
Identification of New Antiseizure Medication Candidates in Preclinical Animal Studies

Abstract
:1. Introduction
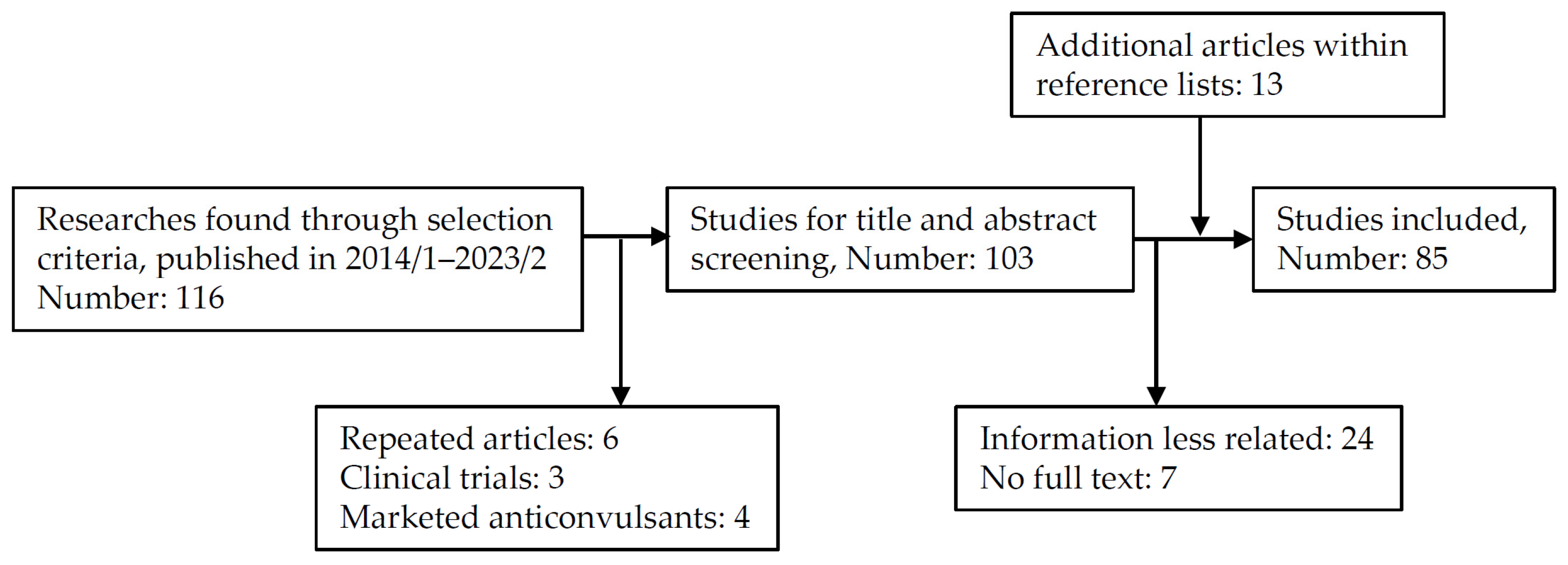
2. Scope of Review
3. Animal Models Used for ASM Screening and Discovery
3.1. Maximal Electroshock Seizure (MES) Model
3.2. Subcutaneous Pentylenetetrazole-Induced Seizure Model
3.3. Kindling Model
3.4. Animal Models for Pharmacoresistant Epilepsy
3.4.1. 6 Hz Psychomotor Seizure Model
3.4.2. Lamotrigine (LTG)-Resistant Amygdala-Kindled Seizure Model
3.4.3. Post-Status Epilepticus Seizure Model
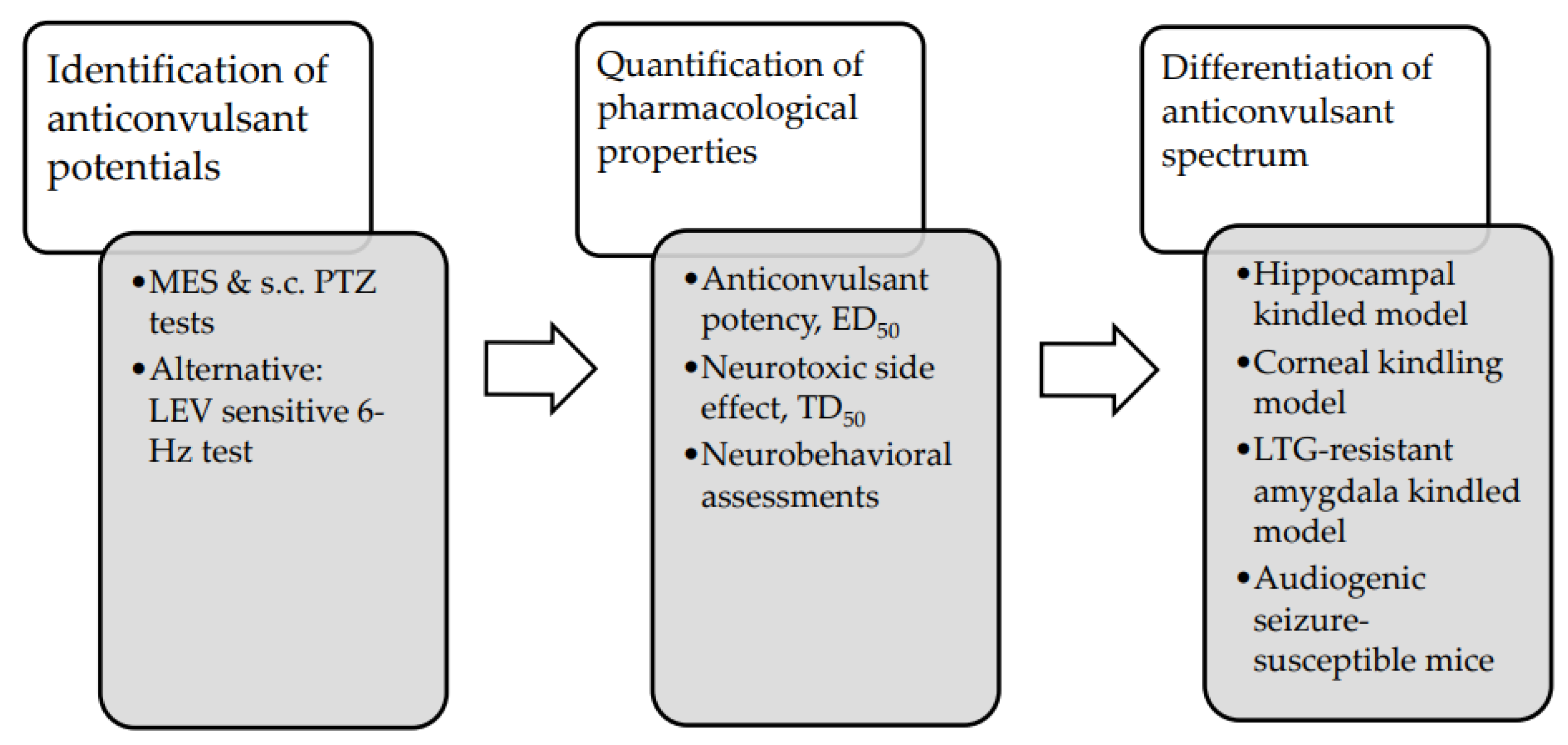
3.5. Evaluation of Antiseizure Potentials of Novel Compounds in Preclinical Animal Studies
3.6. The Potential Benefits and Limitations of Animal Models of Induced Seizures
4. New Antiseizure Candidates
4.1. N-benzyl-2-(2,5-dioxopyrrolidin-1-yl)propanamide (AS-1)
4.2. 2-(2,5-dioxopyrrolidin-1-yl) propanamide derivative (C-11)
4.3. 2-(2′-fluoro-[1,1′-biphenyl]-2-yl)acetamide (DSP-0565)
4.4. S(+)-(2E)-N-(2-hydroxypropyl)-3-phenylprop-2-enamide (KM-568)
4.5. 5-(3-chlorophenyl)-4-hexyl-2,4-dihydro-3H-1,2,4-triazole-3-thione (TP-315)
4.6. Neuroactive Steroid (SGE-516)
4.7. Methylsulfonyl Phenyl Derivative (MTL-1)
4.8. Sebacic Acid
4.9. (S)-3-amino-4-(difluoromethylenyl)-cyclopent-1-ene-1-carboxylic Acid (OV329)
4.10. 5-(8-ethynyl-6-(pyridin-2-yl)-4H-benzo[f] imidazo [1,5-a][1,4]diazepin-3-yl)oxazole (KRM-II-81)
5. The Potentials of Novel ASM Discovery to Surmount Drug Resistance
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADD | After-discharge duration |
| ADT | After-discharge threshold |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| ASM | Antiseizure medication |
| BBB | Blood–brain barrier |
| COX | Cyclooxygenase |
| CYP | Cytochrome P450 |
| DRE | Drug-resistant epilepsy |
| ED50 | Median effective dose |
| ETX | Ethosuximide |
| GABA | γ-Aminobutyric acid |
| GABA-AT | GABA aminotransferase |
| GAERS | Genetic absence epilepsy rat from Strasbourg |
| GTC | Generalized tonic–clonic |
| i.p. | Intraperitoneal |
| i.v. | Intravenous |
| LCS | Lacosamide |
| LEV | Levetiracetam |
| LTG | Lamotrigine |
| MES | Maximal electroshock seizure |
| nAChR | Nicotinic acetylcholine receptor |
| NMDA | N-methyl-D-aspartate |
| PAM | Positive allosteric modulator |
| P-gp | P-glycoprotein |
| PHT | Phenytoin |
| PTZ | pentylenetetrazole |
| SA | Sebacic acid |
| s.c. | Subcutaneous |
| SE | Status epilepticus |
| SRS | Spontaneous recurrent seizure |
| TD50 | Median toxic dose |
| TLE | Temporal lobe epilepsy |
| VGSC | Voltage-gated sodium channel |
| VPA | Valproic acid |
References
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Estimation of the burden of active and life-time epilepsy: A meta-analytic approach. Epilepsia 2010, 51, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E. The epidemiology of epilepsy. Neuroepidemiology 2020, 54, 185–191. [Google Scholar] [CrossRef]
- Rocca, W.A.; Savettieri, G.; Anderson, D.; Meneghini, F.; Grigoletto, F.; Morgante, L.; Reggio, A.; Salemi, G.; Patti, F.; Di Perri, R. Door-to-door prevalence survey of epilepsy in three Sicilian municipalities. Neuroepidemiology 2001, 20, 237–241. [Google Scholar] [CrossRef]
- Brodtkorb, E.; Sjaastad, O. Epilepsy prevalence by individual interview in a Norwegian community. Seizure 2008, 17, 646–650. [Google Scholar] [CrossRef]
- Sander, J.W. The epidemiology of epilepsy revisited. Curr. Opin. Neurol. 2003, 16, 165–170. [Google Scholar] [CrossRef]
- Giussani, G.; Cricelli, C.; Mazzoleni, F.; Cricelli, I.; Pasqua, A.; Pecchioli, S.; Lapi, F.; Beghi, E. Prevalence and incidence of epilepsy in Italy based on a nationwide database. Neuroepidemiology 2015, 43, 228–232. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Sultana, B.; Panzini, M.-A.; Carpentier, A.V.; Comtois, J.; Rioux, B.; Gore, G.; Bauer, P.R.; Kwon, C.-S.; Jetté, N.; Josephson, C.B. Incidence and prevalence of drug-resistant epilepsy: A systematic review and meta-analysis. Neurology 2021, 96, 805–817. [Google Scholar] [CrossRef]
- Kalilani, L.; Sun, X.; Pelgrims, B.; Noack-Rink, M.; Villanueva, V. The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia 2018, 59, 2179–2193. [Google Scholar] [CrossRef] [PubMed]
- Remy, S.; Beck, H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006, 129, 18–35. [Google Scholar] [CrossRef]
- Löscher, W.; Potschka, H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat. Rev. Neurosci. 2005, 6, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Hartz, A.M.; Bauer, B. Drug-resistant epilepsy: Multiple hypotheses, few answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef]
- Broekaart, D.W.; Anink, J.J.; Baayen, J.C.; Idema, S.; De Vries, H.E.; Aronica, E.; Gorter, J.A.; Van Vliet, E.A. Activation of the innate immune system is evident throughout epileptogenesis and is associated with blood-brain barrier dysfunction and seizure progression. Epilepsia 2018, 59, 1931–1944. [Google Scholar] [CrossRef]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef]
- Löscher, W.; Friedman, A. Structural, molecular, and functional alterations of the blood-brain barrier during epileptogenesis and epilepsy: A cause, consequence, or both? Int. J. Mol. Sci. 2020, 21, 591. [Google Scholar] [CrossRef]
- Fang, M.; Xi, Z.-Q.; Wu, Y.; Wang, X.-F. A new hypothesis of drug refractory epilepsy: Neural network hypothesis. Med. Hypotheses 2011, 76, 871–876. [Google Scholar] [CrossRef]
- Löscher, W. The pharmacokinetics of antiepileptic drugs in rats: Consequences for maintaining effective drug levels during prolonged drug administration in rat models of epilepsy. Epilepsia 2007, 48, 1245–1258. [Google Scholar] [CrossRef]
- Klitgaard, H.; Matagne, A.; Lamberty, Y. Use of epileptic animals for adverse effect testing. Epilepsy Res. 2002, 50, 55–65. [Google Scholar] [CrossRef]
- Löscher, W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 2011, 20, 359–368. [Google Scholar] [CrossRef]
- Schmidt, D.; Rogawski, M.A. New strategies for the identification of drugs to prevent the development or progression of epilepsy. Epilepsy Res. 2002, 50, 71–78. [Google Scholar] [CrossRef]
- Barker-Haliski, M.; White, H.S. Validated animal models for antiseizure drug (ASD) discovery: Advantages and potential pitfalls in ASD screening. Neuropharmacology 2020, 167, 107750. [Google Scholar] [CrossRef]
- Stables, J.P.; Bertram, E.H.; White, H.S.; Coulter, D.A.; Dichter, M.A.; Jacobs, M.P.; Loscher, W.; Lowenstein, D.H.; Moshe, S.L.; Noebels, J.L. Models for epilepsy and epileptogenesis: Report from the NIH workshop, Bethesda, Maryland. Epilepsia 2002, 43, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Barker-Haliski, M.; Knox, K.; Zierath, D.; Koneval, Z.; Metcalf, C.; Wilcox, K.S.; White, H.S. Development of an antiepileptogenesis drug screening platform: Effects of everolimus and phenobarbital. Epilepsia 2021, 62, 1677–1688. [Google Scholar] [CrossRef] [PubMed]
- Castel-Branco, M.; Alves, G.; Figueiredo, I.; Falcão, A.; Caramona, M. The maximal electroshock seizure (MES) model in the preclinical assessment of potential new antiepileptic drugs. Methods Find. Exp. Clin. Pharmacol. 2009, 31, 101–106. [Google Scholar] [CrossRef]
- Mahendran, G.; Thamotharan, G.; Sengottuvelu, S.; Bai, V.N. Evaluation of anticonvulsant, sedative, anxiolytic, and phytochemical profile of the methanol extract from the aerial parts of Swertia corymbosa (Griseb.) wight ex CB Clarke. BioMed Res. Int. 2014, 2014, 542385. [Google Scholar] [CrossRef]
- Litchfield, J.J.; Wilcoxon, F. A simplified method of evaluating dose-effect experiments. J. Pharmacol. Exp. Ther. 1949, 96, 99–113. [Google Scholar]
- Krall, R.; Penry, J.; White, B.; Kupferberg, H.; Swinyard, E. Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia 1978, 19, 409–428. [Google Scholar] [CrossRef]
- Shorvon, S.D.; Bermejo, P.E.; Gibbs, A.A.; Huberfeld, G.; Kälviäinen, R. Antiepileptic drug treatment of generalized tonic–clonic seizures: An evaluation of regulatory data and five criteria for drug selection. Epilepsy Behav. 2018, 82, 91–103. [Google Scholar] [CrossRef]
- Löscher, W. Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res. 2016, 126, 157–184. [Google Scholar] [CrossRef]
- Löscher, W.; Hönack, D. Profile of ucb L059, a novel anticonvulsant drug, in models of partial and generalized epilepsy in mice and rats. Eur. J. Pharmacol. 1993, 232, 147–158. [Google Scholar] [CrossRef]
- Rheims, S.; Ryvlin, P. Pharmacotherapy for tonic–clonic seizures. Expert Opin. Pharmacother. 2014, 15, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Rogawski, M.A.; Löscher, W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 2004, 5, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Gunia-Krzyżak, A.; Żesławska, E.; Słoczyńska, K.; Żelaszczyk, D.; Sowa, A.; Koczurkiewicz-Adamczyk, P.; Popiół, J.; Nitek, W.; Pękala, E.; Marona, H. S(+)-(2E)-N-(2-Hydroxypropyl)-3-Phenylprop-2-Enamide (KM-568): A Novel Cinnamamide Derivative with Anticonvulsant Activity in Animal Models of Seizures and Epilepsy. Int. J. Mol. Sci. 2020, 21, 4372. [Google Scholar] [CrossRef] [PubMed]
- Everett, G.M.; Richards, R.K. Comparative anticonvulsive action of 3,5,5-trimethyloxazolidine-2,4-dione (Tridione), dilantin and phenobarbital. J. Pharmacol. Exp. Ther. 1944, 81, 402–407. [Google Scholar] [CrossRef]
- Lennox, W.G. The petit mal epilepsies: Their treatment with tridione. J. Am. Med. Assoc. 1945, 129, 1069–1074. [Google Scholar] [CrossRef]
- Chen, G.; Portman, R.; Ensor, C.R.; Bratton, A.C. The anticonvulsant activity of α-phenyl succinimides. J. Pharmacol. Exp. Ther. 1951, 103, 54–61. [Google Scholar]
- Bazyan, A.; Van Luijtelaar, G. Neurochemical and behavioral features in genetic absence epilepsy and in acutely induced absence seizures. Int. Sch. Res. Not. 2013, 2013, 875834. [Google Scholar] [CrossRef]
- Coenen, A.; Blezer, E.; Van Luijtelaar, E. Effects of the GABA-uptake inhibitor tiagabine on electroencephalogram, spike-wave discharges and behaviour of rats. Epilepsy Res. 1995, 21, 89–94. [Google Scholar] [CrossRef]
- Klitgaard, H.; Matagne, A.; Gobert, J.; Wülfert, E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur. J. Pharmacol. 1998, 353, 191–206. [Google Scholar] [CrossRef]
- Löscher, W.; Schmidt, D. Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epilepsy Res. 1988, 2, 145–181. [Google Scholar] [CrossRef]
- Goddard, G.V.; McIntyre, D.C.; Leech, C.K. A permanent change in brain function resulting from daily electrical stimulation. Exp. Neurol. 1969, 25, 295–330. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.; White, H.S. Key factors in the discovery and development of new antiepileptic drugs. Nat. Rev. Drug Discov. 2010, 9, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Lothman, E.W.; Salerno, R.A.; Perlin, J.B.; Kaiser, D.L. Screening and characterization of antiepileptic drugs with rapidly recurring hippocampal seizures in rats. Epilepsy Res. 1988, 2, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A. Pentylenetetrazol (PTZ) kindling model of epilepsy. Curr. Protoc. Neurosci. 2012, 58, 9.37.1–9.37.12. [Google Scholar] [CrossRef]
- Racine, R.J. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Sato, M.; Racine, R.; McIntyre, D. Kindling: Basic mechanisms and clinical validity. Electroencephalogr. Clin. Neurophysiol. 1990, 76, 459–472. [Google Scholar] [CrossRef]
- Meeker, S.; Beckman, M.; Knox, K.M.; Treuting, P.M.; Barker-Haliski, M. Repeated intraperitoneal administration of low-concentration methylcellulose leads to systemic histologic lesions without loss of preclinical phenotype. J. Pharmacol. Exp. Ther. 2019, 371, 25–35. [Google Scholar] [CrossRef]
- Barker-Haliski, M.L.; Vanegas, F.; Mau, M.J.; Underwood, T.K.; White, H.S. Acute cognitive impact of antiseizure drugs in naive rodents and corneal-kindled mice. Epilepsia 2016, 57, 1386–1397. [Google Scholar] [CrossRef]
- Möller, C.; Wolf, F.; van Dijk, R.M.; Di Liberto, V.; Russmann, V.; Keck, M.; Palme, R.; Hellweg, R.; Gass, P.; Otzdorff, C. Toward evidence-based severity assessment in rat models with repeated seizures: I. Electrical kindling. Epilepsia 2018, 59, 765–777. [Google Scholar] [CrossRef]
- Barton, M.E.; Klein, B.D.; Wolf, H.H.; White, H.S. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001, 47, 217–227. [Google Scholar] [CrossRef]
- Rapacz, A.; Głuch-Lutwin, M.; Mordyl, B.; Filipek, B.; Abram, M.; Kamiński, K. Evaluation of anticonvulsant and analgesic activity of new hybrid compounds derived from N-phenyl-2-(2,5-dioxopyrrolidin-1-yl)-propanamides and –butanamides. Epilepsy Res. 2018, 143, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, K.; Kaminski, R.M. Genetic background of mice strongly influences treatment resistance in the 6 Hz seizure model. Epilepsia 2015, 56, 310–318. [Google Scholar] [CrossRef]
- Potschka, H. Animal models of drug-resistant epilepsy. Epileptic Disord. 2012, 14, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Postma, T.; Krupp, E.; Li, X.-L.; Post, R.M.; Weiss, S. Lamotrigine treatment during amygdala-kindled seizure development fails to inhibit seizures and diminishes subsequent anticonvulsant efficacy. Epilepsia 2000, 41, 1514–1521. [Google Scholar] [PubMed]
- Srivastava, A.K.; White, H.S. Carbamazepine, but not valproate, displays pharmacoresistance in lamotrigine-resistant amygdala kindled rats. Epilepsy Res. 2013, 104, 26–34. [Google Scholar] [CrossRef]
- Löscher, W.; Schmidt, D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia 2006, 47, 1253–1284. [Google Scholar] [CrossRef]
- Bertram, E.H. Temporal lobe epilepsy: Where do the seizures really begin? Epilepsy Behav. 2009, 14, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Stephen, L.J.; Kwan, P.; Brodie, M.J. Does the cause of localisation-related epilepsy influence the response to antiepileptic drug treatment? Epilepsia 2001, 42, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Lothman, E.W.; Bertram, E.H., III. Epileptogenic effects of status epilepticus. Epilepsia 1993, 34, S59–S70. [Google Scholar] [CrossRef]
- Mathern, G.W.; Adelson, P.D.; Cahan, L.D.; Leite, J.P. Hippocampal neuron damage in human epilepsy: Meyer’s hypothesis revisited. Prog. Brain Res. 2002, 135, 237–251. [Google Scholar]
- Glien, M.; Brandt, C.; Potschka, H.; Löscher, W. Effects of the novel antiepileptic drug levetiracetam on spontaneous recurrent seizures in the rat pilocarpine model of temporal lobe epilepsy. Epilepsia 2002, 43, 350–357. [Google Scholar] [CrossRef]
- Brandt, C.; Volk, H.A.; Löscher, W. Striking differences in individual anticonvulsant response to phenobarbital in rats with spontaneous seizures after status epilepticus. Epilepsia 2004, 45, 1488–1497. [Google Scholar] [CrossRef]
- Bethmann, K.; Brandt, C.; Löscher, W. Resistance to phenobarbital extends to phenytoin in a rat model of temporal lobe epilepsy. Epilepsia 2007, 48, 816–826. [Google Scholar] [CrossRef]
- Kamiński, K.; Rapacz, A.; Filipek, B.; Obniska, J. Design, synthesis and anticonvulsant activity of new hybrid compounds derived from N-phenyl-2-(2,5-dioxopyrrolidin-1-yl)-propanamides and-butanamides. Bioorganic Med. Chem. 2016, 24, 2938–2946. [Google Scholar] [CrossRef]
- Kamiński, K.; Rapacz, A.; Łuszczki, J.J.; Latacz, G.; Obniska, J.; Kieć-Kononowicz, K.; Filipek, B. Design, synthesis and biological evaluation of new hybrid anticonvulsants derived from N-benzyl-2-(2,5-dioxopyrrolidin-1-yl) propanamide and 2-(2,5-dioxopyrrolidin-1-yl) butanamide derivatives. Bioorganic Med. Chem. 2015, 23, 2548–2561. [Google Scholar] [CrossRef]
- Rapacz, A.; Kamiński, K.; Obniska, J.; Koczurkiewicz, P.; Pękala, E.; Filipek, B. Analgesic, antiallodynic, and anticonvulsant activity of novel hybrid molecules derived from N-benzyl-2-(2,5-dioxopyrrolidin-1-yl) propanamide and 2-(2,5-dioxopyrrolidin-1-yl) butanamide in animal models of pain and epilepsy. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 567–579. [Google Scholar] [CrossRef]
- Abram, M.; Jakubiec, M.; Reeb, K.; Cheng, M.H.; Gedschold, R.; Rapacz, A.; Mogilski, S.; Socała, K.; Nieoczym, D.; Szafarz, M. Discovery of (R)-N-Benzyl-2-(2,5-dioxopyrrolidin-1-yl) propanamide [(R)-AS-1], a Novel Orally Bioavailable EAAT2 Modulator with Drug-like Properties and Potent Antiseizure Activity In Vivo. J. Med. Chem. 2022, 65, 11703–11725. [Google Scholar] [CrossRef]
- Kamiński, K.; Socała, K.; Zagaja, M.; Andres-Mach, M.; Abram, M.; Jakubiec, M.; Pieróg, M.; Nieoczym, D.; Rapacz, A.; Gawel, K. N-Benzyl-(2,5-dioxopyrrolidin-1-yl) propanamide (AS-1) with Hybrid Structure as a Candidate for a Broad-Spectrum Antiepileptic Drug. Neurotherapeutics 2020, 17, 309–328. [Google Scholar] [CrossRef]
- Abram, M.; Zagaja, M.; Mogilski, S.; Andres-Mach, M.; Latacz, G.; Baś, S.; Łuszczki, J.J.; Kieć-Kononowicz, K.; Kaminski, K. Multifunctional hybrid compounds derived from 2-(2,5-dioxopyrrolidin-1-yl)-3-methoxypropanamides with anticonvulsant and antinociceptive properties. J. Med. Chem. 2017, 60, 8565–8579. [Google Scholar] [CrossRef]
- Góra, M.; Czopek, A.; Rapacz, A.; Gębska, A.; Wójcik-Pszczoła, K.; Pękala, E.; Kamiński, K. Synthesis, Anticonvulsant, and Antinociceptive Activity of New 3-(2-Chlorophenyl)-and 3-(3-Chlorophenyl)-2,5-dioxo-pyrrolidin-1-yl-acetamides. Molecules 2021, 26, 1564. [Google Scholar] [CrossRef]
- Góra, M.; Czopek, A.; Rapacz, A.; Giza, A.; Koczurkiewicz-Adamczyk, P.; Pękala, E.; Obniska, J.; Kamiński, K. Design, Synthesis and Biological Activity of New Amides Derived from 3-Benzhydryl and 3-sec-Butyl-2,5-dioxo-pyrrolidin-1-yl-acetic Acid. ChemMedChem 2021, 16, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Zagaja, M.; Szewczyk, A.; Szala-Rycaj, J.; Raszewski, G.; Chrościńska-Krawczyk, M.; Abram, M.; Kamiński, K.; Andres-Mach, M. C-11, a New Antiepileptic Drug Candidate: Evaluation of the Physicochemical Properties and Impact on the Protective Action of Selected Antiepileptic Drugs in the Mouse Maximal Electroshock-Induced Seizure Model. Molecules 2021, 26, 3144. [Google Scholar] [CrossRef]
- Kamiński, K.; Zagaja, M.; Łuszczki, J.J.; Rapacz, A.; Andres-Mach, M.; Latacz, G.; Kieć-Kononowicz, K. Design, synthesis, and anticonvulsant activity of new hybrid compounds derived from 2-(2,5-dioxopyrrolidin-1-yl) propanamides and 2-(2,5-dioxopyrrolidin-1-yl) butanamides. J. Med. Chem. 2015, 58, 5274–5286. [Google Scholar] [CrossRef]
- Socała, K.; Mogilski, S.; Pieróg, M.; Nieoczym, D.; Abram, M.; Szulczyk, B.; Lubelska, A.; Latacz, G.; Doboszewska, U.; Wlaź, P. KA-11, a novel pyrrolidine-2,5-dione derived broad-spectrum anticonvulsant: Its antiepileptogenic, antinociceptive properties and in vitro characterization. ACS Chem. Neurosci. 2018, 10, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Andres-Mach, M.; Szewczyk, A.; Zagaja, M.; Luszczki, J.; Maj, M.; Rola, R.; Abram, M.; Kaminski, K. Evaluation of the impact of compound C11 a new anticonvulsant candidate on cognitive functions and hippocampal neurogenesis in mouse brain. Neuropharmacology 2020, 163, 107849. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yajima, N.; Kiyoshi, T.; Miura, Y.; Inoue, Y.; Nishimaki, T.; Iwama, S. Identification of 2-(2′-fluoro-[1,1′-biphenyl]-2-yl) acetamide as a Sodium Valproate-like broad spectrum anti-epileptic drug candidate. Bioorganic Med. Chem. Lett. 2019, 29, 138–142. [Google Scholar] [CrossRef]
- Tanaka, T.; Yajima, N.; Kiyoshi, T.; Miura, Y.; Iwama, S. N-alkyl-[1,1′-biphenyl]-2-sulfonamide derivatives as novel broad spectrum anti-epileptic drugs with efficacy equivalent to that of sodium valproate. Bioorganic Med. Chem. Lett. 2017, 27, 4118–4121. [Google Scholar] [CrossRef]
- Gunia-Krzyżak, A.; Pańczyk, K.; Waszkielewicz, A.M.; Marona, H. Cinnamamide Derivatives for central and peripheral nervous system disorders—A review of structure–activity relationships. ChemMedChem 2015, 10, 1302–1325. [Google Scholar] [CrossRef]
- Guan, L.-P.; Wei, C.-X.; Deng, X.-Q.; Sui, X.; Piao, H.-R.; Quan, Z.-S. Synthesis and anticonvulsant activity of N-(2-hydroxyethyl) cinnamamide derivatives. Eur. J. Med. Chem. 2009, 44, 3654–3657. [Google Scholar] [CrossRef]
- Plech, T.; Luszczki, J.J.; Wujec, M.; Flieger, J.; Pizoń, M. Synthesis, characterization and preliminary anticonvulsant evaluation of some 4-alkyl-1,2,4-triazoles. Eur. J. Med. Chem. 2013, 60, 208–215. [Google Scholar] [CrossRef]
- Kaproń, B.; Czarnomysy, R.; Wysokiński, M.; Andrys, R.; Musilek, K.; Angeli, A.; Supuran, C.T.; Plech, T. 1,2,4-Triazole-based anticonvulsant agents with additional ROS scavenging activity are effective in a model of pharmacoresistant epilepsy. J. Enzym. Inhib. Med. Chem. 2020, 35, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Łuszczki, J.J.; Lepiech, J.; Zagaja, M.; Wróblewska-Łuczka, P.; Florek-Łuszczki, M.; Bojar, H.; Walczak, A.; Plech, T. Anticonvulsant and neurotoxic effects of a novel 1,2,4-triazole-3-thione derivative (TPF-34) and its isobolographic interaction profile with classical antiepileptic drugs in mice. Pharmacol. Rep. 2020, 72, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Kaproń, B.; Łuszczki, J.; Paneth, A.; Wujec, M.; Siwek, A.; Karcz, T.; Mordyl, B.; Głuch-Lutwin, M.; Gryboś, A.; Nowak, G. Molecular mechanism of action and safety of 5-(3-chlorophenyl)-4-hexyl-2,4-dihydro-3H-1,2,4-triazole-3-thione-a novel anticonvulsant drug candidate. Int. J. Med. Sci. 2017, 14, 741. [Google Scholar] [CrossRef] [PubMed]
- Makuch-Kocka, A.; Andres-Mach, M.; Zagaja, M.; Śmiech, A.; Pizoń, M.; Flieger, J.; Cielecka-Piontek, J.; Plech, T. Effect of Chronic Administration of 5-(3-chlorophenyl)-4-Hexyl-2,4-Dihydro-3 H-1,2,4-Triazole-3-Thione (TP-315)—A New Anticonvulsant Drug Candidate—On Living Organisms. Int. J. Mol. Sci. 2021, 22, 3358. [Google Scholar] [CrossRef] [PubMed]
- Belelli, D.; Lambert, J.J. Neurosteroids: Endogenous regulators of the GABAA receptor. Nat. Rev. Neurosci. 2005, 6, 565–575. [Google Scholar] [CrossRef]
- Martinez-Botella, G.; Ackley, M.A.; Salituro, F.G.; Doherty, J.J. Natural and synthetic neuroactive steroid modulators of GABAA and NMDA receptors. In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2014; Volume 49, pp. 27–42. [Google Scholar]
- Wafford, K.; Macaulay, A.; Fradley, R.; O’Meara, G.; Reynolds, D.; Rosahl, T. Differentiating the role of γ-aminobutyric acid type A (GABAA) receptor subtypes. Biochem. Soc. Trans. 2004, 32, 553–556. [Google Scholar] [CrossRef]
- Martinez Botella, G.; Salituro, F.G.; Harrison, B.L.; Beresis, R.T.; Bai, Z.; Shen, K.; Belfort, G.M.; Loya, C.M.; Ackley, M.A.; Grossman, S.J. Neuroactive steroids. 1. Positive allosteric modulators of the (γ-Aminobutyric Acid) a receptor: Structure–activity relationships of heterocyclic substitution at C-21. J. Med. Chem. 2015, 58, 3500–3511. [Google Scholar] [CrossRef]
- Hammond, R.S.; Althaus, A.L.; Ackley, M.A.; Maciag, C.; Botella, G.M.; Salituro, F.G.; Robichaud, A.J.; Doherty, J.J. Anticonvulsant profile of the neuroactive steroid, SGE-516, in animal models. Epilepsy Res. 2017, 134, 16–25. [Google Scholar] [CrossRef]
- Althaus, A.L.; McCarren, H.S.; Alqazzaz, A.; Jackson, C.; McDonough, J.H.; Smith, C.D.; Hoffman, E.; Hammond, R.S.; Robichaud, A.J.; Doherty, J.J. The synthetic neuroactive steroid SGE-516 reduces status epilepticus and neuronal cell death in a rat model of soman intoxication. Epilepsy Behav. 2017, 68, 22–30. [Google Scholar] [CrossRef]
- Zarghi, A.; Rao, P.; Knaus, E.E. Design and synthesis of new rofecoxib analogs as selective cyclooxygenase-2 (COX-2) inhibitors: Replacement of the methanesulfonyl pharmacophore by a N-acetylsulfonamido bioisostere. J. Pharm. Pharm. Sci. A Publ. Can. Soc. Pharm. Sci. Soc. Can. Des Sci. Pharm. 2007, 10, 159–167. [Google Scholar]
- Arfaie, S.; Zarghi, A. Design, synthesis and biological evaluation of new (E)-and (Z)-1,2,3-triaryl-2-propen-1-ones as selective COX-2 inhibitors. Eur. J. Med. Chem. 2010, 45, 4013–4017. [Google Scholar] [CrossRef]
- Zarghi, A.; Kakhgi, S.; Hadipoor, A.; Daraee, B.; Dadrass, O.G.; Hedayati, M. Design and synthesis of 1, 3-diarylurea derivatives as selective cyclooxygenase (COX-2) inhibitors. Bioorganic Med. Chem. Lett. 2008, 18, 1336–1339. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.B.; Kumari, S.; Prakash, A.; Yadav, R.; Tiwari, A.K.; Pandey, P.; Tiwari, M. Discovery of novel Methylsulfonyl phenyl derivatives as potent human Cyclooxygenase-2 inhibitors with effective anticonvulsant action: Design, synthesis, in-silico, in-vitro and in-vivo evaluation. Eur. J. Med. Chem. 2018, 151, 520–532. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.A.; Aronica, E.; Vezzani, A.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: Emerging evidence from preclinical and clinical studies. Neuropathol. Appl. Neurobiol. 2018, 44, 91–111. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Kang, X.; Qiu, J.; Du, Y.; Jiang, J. Anti-inflammatory small molecules to treat seizures and epilepsy: From bench to bedside. Trends Pharmacol. Sci. 2016, 37, 463–484. [Google Scholar] [CrossRef]
- Bhukya, G.; Kaki, S.S. Design and Synthesis of Sebacic Acid from Castor Oil by New Alternate Route. Eur. J. Lipid Sci. Technol. 2022, 124, 2100244. [Google Scholar] [CrossRef]
- Enrique, A.V.; Di Ianni, M.E.; Goicoechea, S.; Lazarowski, A.; Valle-Dorado, M.G.; Costa, J.J.L.; Rocha, L.; Girardi, E.; Talevi, A. New anticonvulsant candidates prevent P-glycoprotein (P-gp) overexpression in a pharmacoresistant seizure model in mice. Epilepsy Behav. 2021, 121, 106451. [Google Scholar] [CrossRef]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 107966. [Google Scholar] [CrossRef]
- Tolman, J.A.; Faulkner, M.A. Vigabatrin: A comprehensive review of drug properties including clinical updates following recent FDA approval. Expert Opin. Pharmacother. 2009, 10, 3077–3089. [Google Scholar] [CrossRef]
- Wild, J.M.; Chiron, C.; Ahn, H.; Baulac, M.; Bursztyn, J.; Gandolfo, E.; Goldberg, I.; Goñi, F.J.; Mercier, F.; Nordmann, J.-P. Visual field loss in patients with refractory partial epilepsy treated with vigabatrin: Final results from an open-label, observational, multicentre study. CNS Drugs 2009, 23, 965–982. [Google Scholar] [CrossRef]
- Juncosa, J.I.; Takaya, K.; Le, H.V.; Moschitto, M.J.; Weerawarna, P.M.; Mascarenhas, R.; Liu, D.; Dewey, S.L.; Silverman, R.B. Design and mechanism of (S)-3-amino-4-(difluoromethylenyl) cyclopent-1-ene-1-carboxylic acid, a highly potent γ-aminobutyric acid aminotransferase inactivator for the treatment of addiction. J. Am. Chem. Soc. 2018, 140, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Feja, M.; Meller, S.; Deking, L.S.; Kaczmarek, E.; During, M.J.; Silverman, R.B.; Gernert, M. OV329, a novel highly potent γ-aminobutyric acid aminotransferase inactivator, induces pronounced anticonvulsant effects in the pentylenetetrazole seizure threshold test and in amygdala-kindled rats. Epilepsia 2021, 62, 3091–3104. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S.; Backofen-Wehrhahn, B.; Bankstahl, M.; Gey, L.; Gernert, M.; Löscher, W. Vigabatrin for focal drug delivery in epilepsy: Bilateral microinfusion into the subthalamic nucleus is more effective than intranigral or systemic administration in a rat seizure model. Neurobiol. Dis. 2012, 46, 362–376. [Google Scholar] [CrossRef]
- Witkin, J.M.; Li, G.; Golani, L.K.; Xiong, W.; Smith, J.L.; Ping, X.; Rashid, F.; Jahan, R.; Cook, J.M.; Jin, X. The positive allosteric modulator of α2/3-containing GABAA receptors, KRM-II-81, is active in pharmaco-resistant models of epilepsy and reduces hyperexcitability after traumatic brain injury. J. Pharmacol. Exp. Ther. 2020, 372, 83–94. [Google Scholar] [CrossRef]
- Moerke, M.J.; Li, G.; Golani, L.K.; Cook, J.; Negus, S.S. Effects of the α2/α3-subtype-selective GABAA receptor positive allosteric modulator KRM-II-81 on pain-depressed behavior in rats: Comparison with ketorolac and diazepam. Behav. Pharmacol. 2019, 30, 452. [Google Scholar] [CrossRef] [PubMed]
- Ator, N.A.; Atack, J.R.; Hargreaves, R.J.; Burns, H.D.; Dawson, G.R. Reducing abuse liability of GABAA/benzodiazepine ligands via selective partial agonist efficacy at α1 and α2/3 subtypes. J. Pharmacol. Exp. Ther. 2010, 332, 4–16. [Google Scholar] [CrossRef]
- Pandey, K.P.; Divović, B.; Rashid, F.; Golani, L.K.; Zahn, N.M.; Meyer, M.J.; Arnold, A.L.; Sharmin, D.; Mian, M.Y.; Smith, J.L. Structural analogs of the GABAkine (5-(8-ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazole[1,5-α][1,4]diazepin-3-yl) oxazole)(KRM-II-81) are orally bioavailable anticonvulsants without sedation. J. Pharmacol. Exp. Ther. 2023. [Google Scholar] [CrossRef]
- Witkin, J.M.; Smith, J.L.; Ping, X.; Gleason, S.; Poe, M.; Li, G.; Jin, X.; Hobbs, J.; Schkeryantz, J.; McDermott, J. Bioisosteres of ethyl 8-ethynyl-6-(pyridin-2-yl)-4H-benzo[f]imidazo [1,5-a][1,4] diazepine-3-carboxylate (HZ-166) as novel alpha 2, 3 selective potentiators of GABAA receptors: Improved bioavailability enhances anticonvulsant efficacy. Neuropharmacology 2018, 137, 332–343. [Google Scholar] [CrossRef]
- Knutson, D.E.; Smith, J.L.; Ping, X.; Jin, X.; Golani, L.K.; Li, G.; Tiruveedhula, V.P.B.; Rashid, F.; Mian, M.Y.; Jahan, R. Imidazodiazepine anticonvulsant, KRM-II-81, produces novel, non-diazepam-like antiseizure effects. ACS Chem. Neurosci. 2020, 11, 2624–2637. [Google Scholar] [CrossRef]
- Golyala, A.; Kwan, P. Drug development for refractory epilepsy: The past 25 years and beyond. Seizure 2017, 44, 147–156. [Google Scholar] [CrossRef]
- Kaur, H.; Kumar, B.; Medhi, B. Antiepileptic drugs in development pipeline: A recent update. Eneurologicalsci 2016, 4, 42–51. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Model | Induction Methods | Clinical Relevance | Advantages | Disadvantages | Reference |
|---|---|---|---|---|---|
| Acute seizure model | |||||
| Maximal electroshock seizure model | Corneal or auricular stimulation with suprathreshold electric current | Generalized tonic–clonic seizures | High throughput and time-efficient for ASM screening Reproducibility Low cost Easy implementation | Lack of predictive validity for some ASMs, such as levetiracetam, tiagabine, and vigabatrin Species difference Limited mechanistic insights | [24,25,27,28,30,31] |
| Subcutaneous pentylenetetrazole-induced seizure model | Subcutaneous pentylenetetrazole injection | Generalized myoclonus and spike-wave seizures | Inexpensive and time-efficient for ASM screening Simplicity Standardization of induction Rapid onset of seizures | Low predictive validity in ASM effectiveness Lack of clinical relevance for all seizure types Limited mechanistic insights | [29,33,37,38,39] |
| Chronic seizure model | |||||
| Kindling model | Repeated seizure induction via electrical/chemical stimuli | Focal seizures | High predictive validity for focal seizures Recapitulation of epileptogenesis Etiologically relevant model Translational relevance | Expensive and time-consuming Individual variability Technique demand Complexity of data interpretation | [32,41,42,44,46,47,48,49] |
| Models for Pharmacoresistant Epilepsy | Induction Methods | Clinical Relevance | Advantages | Disadvantages | Reference |
| 6 Hz psychomotor seizure model | 6 Hz, 3 s currents with intensity of 22 mA to 44 mA delivered to anesthetized corneas | Focal psychomotor seizures Pharmacoresistant seizures evoked by 44 mA | Differentiation of new ASMs against drug-resistant seizures Rapid and inexpensive test Simple stimulation protocol Reproducibility | Species difference Limited clinical relevance | [51,52,53] |
| Lamotrigine-resistant amygdala-kindled seizure model | Treatment of a low dose of LTG prior to each stimulation during kindling acquisition | Drug-resistant chronic focal seizures | Differentiation of novel ASMs against pharmacoresistant seizures Investigating mechanisms of drug resistance | The tolerability of some ASMs differs compared to LTG-naive mice Limited clinical relevance Complex drug resistance mechanism | [54,55] |
| Post-status epilepticus seizure model | Status epilepticus evoked by either electrical stimulation or chemical stimulation | Human mesial temporal lobe epilepsy | Recapitulation of human TLE High translational relevance with SRS Exploration of pathophysiology of epileptogenesis | High mortality Variable intensity of evoked status epilepticus and consequential seizures Labor intensive and time-consuming Interpretation of SRS | [59,60,62,63] |
| Drug Name | Chemical Formula | Antiseizure Effects on Models of | Postulated Antiseizure Mechanism | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Maximal Electroshock Seizure | s.c. PTZ-Induced Seizure | Amygdala- or Hippocampal-Kindled Rat | PTZ-Induced Kindling | 6 Hz Psychomotor Seizure | LTG-Resistant Amygdala-Kindled Seizure | Miscellaneous Batteryof Seizures | ||||
| AS-1 |  | + | + | ND | + | + | ND | - | Unclear | [51,64,65,66,67,68] |
| C-11 |  | + | + | ND | + | + | ND | - | Inhibition of VGSC | [72,73,74] |
| DSP-0565 | 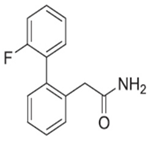 | + | + | + | ND | + | ND | - | Unclear | [76,77] |
| KM-568 | 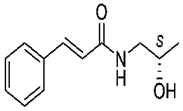 | + | + | + | ND | + | + | Corneal kindling | Unclear | [33] |
| TP-315 | 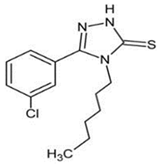 | + | ND | ND | ND | + | ND | - | Inhibition of VGSC | [80,81,82,83] |
| SGE-516 | 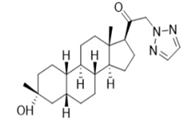 | ND | + | ND | ND | + | ND | Corneal kindling, Li-Pilo-evoked status epilepticus | Potentiating γ and δ subunit-containing GABAergic transmission | [85,86,89,90] |
| MTL-1 | 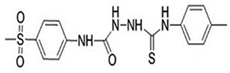 | ND | + | ND | + | ND | ND | - | COX-2 inhibitor against neuroinflammation | [92,93,94] |
| Sebacicacid | 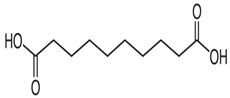 | + | + | ND | ND | ND | ND | - | Attenuating overexpression of P-glycoprotein | [97,98] |
| OV329 |  | ND | ND | + | ND | ND | ND | i.v. PTZ-induced seizure | Inactivator of GABA aminotransferase | [102,103,104] |
| KRM-II-81 | 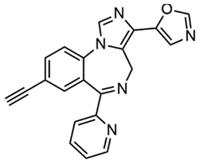 | + | + | + | ND | + | + | Kainate-induced chronic epilepsy | Selective modulator of α2/3-containing GABAA receptors | [105,106,107,108,109] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-S.; Wu, M.-C.; Lai, M.-C.; Wu, S.-N.; Huang, C.-W. Identification of New Antiseizure Medication Candidates in Preclinical Animal Studies. Int. J. Mol. Sci. 2023, 24, 13143. https://doi.org/10.3390/ijms241713143
Yang C-S, Wu M-C, Lai M-C, Wu S-N, Huang C-W. Identification of New Antiseizure Medication Candidates in Preclinical Animal Studies. International Journal of Molecular Sciences. 2023; 24(17):13143. https://doi.org/10.3390/ijms241713143
Chicago/Turabian StyleYang, Chih-Sheng, Man-Chun Wu, Ming-Chi Lai, Sheng-Nan Wu, and Chin-Wei Huang. 2023. "Identification of New Antiseizure Medication Candidates in Preclinical Animal Studies" International Journal of Molecular Sciences 24, no. 17: 13143. https://doi.org/10.3390/ijms241713143
APA StyleYang, C.-S., Wu, M.-C., Lai, M.-C., Wu, S.-N., & Huang, C.-W. (2023). Identification of New Antiseizure Medication Candidates in Preclinical Animal Studies. International Journal of Molecular Sciences, 24(17), 13143. https://doi.org/10.3390/ijms241713143