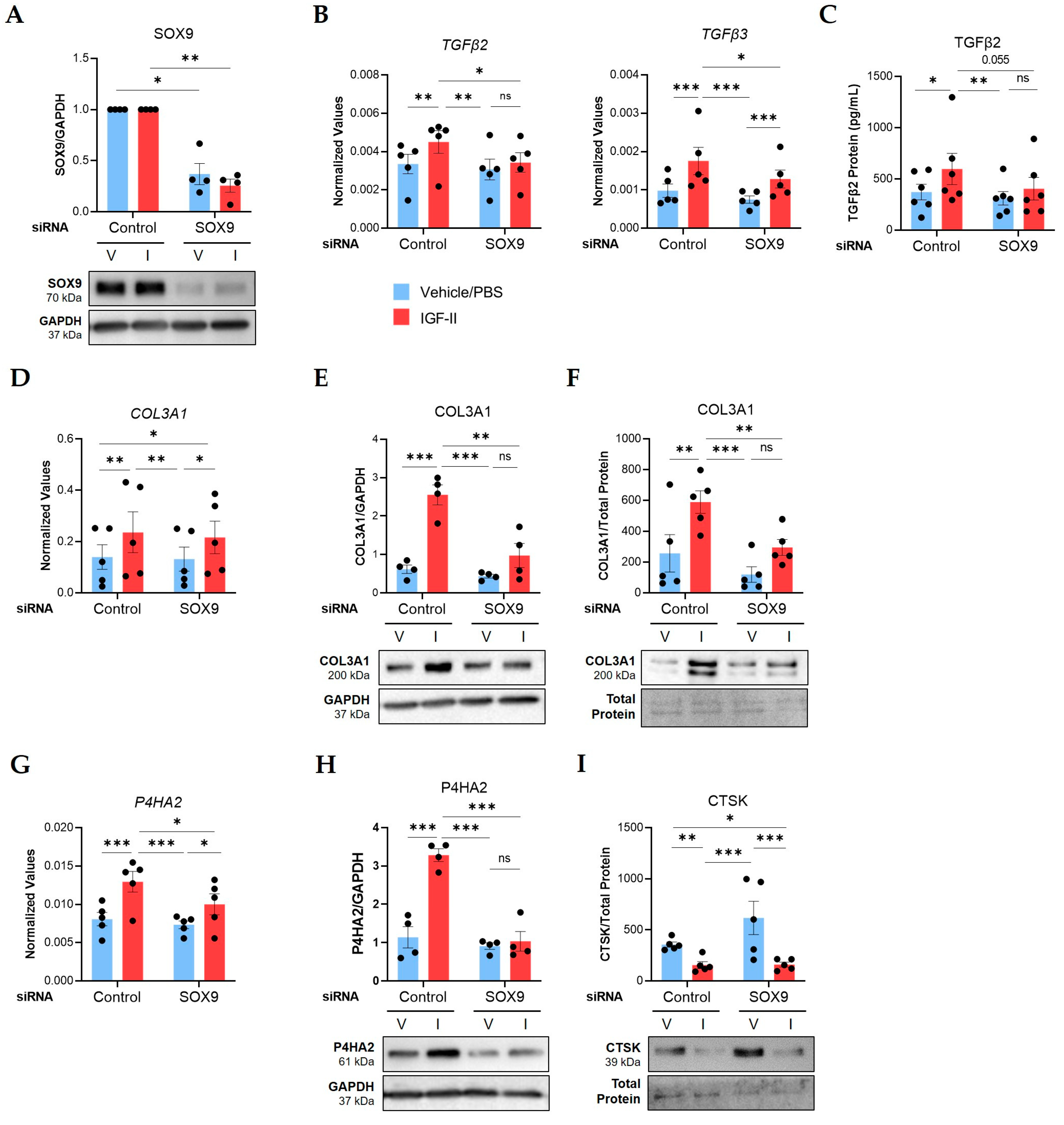
2.4. SOX9 Mediates IGF-II Induction of TGFβ2, TGFβ3, COL3A1, and P4HA2 in NL Fibroblasts
3. Discussion
3.1. The IGF System in Pulmonary Fibrosis
3.2. SOX9 in Pulmonary Fibrosis
3.3. The IGF-II-SOX9 Pathway in Human Lung Fibroblasts
4. Materials and Methods
4.1. In Vitro Primary Human Lung Fibroblast Cell Culture
4.2. Ex Vivo Human Lung Tissue Culture and IGF-II Stimulation
4.3. Donor Information
4.4. Culture and Treatment of Human Lung Fibroblasts
4.5. Subcellullar Fractionation of Human Lung Fibroblasts
4.6. Small Interfering RNA Transfection
4.7. RNA Extraction, cDNA, and qPCR
4.8. Immunoblotting
4.9. ELISA
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and Cellular Mechanisms of Pulmonary Fibrosis. Fibrogenesis Tissue Repair 2012, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Steen, V.D.; Medsger, T.A. Changes in Causes of Death in Systemic Sclerosis, 1972–2002. Ann. Rheum. Dis. 2007, 66, 940–944. [Google Scholar] [CrossRef]
- Herzog, E.L.; Mathur, A.; Tager, A.M.; Feghali-Bostwick, C.; Schneider, F.; Varga, J. Interstitial Lung Disease Associated With Systemic Sclerosis and Idiopathic Pulmonary Fibrosis. Arthritis Rheumatol. Hoboken NJ 2014, 66, 1967–1978. [Google Scholar] [CrossRef] [Green Version]
- Cottin, V.; Brown, K.K. Interstitial Lung Disease Associated with Systemic Sclerosis (SSc-ILD). Respir. Res. 2019, 20, 13. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Ruaro, B.; Montagna, P.; Brizzolara, R.; Stratta, E.; Trombetta, A.C.; Scabini, S.; Tavilla, P.P.; Parodi, A.; Corallo, C.; et al. Effects of Selexipag and Its Active Metabolite in Contrasting the Profibrotic Myofibroblast Activity in Cultured Scleroderma Skin Fibroblasts. Arthritis Res. Ther. 2018, 20, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Highland, K.B.; Distler, O.; Kuwana, M.; Allanore, Y.; Assassi, S.; Azuma, A.; Bourdin, A.; Denton, C.P.; Distler, J.H.W.; Hoffmann-Vold, A.M.; et al. Efficacy and Safety of Nintedanib in Patients with Systemic Sclerosis-Associated Interstitial Lung Disease Treated with Mycophenolate: A Subgroup Analysis of the SENSCIS Trial. Lancet Respir. Med. 2021, 9, 96–106. [Google Scholar] [CrossRef]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Goldin, J.; Kim, G.; Kuwana, M.; Allanore, Y.; Matucci-Cerinic, M.; Distler, O.; Shima, Y.; et al. Tocilizumab in Systemic Sclerosis: A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Respir. Med. 2020, 8, 963–974. [Google Scholar] [CrossRef]
- Campochiaro, C.; De Luca, G.; Lazzaroni, M.-G.; Armentaro, G.; Spinella, A.; Vigone, B.; Ruaro, B.; Stanziola, A.; Benfaremo, D.; De Lorenzis, E.; et al. Real-Life Efficacy and Safety of Nintedanib in Systemic Sclerosis-Interstitial Lung Disease: Data from an Italian Multicentre Study. RMD Open 2023, 9, e002850. [Google Scholar] [CrossRef]
- Kafle, S.; Thapa Magar, M.; Patel, P.; Poudel, A.; Cancarevic, I. Systemic Sclerosis Associated Interstitial Lung Disease and Nintedanib: A Rare Disease and a Promising Drug. Cureus 2021, 13, e16404. [Google Scholar] [CrossRef] [PubMed]
- Mouawad, J.E.; Feghali-Bostwick, C. The Molecular Mechanisms of Systemic Sclerosis-Associated Lung Fibrosis. Int. J. Mol. Sci. 2023, 24, 2963. [Google Scholar] [CrossRef]
- Mattoo, H.; Pillai, S. Idiopathic Pulmonary Fibrosis and Systemic Sclerosis: Pathogenic Mechanisms and Therapeutic Interventions. Cell. Mol. Life Sci. 2021, 78, 5527–5542. [Google Scholar] [CrossRef] [PubMed]
- Mirsaeidi, M.; Barletta, P.; Glassberg, M.K. Systemic Sclerosis Associated Interstitial Lung Disease: New Directions in Disease Management. Front. Med. 2019, 6, 248. [Google Scholar]
- Garrett, S.M.; Baker Frost, D.; Feghali-Bostwick, C. The Mighty Fibroblast and Its Utility in Scleroderma Research. J. Scleroderma Relat. Disord. 2017, 2, 100–107. [Google Scholar] [CrossRef]
- Burgstaller, G.; Oehrle, B.; Gerckens, M.; White, E.S.; Schiller, H.B.; Eickelberg, O. The Instructive Extracellular Matrix of the Lung: Basic Composition and Alterations in Chronic Lung Disease. Eur. Respir. J. 2017, 50, 1601805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, Q.; Liu, Z.; Zuo, H.; Yang, Z.; Qu, J. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Front. Pharmacol. 2022, 12, 797292. [Google Scholar] [PubMed]
- Hsu, E.; Feghali-Bostwick, C.A. Insulin-Like Growth Factor-II Is Increased in Systemic Sclerosis-Associated Pulmonary Fibrosis and Contributes to the Fibrotic Process via Jun N-Terminal Kinase- and Phosphatidylinositol-3 Kinase-Dependent Pathways. Am. J. Pathol. 2008, 172, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Garrett, S.M.; Hsu, E.; Thomas, J.M.; Pilewski, J.M.; Feghali-Bostwick, C. Insulin-like Growth Factor (IGF)-II- Mediated Fibrosis in Pathogenic Lung Conditions. PLoS ONE 2019, 14, e0225422. [Google Scholar] [CrossRef]
- Livingstone, C.; Borai, A. Insulin-like Growth Factor-II: Its Role in Metabolic and Endocrine Disease. Clin. Endocrinol. (Oxf.) 2014, 80, 773–781. [Google Scholar] [CrossRef]
- Livingstone, C. IGF2 and Cancer. Endocr. Relat. Cancer 2013, 20, R321–R339. [Google Scholar] [CrossRef] [Green Version]
- Blyth, A.J.; Kirk, N.S.; Forbes, B.E. Understanding IGF-II Action through Insights into Receptor Binding and Activation. Cells 2020, 9, 2276. [Google Scholar] [CrossRef]
- Humbel, R.E. Review Insulin-Like Growth Factors I and II. In EJB Reviews 1990; Christen, P., Hofmann, E., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 109–126. ISBN 978-3-642-76168-3. [Google Scholar]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-Cell RNA-Seq Reveals Ectopic and Aberrant Lung-Resident Cell Populations in Idiopathic Pulmonary Fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-Cell RNA Sequencing Reveals Profibrotic Roles of Distinct Epithelial and Mesenchymal Lineages in Pulmonary Fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.E.; Ahangari, F.; Li, Q.; Jain, S.; Verleden, S.E.; Herazo-Maya, J.; Vukmirovic, M.; DeIuliis, G.; Tzouvelekis, A.; Tanabe, N.; et al. Transcriptional Regulatory Model of Fibrosis Progression in the Human Lung. JCI Insight 2019, 4, e131597. [Google Scholar] [CrossRef]
- IPF Cell Atlas. Available online: http://www.ipfcellatlas.com/ (accessed on 21 March 2023).
- Renaud, L.; Waldrep, K.M.; da Silveira, W.A.; Pilewski, J.M.; Feghali-Bostwick, C.A. First Characterization of the Transcriptome of Lung Fibroblasts of SSc Patients and Healthy Donors of African Ancestry. Int. J. Mol. Sci. 2023, 24, 3645. [Google Scholar] [CrossRef] [PubMed]
- Mouawad, J.E.; Sharma, S.; Renaud, L.; Pilewski, J.M.; Nadig, S.N.; Feghali-Bostwick, C. Reduced Cathepsin L Expression and Secretion into the Extracellular Milieu Contribute to Lung Fibrosis in Systemic Sclerosis. Rheumatology 2022, 62, keac411. [Google Scholar] [CrossRef]
- Pritchett, J.; Athwal, V.; Roberts, N.; Hanley, N.A.; Hanley, K.P. Understanding the Role of SOX9 in Acquired Diseases: Lessons from Development. Trends Mol. Med. 2011, 17, 166–174. [Google Scholar] [CrossRef]
- Haseeb, A.; Lefebvre, V. The SOXE Transcription Factors—SOX8, SOX9 and SOX10—Share a Bi-Partite Transactivation Mechanism. Nucleic Acids Res. 2019, 47, 6917–6931. [Google Scholar] [CrossRef]
- Grimm, D.; Bauer, J.; Wise, P.; Krüger, M.; Simonsen, U.; Wehland, M.; Infanger, M.; Corydon, T.J. The Role of SOX Family Members in Solid Tumours and Metastasis. Semin. Cancer Biol. 2020, 67, 122–153. [Google Scholar] [CrossRef]
- Schock, E.N.; LaBonne, C. Sorting Sox: Diverse Roles for Sox Transcription Factors During Neural Crest and Craniofacial Development. Front. Physiol. 2020, 11, 606889. [Google Scholar]
- Staab-Weijnitz, C.A. Fighting the Fiber: Targeting Collagen in Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 363–381. [Google Scholar] [CrossRef]
- McKleroy, W.; Lee, T.-H.; Atabai, K. Always Cleave up Your Mess: Targeting Collagen Degradation to Treat Tissue Fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L709–L721. [Google Scholar] [CrossRef] [Green Version]
- Fonović, M.; Turk, B. Cysteine Cathepsins and Extracellular Matrix Degradation. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 2560–2570. [Google Scholar] [CrossRef]
- Morse, C.; Tabib, T.; Sembrat, J.; Buschur, K.L.; Bittar, H.T.; Valenzi, E.; Jiang, Y.; Kass, D.J.; Gibson, K.; Chen, W.; et al. Proliferating SPP1/MERTK-Expressing Macrophages in Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2019, 54, 1802441. [Google Scholar] [CrossRef] [PubMed]
- Gajjala, P.R.; Kasam, R.K.; Soundararajan, D.; Sinner, D.; Huang, S.K.; Jegga, A.G.; Madala, S.K. Dysregulated Overexpression of Sox9 Induces Fibroblast Activation in Pulmonary Fibrosis. JCI Insight 2021, 6, e152503. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.; Wirth, J.; Meyer, J.; Zabel, B.; Held, M.; Zimmer, J.; Pasantes, J.; Bricarelli, F.D.; Keutel, J.; Hustert, E.; et al. Autosomal Sex Reversal and Campomelic Dysplasia Are Caused by Mutations in and around the SRY-Related Gene SOX9. Cell 1994, 79, 1111–1120. [Google Scholar] [CrossRef]
- Williams, C.A.C.; Soufi, A.; Pollard, S.M. Post-Translational Modification of SOX Family Proteins: Key Biochemical Targets in Cancer? Semin. Cancer Biol. 2020, 67, 30–38. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of Action of Nintedanib in the Treatment of Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruwanpura, S.M.; Thomas, B.J.; Bardin, P.G. Pirfenidone: Molecular Mechanisms and Potential Clinical Applications in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2020, 62, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, M.; Granata, R.; Ghigo, E. The IGF System. Acta Diabetol. 2011, 48, 1–9. [Google Scholar] [CrossRef]
- Hung, C.F.; Rohani, M.G.; Lee, S.-S.; Chen, P.; Schnapp, L.M. Role of IGF-1 Pathway in Lung Fibroblast Activation. Respir. Res. 2013, 14, 102. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro-Hermida, S.; López, I.P.; Alfaro-Arnedo, E.; Torrens, R.; Iñiguez, M.; Alvarez-Erviti, L.; Ruíz-Martínez, C.; Pichel, J.G. IGF1R Deficiency Attenuates Acute Inflammatory Response in a Bleomycin-Induced Lung Injury Mouse Model. Sci. Rep. 2017, 7, 4290. [Google Scholar] [CrossRef] [Green Version]
- Renaud, L.; da Silveira, W.A.; Takamura, N.; Hardiman, G.; Feghali-Bostwick, C. Prominence of IL6, IGF, TLR, and Bioenergetics Pathway Perturbation in Lung Tissues of Scleroderma Patients With Pulmonary Fibrosis. Front. Immunol. 2020, 11, 383. [Google Scholar] [CrossRef]
- Nguyen, X.-X.; Muhammad, L.; Nietert, P.J.; Feghali-Bostwick, C. IGFBP-5 Promotes Fibrosis via Increasing Its Own Expression and That of Other Pro-Fibrotic Mediators. Front. Endocrinol. 2018, 9, 601. [Google Scholar] [CrossRef]
- Pilewski, J.M.; Liu, L.; Henry, A.C.; Knauer, A.V.; Feghali-Bostwick, C.A. Insulin-like Growth Factor Binding Proteins 3 and 5 Are Overexpressed in Idiopathic Pulmonary Fibrosis and Contribute to Extracellular Matrix Deposition. Am. J. Pathol. 2005, 166, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, E.; Shi, H.; Jordan, R.M.; Lyons-Weiler, J.; Pilewski, J.M.; Feghali-Bostwick, C.A. Lung Tissues in Patients with Systemic Sclerosis Have Gene Expression Patterns Unique to Pulmonary Fibrosis and Pulmonary Hypertension. Arthritis Rheum. 2011, 63, 783–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuoka, H.; Zhou, Z.; Pilewski, J.M.; Oury, T.D.; Choi, A.M.K.; Feghali-Bostwick, C.A. Insulin-Like Growth Factor-Binding Protein-5 Induces Pulmonary Fibrosis and Triggers Mononuclear Cellular Infiltration. Am. J. Pathol. 2006, 169, 1633–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuoka, H.; Hsu, E.; Ruiz, X.D.; Steinman, R.A.; Choi, A.M.K.; Feghali-Bostwick, C.A. The Fibrotic Phenotype Induced by IGFBP-5 Is Regulated by MAPK Activation and Egr-1-Dependent and -Independent Mechanisms. Am. J. Pathol. 2009, 175, 605–615. [Google Scholar] [CrossRef] [Green Version]
- Yasuoka, H.; Jukic, D.M.; Zhou, Z.; Choi, A.M.K.; Feghali-Bostwick, C.A. Insulin-like Growth Factor Binding Protein 5 Induces Skin Fibrosis: A Novel Murine Model for Dermal Fibrosis. Arthritis Rheum. 2006, 54, 3001–3010. [Google Scholar] [CrossRef]
- Yasuoka, H.; Larregina, A.T.; Yamaguchi, Y.; Feghali-Bostwick, C.A. Human Skin Culture as an Ex Vivo Model for Assessing the Fibrotic Effects of Insulin-Like Growth Factor Binding Proteins. Open Rheumatol. J. 2008, 2, 17–22. [Google Scholar] [CrossRef] [Green Version]
- Yasuoka, H.; Yamaguchi, Y.; Feghali-Bostwick, C.A. The Pro-Fibrotic Factor IGFBP-5 Induces Lung Fibroblast and Mononuclear Cell Migration. Am. J. Respir. Cell Mol. Biol. 2009, 41, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.A.; Guo, L.-W.; Shi, X.-D.; Kundi, R.; Sovinski, G.; Seedial, S.; Liu, B.; Kent, K.C. Preferential Secretion of Collagen Type 3 versus Type 1 from Adventitial Fibroblasts Stimulated by TGF-β/Smad3-Treated Medial Smooth Muscle Cells. Cell. Signal. 2013, 25, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Huang, Z.; Liang, W.-C.; Yin, J.; Lin, W.Y.; Wu, J.; Vernes, J.-M.; Lutman, J.; Caplazi, P.; Jeet, S.; et al. TGFβ2 and TGFβ3 Isoforms Drive Fibrotic Disease Pathogenesis. Sci. Transl. Med. 2021, 13, eabe0407. [Google Scholar] [CrossRef]
- Rappu, P.; Salo, A.M.; Myllyharju, J.; Heino, J. Role of Prolyl Hydroxylation in the Molecular Interactions of Collagens. Essays Biochem. 2019, 63, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Reddy, D.P.K.; Xue, C.; Liu, X.; Chen, X.; Li, J.; Ling, X.; Zheng, S. Profiling of MiR-205/P4HA3 Following Angiotensin II-Induced Atrial Fibrosis: Implications for Atrial Fibrillation. Front. Cardiovasc. Med. 2021, 8. [Google Scholar]
- Li, J.; Ghazwani, M.; Zhang, Y.; Lu, J.; Li, J.; Fan, J.; Gandhi, C.R.; Li, S. MiR-122 Regulates Collagen Production via Targeting Hepatic Stellate Cells and Suppressing P4HA1 Expression. J. Hepatol. 2013, 58, 522–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Xu, W.; Chen, H.; Warburton, D.; Dong, R.; Qian, B.; Selman, M.; Gauldie, J.; Kolb, M.; Shi, W. A Novel Profibrotic Mechanism Mediated by TGF-β-Stimulated Collagen Prolyl Hydroxylase Expression in Fibrotic Lung Mesenchymal Cells. J. Pathol. 2015, 236, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, Y.; Kitani, A.; Hara, M.; Harigai, M.; Hirose, T.; Suzuki, K.; Kawakami, M.; Hidaka, T.; Ishizuka, T.; Kawagoe, M. Cytokine Regulation of Prolyl 4-Hydroxylase Production in Skin Fibroblast Cultures from Patients with Systemic Sclerosis: Contribution to Collagen Synthesis and Fibrosis. J. Rheumatol. 1992, 19, 1195–1201. [Google Scholar] [PubMed]
- Castello-Cros, R.; Whitaker-Menezes, D.; Molchansky, A.; Purkins, G.; Soslowsky, L.J.; Beason, D.P.; Sotgia, F.; Iozzo, R.V.; Lisanti, M.P. Scleroderma-like Properties of Skin from Caveolin-1-Deficient Mice. Cell Cycle 2011, 10, 2140–2150. [Google Scholar] [CrossRef] [Green Version]
- Keiser, H.R.; Stein, H.D.; Sjoerdsma, A. Increased Protocollagen Proline Hydroxylase Activity in Sclerodermatous Skin. Arch. Dermatol. 1971, 104, 57–60. [Google Scholar] [CrossRef]
- Vasta, J.D.; Raines, R.T. Collagen Prolyl 4-Hydroxylase as a Therapeutic Target. J. Med. Chem. 2018, 61, 10403–10411. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, S.; Li, W. LOX/LOXL in Pulmonary Fibrosis: Potential Therapeutic Targets. J. Drug Target. 2019, 27, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Aumiller, V.; Strobel, B.; Romeike, M.; Schuler, M.; Stierstorfer, B.E.; Kreuz, S. Comparative Analysis of Lysyl Oxidase (like) Family Members in Pulmonary Fibrosis. Sci. Rep. 2017, 7, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, X.-X.; Nishimoto, T.; Takihara, T.; Mlakar, L.; Bradshaw, A.D.; Feghali-Bostwick, C. Lysyl Oxidase Directly Contributes to Extracellular Matrix Production and Fibrosis in Systemic Sclerosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L29–L40. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Findlay, A.; Stolp, J.; Rayner, B.; Ask, K.; Jarolimek, W. Pan-Lysyl Oxidase Inhibitor PXS-5505 Ameliorates Multiple-Organ Fibrosis by Inhibiting Collagen Crosslinks in Rodent Models of Systemic Sclerosis. Int. J. Mol. Sci. 2022, 23, 5533. [Google Scholar] [CrossRef]
- Huang, M.; Cai, G.; Baugh, L.M.; Liu, Z.; Smith, A.; Watson, M.; Popovich, D.; Zhang, T.; Stawski, L.S.; Trojanowska, M.; et al. Systemic Sclerosis Dermal Fibroblasts Induce Cutaneous Fibrosis Through Lysyl Oxidase–like 4: New Evidence From Three-Dimensional Skin-like Tissues. Arthritis Rheumatol. 2020, 72, 791–801. [Google Scholar] [CrossRef]
- Vallet, S.D.; Ricard-Blum, S. Lysyl Oxidases: From Enzyme Activity to Extracellular Matrix Cross-Links. Essays Biochem. 2019, 63, 349–364. [Google Scholar] [CrossRef]
- Matsuo, A.; Tanida, R.; Yanagi, S.; Tsubouchi, H.; Miura, A.; Shigekusa, T.; Matsumoto, N.; Nakazato, M. Significance of Nuclear LOXL2 Inhibition in Fibroblasts and Myofibroblasts in the Fibrotic Process of Acute Respiratory Distress Syndrome. Eur. J. Pharmacol. 2021, 892, 173754. [Google Scholar] [CrossRef]
- Yang, J.; Savvatis, K.; Kang, J.S.; Fan, P.; Zhong, H.; Schwartz, K.; Barry, V.; Mikels-Vigdal, A.; Karpinski, S.; Kornyeyev, D.; et al. Targeting LOXL2 for Cardiac Interstitial Fibrosis and Heart Failure Treatment. Nat. Commun. 2016, 7, 13710. [Google Scholar] [CrossRef]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef]
- Lecaille, F.; Brömme, D.; Lalmanach, G. Biochemical Properties and Regulation of Cathepsin K Activity. Biochimie 2008, 90, 208–226. [Google Scholar] [CrossRef]
- Quintanilla-Dieck, M.J.; Codriansky, K.; Keady, M.; Bhawan, J.; Rünger, T.M. Expression and Regulation of Cathepsin K in Skin Fibroblasts. Exp. Dermatol. 2009, 18, 596–602. [Google Scholar] [CrossRef]
- Rünger, T.M.; Quintanilla-Dieck, M.J.; Bhawan, J. Role of Cathepsin K in the Turnover of the Dermal Extracellular Matrix during Scar Formation. J. Invest. Dermatol. 2007, 127, 293–297. [Google Scholar] [CrossRef] [Green Version]
- Marques, A.R.A.; Di Spiezio, A.; Thießen, N.; Schmidt, L.; Grötzinger, J.; Lüllmann-Rauch, R.; Damme, M.; Storck, S.E.; Pietrzik, C.U.; Fogh, J.; et al. Enzyme Replacement Therapy with Recombinant Pro-CTSD (Cathepsin D) Corrects Defective Proteolysis and Autophagy in Neuronal Ceroid Lipofuscinosis. Autophagy 2020, 16, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Gu, L.; Li, A.; Lin, J.; Gan, Y.; He, C.; Xiao, R.; Liao, J.; Li, Y.; Guo, S. Knockdown of SOX9 Alleviates Tracheal Fibrosis through the Wnt/β-Catenin Signaling Pathway. J. Mol. Med. Berl. Ger. 2022, 100, 1659–1670. [Google Scholar] [CrossRef]
- Wang, C.; Deng, J.; Deng, H.; Kang, Z.; Huang, Z.; Ding, Z.; Dong, L.; Chen, J.; Zhang, J.; Zang, Y. A Novel Sox9/LncRNA H19 Axis Contributes to Hepatocyte Death and Liver Fibrosis. Toxicol. Sci. 2020, 177, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Scharf, G.M.; Kilian, K.; Cordero, J.; Wang, Y.; Grund, A.; Hofmann, M.; Froese, N.; Wang, X.; Kispert, A.; Kist, R.; et al. Inactivation of Sox9 in Fibroblasts Reduces Cardiac Fibrosis and Inflammation. JCI Insight 2019, 5, e126721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, S.; Jokl, E.; Pritchett, J.; Martin, K.; Su, K.; Simpson, K.; Birchall, L.; Mullan, A.F.; Athwal, V.S.; Doherty, D.T.; et al. SOX9 Is Required for Kidney Fibrosis and Activates NAV3 to Drive Renal Myofibroblast Function. Sci. Signal. 2021, 14, eabb4282. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cai, H.; Deng, J.; Tu, X.; Sun, Y.; Huang, Z.; Ding, Z.; Dong, L.; Chen, J.; Zang, Y.; et al. TGF-β-Mediated Upregulation of Sox9 in Fibroblast Promotes Renal Fibrosis. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 520–532. [Google Scholar] [CrossRef]
- Roberson, E.D.O.; Carns, M.; Cao, L.; Aren, K.; Goldberg, I.A.; Morales-Heil, D.J.; Korman, B.D.; Atkinson, J.P.; Varga, J. Alterations of the Primary Cilia Gene SPAG17 and SOX9 Locus Noncoding RNAs Identified by RNA-Sequencing Analysis in Patients With Systemic Sclerosis. Arthritis Rheumatol. Hoboken NJ 2023, 75, 108–119. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, Y.; Hou, B.; Wang, Y.; Deng, D.; Fu, Z.; Xu, Z. A SOX9-AS1/MiR-5590-3p/SOX9 Positive Feedback Loop Drives Tumor Growth and Metastasis in Hepatocellular Carcinoma through the Wnt/β-Catenin Pathway. Mol. Oncol. 2019, 13, 2194–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Cancio, M.; Audi, L.; Carrascosa, A.; Toran, N.; Andaluz, P.; Esteban, C.; Granada, M.L. Vitamin D and Growth Hormone Regulate Growth Hormone/Insulin-like Growth Factor (GH-IGF) Axis Gene Expression in Human Fetal Epiphyseal Chondrocytes. Growth Horm. IGF Res. Off. J. Growth Horm. Res. Soc. Int. IGF Res. Soc. 2009, 19, 232–237. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, B.; Zhao, J.; Pan, W.; Xu, J.; Chen, S. IGF-I Induces Adipose Derived Mesenchymal Cell Chondrogenic Differentiation in Vitro and Enhances Chondrogenesis in Vivo. Vitr. Cell. Dev. Biol. Anim. 2016, 52, 356–364. [Google Scholar] [CrossRef]
- Hamamura, K.; Zhang, P.; Yokota, H. IGF2-Driven PI3 Kinase and TGFβ Signaling Pathways in Chondrogenesis. Cell Biol. Int. 2008, 32, 1238–1246. [Google Scholar] [CrossRef] [Green Version]
- Turvey, S.J.; McPhillie, M.J.; Kearney, M.T.; Muench, S.P.; Simmons, K.J.; Fishwick, C.W.G. Recent Developments in the Structural Characterisation of the IR and IGF1R: Implications for the Design of IR–IGF1R Hybrid Receptor Modulators. RSC Med. Chem. 2022, 13, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Slaaby, R. Specific Insulin/IGF1 Hybrid Receptor Activation Assay Reveals IGF1 as a More Potent Ligand than Insulin. Sci. Rep. 2015, 5, 7911. [Google Scholar] [CrossRef] [Green Version]
- Federici, M.; Zucaro, L.; Porzio, O.; Massoud, R.; Borboni, P.; Lauro, D.; Sesti, G. Increased Expression of Insulin/Insulin-like Growth Factor-I Hybrid Receptors in Skeletal Muscle of Noninsulin-Dependent Diabetes Mellitus Subjects. J. Clin. Invest. 1996, 98, 2887–2893. [Google Scholar] [CrossRef] [Green Version]
- Pandini, G.; Vigneri, R.; Costantino, A.; Frasca, F.; Ippolito, A.; Fujita-Yamaguchi, Y.; Siddle, K.; Goldfine, I.D.; Belfiore, A. Insulin and Insulin-like Growth Factor-I (IGF-I) Receptor Overexpression in Breast Cancers Leads to Insulin/IGF-I Hybrid Receptor Overexpression: Evidence for a Second Mechanism of IGF-I Signaling. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 1935–1944. [Google Scholar]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin Receptor Isoforms and Insulin Receptor/Insulin-Like Growth Factor Receptor Hybrids in Physiology and Disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Danopoulos, S.; Alonso, I.; Thornton, M.E.; Grubbs, B.H.; Bellusci, S.; Warburton, D.; Al Alam, D. Human Lung Branching Morphogenesis Is Orchestrated by the Spatiotemporal Distribution of ACTA2, SOX2, and SOX9. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L144–L149. [Google Scholar] [CrossRef]
- Silva, D.; Venihaki, M.; Guo, W.-H.; Lopez, M.F. Igf2 Deficiency Results in Delayed Lung Development at the End of Gestation. Endocrinology 2006, 147, 5584–5591. [Google Scholar] [CrossRef]
- Song, H.; Park, K.-H. Regulation and Function of SOX9 during Cartilage Development and Regeneration. Semin. Cancer Biol. 2020, 67, 12–23. [Google Scholar] [CrossRef]
- Liu, C.-F.; Lefebvre, V. The Transcription Factors SOX9 and SOX5/SOX6 Cooperate Genome-Wide through Super-Enhancers to Drive Chondrogenesis. Nucleic Acids Res. 2015, 43, 8183–8203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raftery, R.M.; Gonzalez Vazquez, A.G.; Chen, G.; O’Brien, F.J. Activation of the SOX-5, SOX-6, and SOX-9 Trio of Transcription Factors Using a Gene-Activated Scaffold Stimulates Mesenchymal Stromal Cell Chondrogenesis and Inhibits Endochondral Ossification. Adv. Healthc. Mater. 2020, 9, 1901827. [Google Scholar] [CrossRef]
- Coricor, G.; Serra, R. TGF-β Regulates Phosphorylation and Stabilization of Sox9 Protein in Chondrocytes through P38 and Smad Dependent Mechanisms. Sci. Rep. 2016, 6, 38616. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y.; Tsuda, M.; Takahashi, S.; Taniguchi, N.; Esteban, C.R.; Zemmyo, M.; Furumatsu, T.; Lotz, M.; Izpisúa Belmonte, J.C.; Asahara, H. Transcriptional Coactivator PGC-1alpha Regulates Chondrogenesis via Association with Sox9. Proc. Natl. Acad. Sci. USA 2005, 102, 2414–2419. [Google Scholar] [CrossRef]
- Chavez, R.; Coricor, G.; Perez, J.; Seo, H.-S.; Serra, R. SOX9 Protein Is Stabilized by TGF-β and Regulates PAPSS2 MRNA Expression in Chondrocytes. Osteoarthr. Cartil. 2017, 25, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, Y.; Zhao, S.; Wang, X.; Lu, K.; Xiao, H. Effect of Sox9 on TGF-Β1-Mediated Atrial Fibrosis. Acta Biochim. Biophys. Sin. 2021, 53, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Saito, D.; Yamada, H.; Ishisaki, A.; Kamo, M. TGF-Β1 Induces N-Cadherin Expression by Upregulating Sox9 Expression and Promoting Its Nuclear Translocation in Human Oral Squamous Cell Carcinoma Cells. Oncol. Lett. 2020, 20, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.; Liu, N.; Sun, Z.; Jiang, Y.; Jiang, T.; Xv, M.; Jia, L.; Tu, Y.; Wang, L. TGF-β Signaling Promotes Glioma Progression Through Stabilizing Sox9. Front. Immunol. 2021, 11, 592080. [Google Scholar] [CrossRef]
- Zhang, S.; Che, D.; Yang, F.; Chi, C.; Meng, H.; Shen, J.; Qi, L.; Liu, F.; Lv, L.; Li, Y.; et al. Tumor-Associated Macrophages Promote Tumor Metastasis via the TGF-β/SOX9 Axis in Non-Small Cell Lung Cancer. Oncotarget 2017, 8, 99801–99815. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Xu, X.; Chen, X.; Hao, C.; Ji, Z.; Zuo, P.; Yang, M.; Ma, G.; Li, Y. Upregulation of Key Genes Eln and Tgfb3 Were Associated with the Severity of Cardiac Hypertrophy. BMC Genom. 2022, 23, 592. [Google Scholar] [CrossRef]
- Maulik, S.K.; Mishra, S. Hypertrophy to Failure: What Goes Wrong with the Fibers of the Heart? Indian Heart, J. 2015, 67, 66–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athwal, V.S.; Pritchett, J.; Martin, K.; Llewellyn, J.; Scott, J.; Harvey, E.; Zaitoun, A.M.; Mullan, A.F.; Zeef, L.A.H.; Friedman, S.L.; et al. SOX9 Regulated Matrix Proteins Are Increased in Patients Serum and Correlate with Severity of Liver Fibrosis. Sci. Rep. 2018, 8, 17905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal Lung Development and Cleft Palate in Mice Lacking TGF–Β3 Indicates Defects of Epithelial–Mesenchymal Interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef]
- Herrera, J.A.; Dingle, L.; Montero, M.A.; Venkateswaran, R.V.; Blaikley, J.F.; Lawless, C.; Schwartz, M.A. The UIP/IPF Fibroblastic Focus Is a Collagen Biosynthesis Factory Embedded in a Distinct Extracellular Matrix. JCI Insight 2022, 7, e156115. [Google Scholar] [CrossRef]
- Evans, P.; Etherington, D.J. Characterisation of Cathepsin B and Collagenolytic Cathepsin from Human Placenta. Eur. J. Biochem. 1978, 83, 87–97. [Google Scholar] [CrossRef]
- Bühling, F.; Waldburg, N.; Reisenauer, A.; Heimburg, A.; Golpon, H.; Welte, T. Lysosomal Cysteine Proteases in the Lung: Role in Protein Processing and Immunoregulation. Eur. Respir. J. 2004, 23, 620–628. [Google Scholar] [CrossRef] [Green Version]
- Kafienah, W.; Brömme, D.; Buttle, D.J.; Croucher, L.J.; Hollander, A.P. Human Cathepsin K Cleaves Native Type I and II Collagens at the N-Terminal End of the Triple Helix. Biochem. J. 1998, 331, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Korenč, M.; Lenarčič, B.; Novinec, M. Human Cathepsin L, a Papain-like Collagenase without Proline Specificity. FEBS J. 2015, 282, 4328–4340. [Google Scholar] [CrossRef]
- Rasmussen, D.G.K.; Sand, J.M.B.; Karsdal, M.A.; Genovese, F. Development of a Novel Enzyme-Linked Immunosorbent Assay Targeting a Neo-Epitope Generated by Cathepsin-Mediated Turnover of Type III Collagen and Its Application in Chronic Obstructive Pulmonary Disease. PLoS ONE 2017, 12, e0170023. [Google Scholar] [CrossRef] [Green Version]
- She, Z.-Y.; Yang, W.-X. SOX Family Transcription Factors Involved in Diverse Cellular Events during Development. Eur. J. Cell Biol. 2015, 94, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Smits, P.; Li, P.; Mandel, J.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B.; Lefebvre, V. The Transcription Factors L-Sox5 and Sox6 Are Essential for Cartilage Formation. Dev. Cell 2001, 1, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Shakhova, O.; Cheng, P.; Mishra, P.J.; Zingg, D.; Schaefer, S.M.; Debbache, J.; Häusel, J.; Matter, C.; Guo, T.; Davis, S.; et al. Antagonistic Cross-Regulation between Sox9 and Sox10 Controls an Anti-Tumorigenic Program in Melanoma. PLoS Genet. 2015, 11, e1004877. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Liang, R.; Liu, C.; Liu, J.A.; Cheung, M.P.L.; Liu, X.; Man, O.Y.; Guan, X.-Y.; Lung, H.L.; Cheung, M. SOX9 Is a Dose-Dependent Metastatic Fate Determinant in Melanoma. J. Exp. Clin. Cancer Res. CR 2019, 38, 17. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.-F.; Kuo, S.-H. Inhibition of SOX9 May Be an Effective Target for Increasing Radiosensitivity in Gastrointestinal Cancer. Ann. Transl. Med. 2017, 5, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Shepherd, J.; Zhao, D.; Bollu, L.R.; Tahaney, W.M.; Hill, J.; Zhang, Y.; Mazumdar, A.; Brown, P.H. SOX9 Is Essential for Triple-Negative Breast Cancer Cell Survival and Metastasis. Mol. Cancer Res. 2020, 18, 1825–1838. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Sahoo, R.K.; Biswal, B.K. SOX9 as an Emerging Target for Anticancer Drugs and a Prognostic Biomarker for Cancer Drug Resistance. Drug Discov. Today 2022, 27, 2541–2550. [Google Scholar] [CrossRef]
- Holly, J.M.P.; Biernacka, K.; Perks, C.M. The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer. Cells 2019, 8, 1207. [Google Scholar] [CrossRef] [Green Version]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waldrep, K.M.; Rodgers, J.I.; Garrett, S.M.; Wolf, B.J.; Feghali-Bostwick, C.A. The Role of SOX9 in IGF-II-Mediated Pulmonary Fibrosis. Int. J. Mol. Sci. 2023, 24, 11234. https://doi.org/10.3390/ijms241411234
Waldrep KM, Rodgers JI, Garrett SM, Wolf BJ, Feghali-Bostwick CA. The Role of SOX9 in IGF-II-Mediated Pulmonary Fibrosis. International Journal of Molecular Sciences. 2023; 24(14):11234. https://doi.org/10.3390/ijms241411234
Chicago/Turabian StyleWaldrep, Kristy M., Jessalyn I. Rodgers, Sara M. Garrett, Bethany J. Wolf, and Carol A. Feghali-Bostwick. 2023. "The Role of SOX9 in IGF-II-Mediated Pulmonary Fibrosis" International Journal of Molecular Sciences 24, no. 14: 11234. https://doi.org/10.3390/ijms241411234
APA StyleWaldrep, K. M., Rodgers, J. I., Garrett, S. M., Wolf, B. J., & Feghali-Bostwick, C. A. (2023). The Role of SOX9 in IGF-II-Mediated Pulmonary Fibrosis. International Journal of Molecular Sciences, 24(14), 11234. https://doi.org/10.3390/ijms241411234