
GSK3α Regulates Temporally Dynamic Changes in Ribosomal Proteins upon Amino Acid Starvation in Cancer Cells

 ,
,  ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
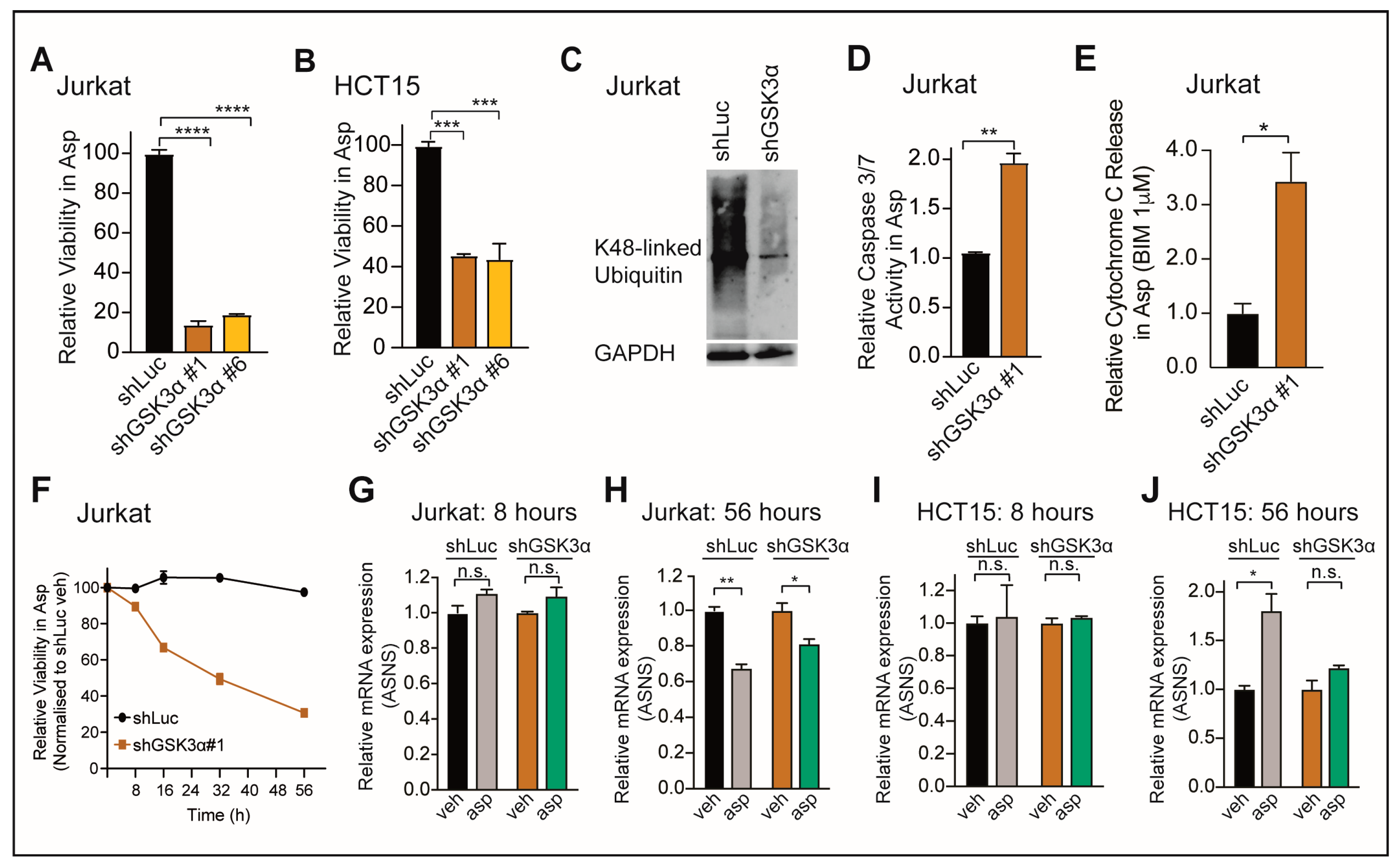
2.1. Loss of GSK3α Induces Asparaginase Cytotoxicity Independent from ASNS Expression in Resistant Cancer Cells
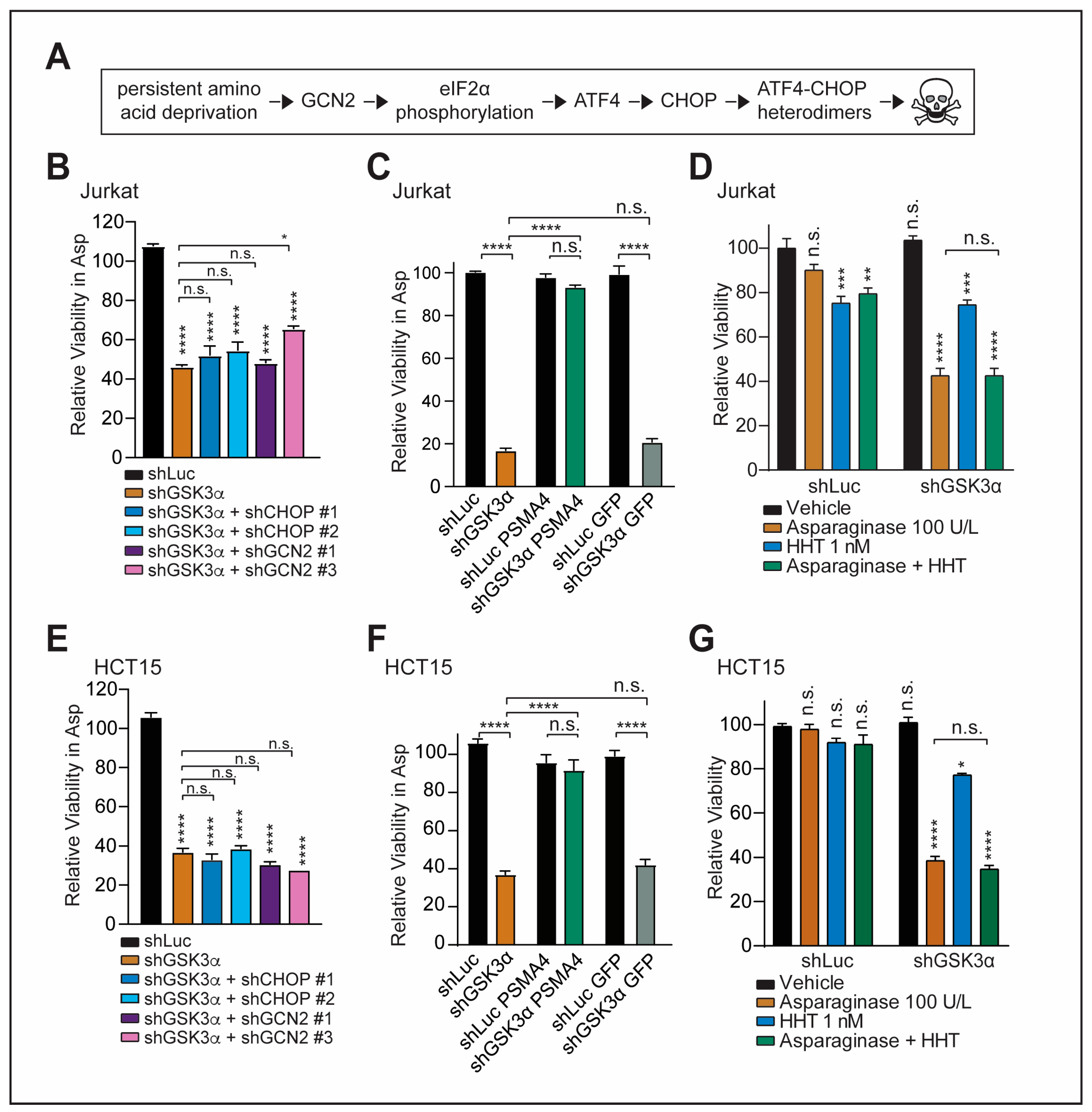
2.2. GSK3α-Mediated Response to Chronic Amino Acid Deprivation Is Independent of the GCN2-CHOP Axis
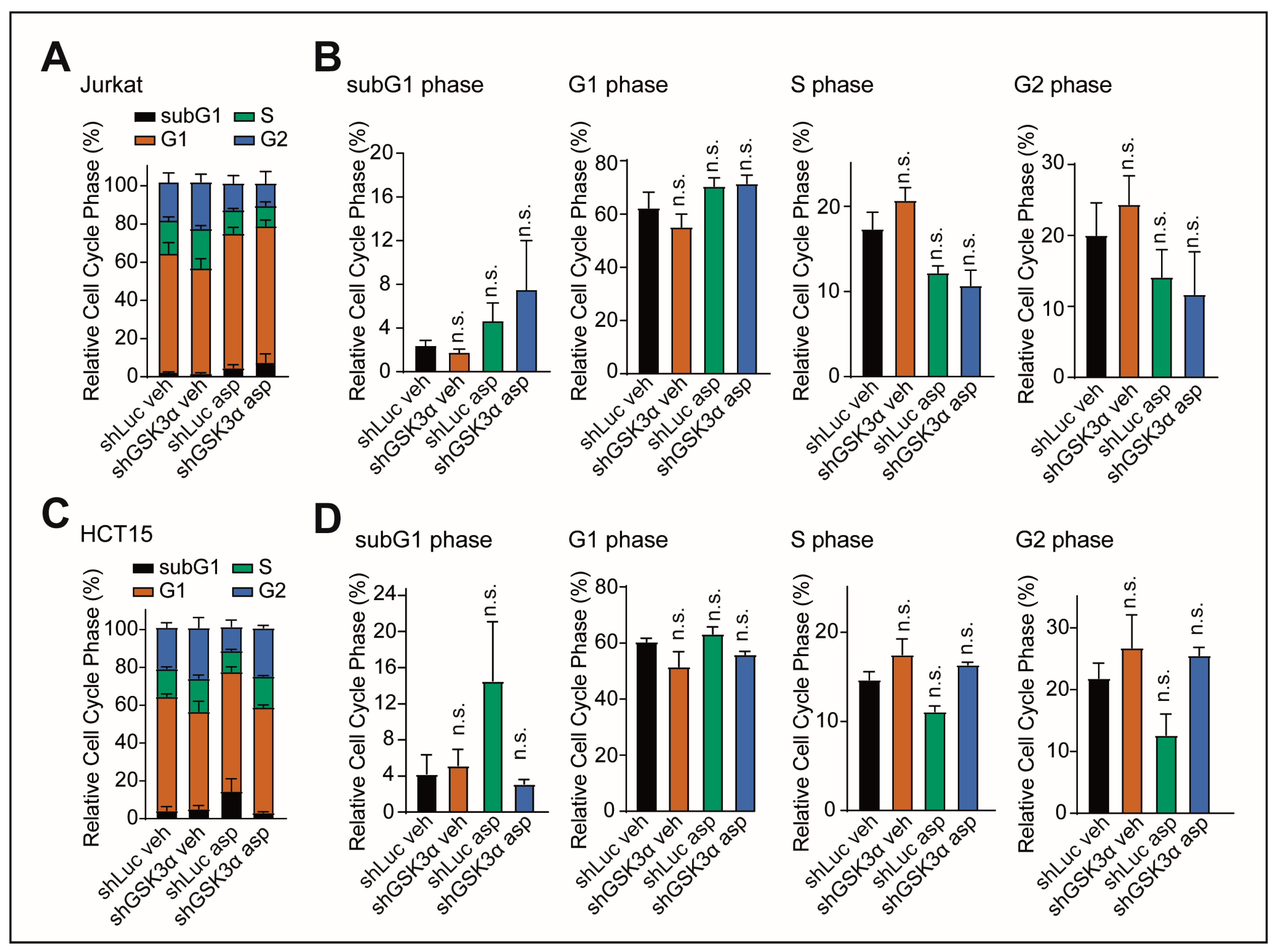
2.3. Cell Death upon Inhibition of GSK3α Is Not Mediated by Changes in Cell Cycle
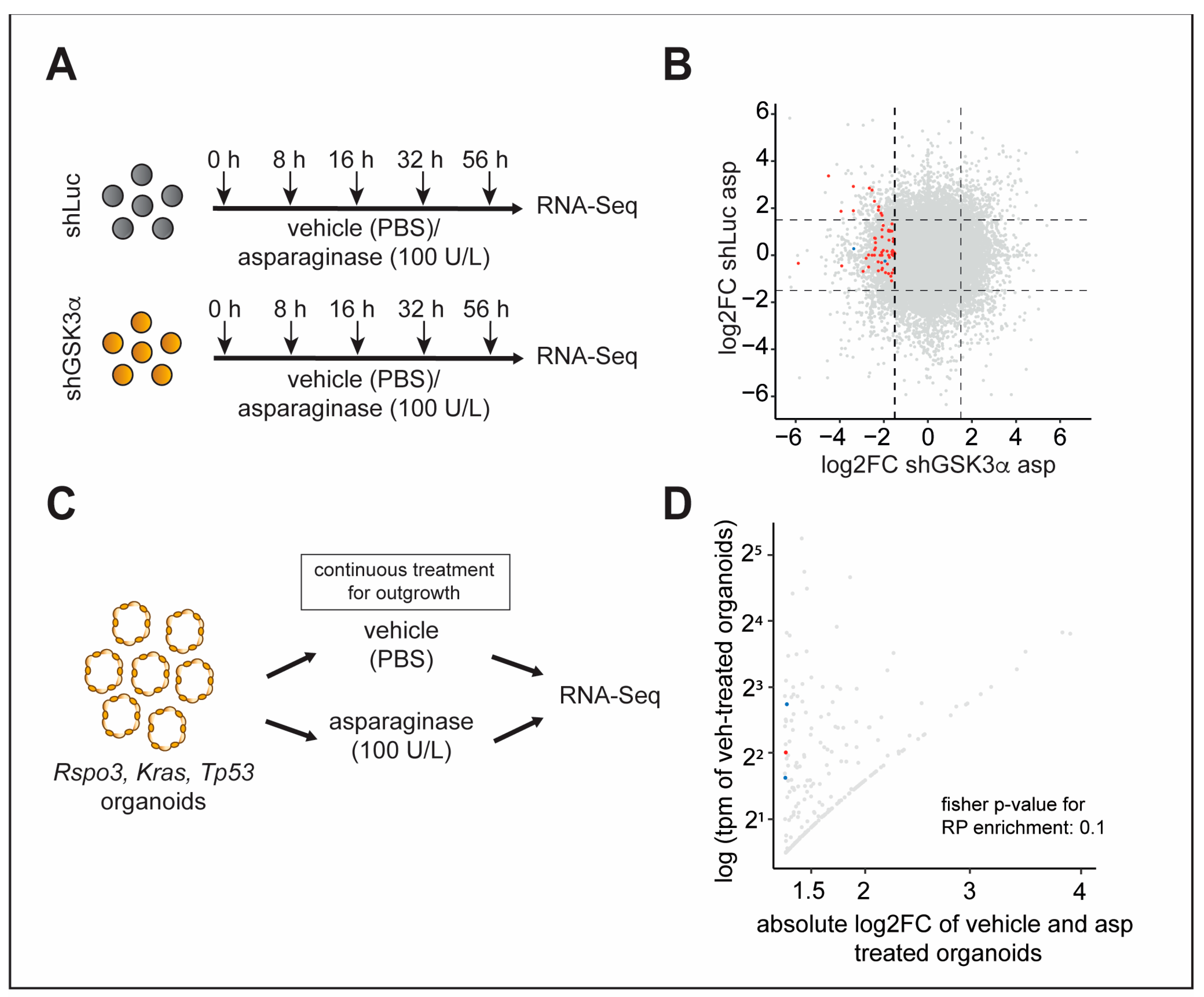
2.4. Inhibition of GSK3α Leads to Temporally Dynamic Downregulation of Distinct Ribosomal Proteins in the Presence of Asparagine Deprivation
2.5. Inhibition of Specific Ribosomal Proteins Promotes Cellular Fitness upon GSK3α Inhibition and Asparagine Starvation
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Cell Culture, and Organoids
4.2. Lentiviral Transduction
4.3. Short Hairpin RNA (shRNA), Single-Guide RNA (sgRNA), and Expression Plasmids
4.4. Assessment of Chemotherapy Response
4.5. Quantitative Reverse Transcriptase PCR (qRT-PCR)
4.6. Western Blot Analysis
4.7. Cell Cycle Analysis
4.8. Caspase 3/7 Activity and BH3 Profiling
4.9. Timeseries RNA Sequencing and Bioinformatic Analysis
4.10. Organoids RNA Sequencing and Bioinformatic Analysis
4.11. CRISPR/Cas9 Loss of Function Screen
4.12. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeAngelo, D.J.; Stevenson, K.E.; Dahlberg, S.E.; Silverman, L.B.; Couban, S.; Supko, J.G.; Amrein, P.C.; Ballen, K.K.; Seftel, M.D.; Turner, A.R.; et al. Long-term outcome of a pediatric-inspired regimen used for adults aged 18-50 years with newly diagnosed acute lymphoblastic leukemia. Leukemia 2015, 29, 526–534. [Google Scholar] [CrossRef]
- Place, A.E.; Stevenson, K.E.; Vrooman, L.M.; Harris, M.H.; Hunt, S.K.; O’Brien, J.E.; Supko, J.G.; Asselin, B.L.; Athale, U.H.; Clavell, L.A.; et al. Intravenous pegylated asparaginase versus intramuscular native Escherichia coli L-asparaginase in newly diagnosed childhood acute lymphoblastic leukaemia (DFCI 05-001): A randomised, open-label phase 3 trial. Lancet Oncol. 2015, 16, 1677–1690. [Google Scholar] [CrossRef] [PubMed]
- Wetzler, M.; Sanford, B.L.; Kurtzberg, J.; DeOliveira, D.; Frankel, S.R.; Powell, B.L.; Kolitz, J.E.; Bloomfield, C.D.; Larson, R.A. Effective asparagine depletion with pegylated asparaginase results in improved outcomes in adult acute lymphoblastic leukemia: Cancer and Leukemia Group B Study 9511. Blood 2007, 109, 4164–4167. [Google Scholar] [CrossRef] [PubMed]
- Pession, A.; Valsecchi, M.G.; Masera, G.; Kamps, W.A.; Magyarosy, E.; Rizzari, C.; van Wering, E.R.; Lo Nigro, L.; van der Does, A.; Locatelli, F.; et al. Long-term results of a randomized trial on extended use of high dose L-asparaginase for standard risk childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2005, 23, 7161–7167. [Google Scholar] [CrossRef]
- Acebron, S.P.; Karaulanov, E.; Berger, B.S.; Huang, Y.L.; Niehrs, C. Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol. Cell 2014, 54, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Hinze, L.; Labrosse, R.; Degar, J.; Han, T.; Schatoff, E.M.; Schreek, S.; Karim, S.; McGuckin, C.; Sacher, J.R.; Wagner, F.; et al. Exploiting the Therapeutic Interaction of WNT Pathway Activation and Asparaginase for Colorectal Cancer Therapy. Cancer Discov. 2020, 10, 1690–1705. [Google Scholar] [CrossRef] [PubMed]
- Hinze, L.; Pfirrmann, M.; Karim, S.; Degar, J.; McGuckin, C.; Vinjamur, D.; Sacher, J.; Stevenson, K.E.; Neuberg, D.S.; Orellana, E.; et al. Synthetic Lethality of Wnt Pathway Activation and Asparaginase in Drug-Resistant Acute Leukemias. Cancer Cell 2019, 35, 664–676.e7. [Google Scholar] [CrossRef] [PubMed]
- Hinze, L.; Schreek, S.; Zeug, A.; Ibrahim, N.K.; Fehlhaber, B.; Loxha, L.; Cinar, B.; Ponimaskin, E.; Degar, J.; McGuckin, C.; et al. Supramolecular assembly of GSK3α as a cellular response to amino acid starvation. Mol. Cell 2022, 82, 2858–2870.e8. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S.; Bröer, A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J. 2017, 474, 1935–1963. [Google Scholar] [CrossRef]
- Wek, S.A.; Zhu, S.; Wek, R.C. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol. Cell. Biol. 1995, 15, 4497–4506. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.T.; Masson, G.R. GCN2: Roles in tumour development and progression. Biochem. Soc. Trans. 2022, 50, 737–745. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Ye, J.; Kumanova, M.; Hart, L.S.; Sloane, K.; Zhang, H.; De Panis, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Ron, D.; Koumenis, C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010, 29, 2082–2096. [Google Scholar] [CrossRef] [PubMed]
- Nikonorova, I.A.; Mirek, E.T.; Signore, C.C.; Goudie, M.P.; Wek, R.C.; Anthony, T.G. Time-resolved analysis of amino acid stress identifies eIF2 phosphorylation as necessary to inhibit mTORC1 activity in liver. J. Biol. Chem. 2018, 293, 5005–5015. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Sadighi Akha, A.A.; Raden, D.; Kaufman, R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, e374. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Siu, F.; Bain, P.J.; LeBlanc-Chaffin, R.; Chen, H.; Kilberg, M.S. ATF4 is a mediator of the nutrient-sensing response pathway that activates the human asparagine synthetase gene. J. Biol. Chem. 2002, 277, 24120–24127. [Google Scholar] [CrossRef]
- Chen, H.; Pan, Y.X.; Dudenhausen, E.E.; Kilberg, M.S. Amino acid deprivation induces the transcription rate of the human asparagine synthetase gene through a timed program of expression and promoter binding of nutrient-responsive basic region/leucine zipper transcription factors as well as localized histone acetylation. J. Biol. Chem. 2004, 279, 50829–50839. [Google Scholar] [PubMed]
- Wang, Y.; Ning, Y.; Alam, G.N.; Jankowski, B.M.; Dong, Z.; Nör, J.E.; Polverini, P.J. Amino acid deprivation promotes tumor angiogenesis through the GCN2/ATF4 pathway. Neoplasia 2013, 15, 989–997. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef]
- Lindström, M.S. Emerging functions of ribosomal proteins in gene-specific transcription and translation. Biochem. Biophys. Res. Commun. 2009, 379, 167–170. [Google Scholar] [CrossRef]
- Rodnina, M.V.; Wintermeyer, W. Recent mechanistic insights into eukaryotic ribosomes. Curr. Opin. Cell Biol. 2009, 21, 435–443. [Google Scholar] [CrossRef]
- Weisberg, R.A. Transcription by moonlight: Structural basis of an extraribosomal activity of ribosomal protein S10. Mol. Cell 2008, 32, 747–748. [Google Scholar] [CrossRef]
- Wool, I.G. Extraribosomal functions of ribosomal proteins. Trends Biochem. Sci. 1996, 21, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nag, S.; Zhang, X.; Wang, M.H.; Wang, H.; Zhou, J.; Zhang, R. Ribosomal proteins and human diseases: Pathogenesis, molecular mechanisms, and therapeutic implications. Med. Res. Rev. 2015, 35, 225–285. [Google Scholar] [CrossRef]
- Warner, J.R.; McIntosh, K.B. How common are extraribosomal functions of ribosomal proteins? Mol. Cell 2009, 34, 3–11. [Google Scholar] [CrossRef]
- Kang, J.; Brajanovski, N.; Chan, K.T.; Xuan, J.; Pearson, R.B.; Sanij, E. Ribosomal proteins and human diseases: Molecular mechanisms and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 323. [Google Scholar] [CrossRef]
- Komili, S.; Farny, N.G.; Roth, F.P.; Silver, P.A. Functional specificity among ribosomal proteins regulates gene expression. Cell 2007, 131, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Taelman, V.F.; Dobrowolski, R.; Plouhinec, J.L.; Fuentealba, L.C.; Vorwald, P.P.; Gumper, I.; Sabatini, D.D.; De Robertis, E.M. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 2010, 143, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Hermanova, I.; Zaliova, M.; Trka, J.; Starkova, J. Low expression of asparagine synthetase in lymphoid blasts precludes its role in sensitivity to L-asparaginase. Exp. Hematol. 2012, 40, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Holleman, A.; Cheok, M.H.; den Boer, M.L.; Yang, W.; Veerman, A.J.; Kazemier, K.M.; Pei, D.; Cheng, C.; Pui, C.H.; Relling, M.V.; et al. Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N. Engl. J. Med. 2004, 351, 533–542. [Google Scholar] [CrossRef]
- Stams, W.A.; den Boer, M.L.; Beverloo, H.B.; Meijerink, J.P.; Stigter, R.L.; van Wering, E.R.; Janka-Schaub, G.E.; Slater, R.; Pieters, R. Sensitivity to L-asparaginase is not associated with expression levels of asparagine synthetase in t(12;21)+ pediatric ALL. Blood 2003, 101, 2743–2747. [Google Scholar] [CrossRef]
- Appel, I.M.; den Boer, M.L.; Meijerink, J.P.; Veerman, A.J.; Reniers, N.C.; Pieters, R. Up-regulation of asparagine synthetase expression is not linked to the clinical response L-asparaginase in pediatric acute lymphoblastic leukemia. Blood 2006, 107, 4244–4249. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Qiu, H.; Garcia-Barrio, M.; Anderson, J.; Hinnebusch, A.G. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 2000, 6, 269–279. [Google Scholar] [CrossRef]
- Nakamura, A.; Nambu, T.; Ebara, S.; Hasegawa, Y.; Toyoshima, K.; Tsuchiya, Y.; Tomita, D.; Fujimoto, J.; Kurasawa, O.; Takahara, C.; et al. Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc. Natl. Acad. Sci. USA 2018, 115, E7776–E7785. [Google Scholar] [CrossRef]
- Wortel, I.M.N.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef]
- Choi, W.H.; de Poot, S.A.; Lee, J.H.; Kim, J.H.; Han, D.H.; Kim, Y.K.; Finley, D.; Lee, M.J. Open-gate mutants of the mammalian proteasome show enhanced ubiquitin-conjugate degradation. Nat. Commun. 2016, 7, 10963. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, J.C.; Zavolan, M. Patterns of ribosomal protein expression specify normal and malignant human cells. Genome Biol. 2016, 17, 236. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Zhao, Y.; He, H.; Sun, Y. Ribosomal protein S27-like and S27 interplay with p53-MDM2 axis as a target, a substrate and a regulator. Oncogene 2011, 30, 1798–1811. [Google Scholar] [CrossRef]
- Bai, D.; Zhang, J.; Xiao, W.; Zheng, X. Regulation of the HDM2-p53 pathway by ribosomal protein L6 in response to ribosomal stress. Nucleic Acids Res. 2014, 42, 1799–1811. [Google Scholar] [CrossRef] [PubMed]
- Vabulas, R.M.; Hartl, F.U. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 2005, 310, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; Münch, C.; Hanssum, A.; Bertolotti, A. Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell 2012, 48, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Lindström, M.S.; Zhang, Y. Ribosomal protein S9 is a novel B23/NPM-binding protein required for normal cell proliferation. J. Biol. Chem. 2008, 283, 15568–15576. [Google Scholar] [CrossRef] [PubMed]
- Oliver, E.R.; Saunders, T.L.; Tarlé, S.A.; Glaser, T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development 2004, 131, 3907–3920. [Google Scholar] [CrossRef]
- Mazumder, B.; Sampath, P.; Seshadri, V.; Maitra, R.K.; DiCorleto, P.E.; Fox, P.L. Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell 2003, 115, 187–198. [Google Scholar] [CrossRef]
- Horos, R.; Ijspeert, H.; Pospisilova, D.; Sendtner, R.; Andrieu-Soler, C.; Taskesen, E.; Nieradka, A.; Cmejla, R.; Sendtner, M.; Touw, I.P.; et al. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood 2012, 119, 262–272. [Google Scholar] [CrossRef]
- Kondrashov, N.; Pusic, A.; Stumpf, C.R.; Shimizu, K.; Hsieh, A.C.; Ishijima, J.; Shiroishi, T.; Barna, M. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 2011, 145, 383–397. [Google Scholar] [CrossRef]
- Sulima, S.O.; Kampen, K.R.; De Keersmaecker, K. Cancer Biogenesis in Ribosomopathies. Cells 2019, 8, 229. [Google Scholar] [CrossRef]
- Narla, A.; Ebert, B.L. Ribosomopathies: Human disorders of ribosome dysfunction. Blood 2010, 115, 3196–3205. [Google Scholar] [CrossRef]
- Hannan, K.M.; Sanij, E.; Rothblum, L.I.; Hannan, R.D.; Pearson, R.B. Dysregulation of RNA polymerase I transcription during disease. Biochim. Biophys. Acta 2013, 1829, 342–360. [Google Scholar] [CrossRef]
- Aspesi, A.; Ellis, S.R. Rare ribosomopathies: Insights into mechanisms of cancer. Nat. Rev. Cancer 2019, 19, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Dameshek, W. Riddle: What do aplastic anemia, paroxysmal nocturnal hemoglobinuria (PNH) and “hypoplastic” leukemia have in common? Blood 1967, 30, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.W.; Green, R. Ribosomopathies: There’s strength in numbers. Science 2017, 358, eaan2755. [Google Scholar] [CrossRef]
- Guerra-Moreno, A.; Isasa, M.; Bhanu, M.K.; Waterman, D.P.; Eapen, V.V.; Gygi, S.P.; Hanna, J. Proteomic Analysis Identifies Ribosome Reduction as an Effective Proteotoxic Stress Response. J. Biol. Chem. 2015, 290, 29695–29706. [Google Scholar] [CrossRef]
- Albert, B.; Kos-Braun, I.C.; Henras, A.K.; Dez, C.; Rueda, M.P.; Zhang, X.; Gadal, O.; Kos, M.; Shore, D. A ribosome assembly stress response regulates transcription to maintain proteome homeostasis. eLife 2019, 8, e45002. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Sanson, K.R.; Hanna, R.E.; Hegde, M.; Donovan, K.F.; Strand, C.; Sullender, M.E.; Vaimberg, E.W.; Goodale, A.; Root, D.E.; Piccioni, F.; et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 2018, 9, 5416. [Google Scholar] [CrossRef]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2011, 12, 51–58. [Google Scholar] [CrossRef]
- Ariës, I.M.; Bodaar, K.; Karim, S.A.; Chonghaile, T.N.; Hinze, L.; Burns, M.A.; Pfirrmann, M.; Degar, J.; Landrigan, J.T.; Balbach, S.; et al. PRC2 loss induces chemoresistance by repressing apoptosis in T cell acute lymphoblastic leukemia. J. Exp. Med. 2018, 215, 3094–3114. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. In Proceedings of the 9th Annual Genomics of Energy & Environment Meeting, Walnut Creek, CA, USA, 17–20 March 2014. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2021, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Komori, H.K.; LaMere, S.; Podshivalova, K.; Salomon, D.R. Finding the active genes in deep RNA-seq gene expression studies. BMC Genom. 2013, 14, 778. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loxha, L.; Ibrahim, N.K.; Stasche, A.S.; Cinar, B.; Dolgner, T.; Niessen, J.; Schreek, S.; Fehlhaber, B.; Forster, M.; Stanulla, M.; et al. GSK3α Regulates Temporally Dynamic Changes in Ribosomal Proteins upon Amino Acid Starvation in Cancer Cells. Int. J. Mol. Sci. 2023, 24, 13260. https://doi.org/10.3390/ijms241713260
Loxha L, Ibrahim NK, Stasche AS, Cinar B, Dolgner T, Niessen J, Schreek S, Fehlhaber B, Forster M, Stanulla M, et al. GSK3α Regulates Temporally Dynamic Changes in Ribosomal Proteins upon Amino Acid Starvation in Cancer Cells. International Journal of Molecular Sciences. 2023; 24(17):13260. https://doi.org/10.3390/ijms241713260
Chicago/Turabian StyleLoxha, Lorent, Nurul Khalida Ibrahim, Anna Sophie Stasche, Büsra Cinar, Tim Dolgner, Julia Niessen, Sabine Schreek, Beate Fehlhaber, Michael Forster, Martin Stanulla, and et al. 2023. "GSK3α Regulates Temporally Dynamic Changes in Ribosomal Proteins upon Amino Acid Starvation in Cancer Cells" International Journal of Molecular Sciences 24, no. 17: 13260. https://doi.org/10.3390/ijms241713260
APA StyleLoxha, L., Ibrahim, N. K., Stasche, A. S., Cinar, B., Dolgner, T., Niessen, J., Schreek, S., Fehlhaber, B., Forster, M., Stanulla, M., & Hinze, L. (2023). GSK3α Regulates Temporally Dynamic Changes in Ribosomal Proteins upon Amino Acid Starvation in Cancer Cells. International Journal of Molecular Sciences, 24(17), 13260. https://doi.org/10.3390/ijms241713260