
Features of Congenital Arthrogryposis Due to Abnormalities in Collagen Homeostasis, a Scoping Review


Abstract
:1. Introduction
- Are there common phenotypic features of the CA caused by collagen defects?
- Are there mechanistic similarities in collagen formation or modification defects that lead to CA?
- Could collagen or collagen-related genes be potential targets for CA therapy development?
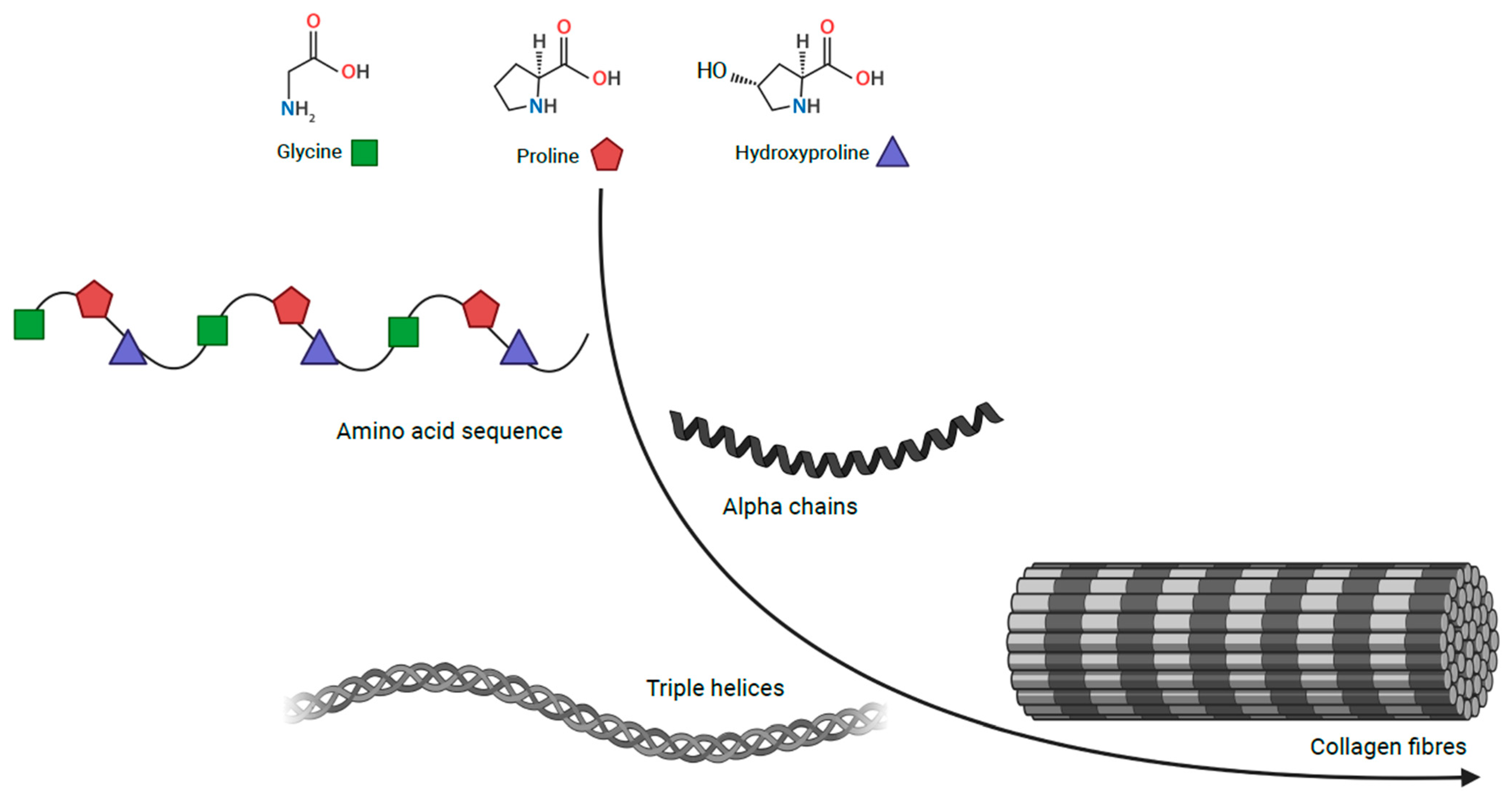
2. Inherited Defects in Collagen and Collagen-Related Genes
2.1. Disorders Related to Collagen Defects
Collagen VI Related Diseases
2.2. Collagen XXV Related Diseases
2.3. Disorders Related to Collagen Modifying Defects
LH2 and FKBP65 Related Diseases
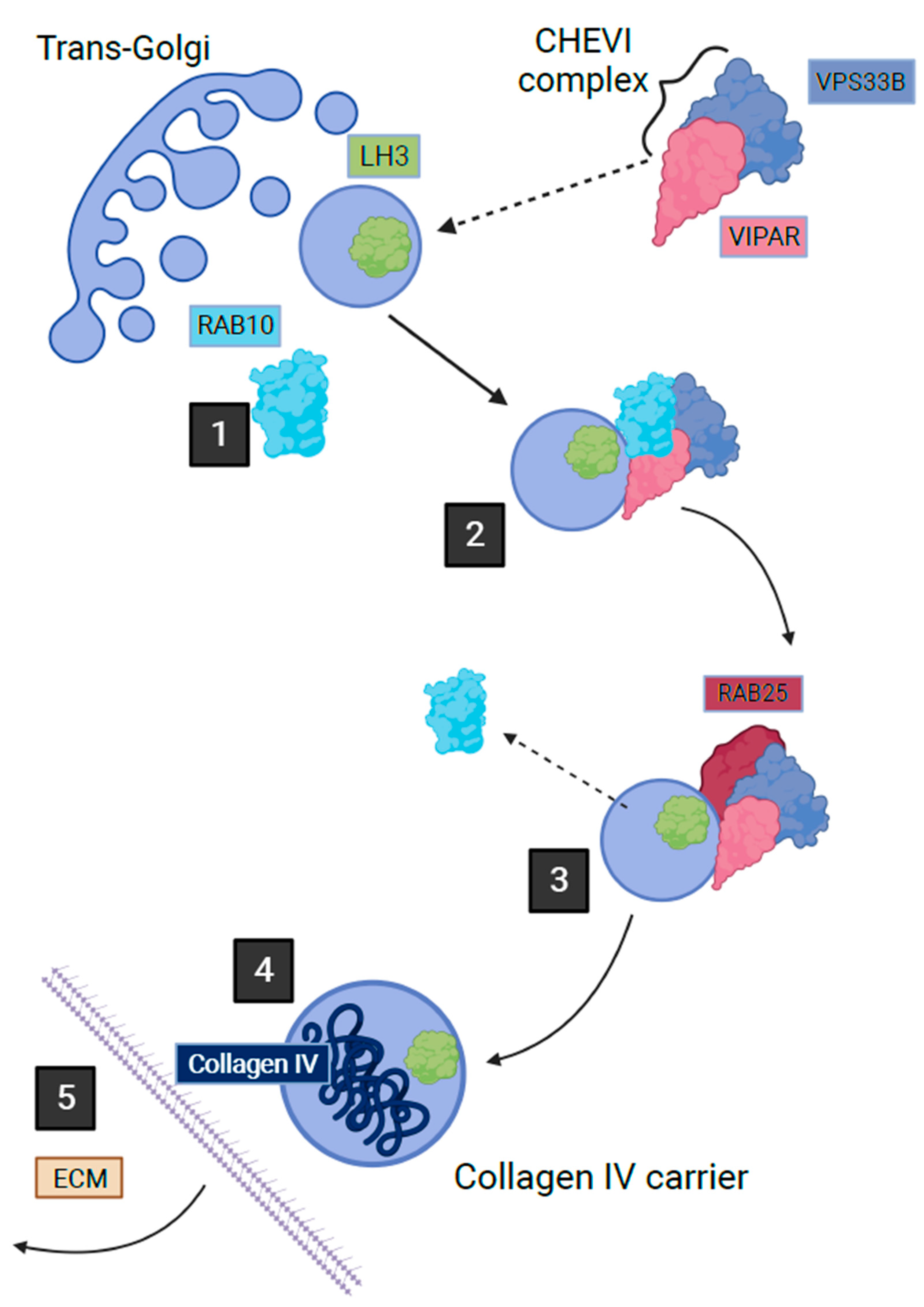
2.4. VPS33B, VIPAR and LHS Related Diseases
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bamshad, M.; Van Heest, A.E.; Pleasure, D. Arthrogryposis: A Review and Update. J. Bone Joint Surg. Am. 2009, 91 (Suppl. S4), 40–46. [Google Scholar] [CrossRef]
- Hall, J.G.; Kimber, E.; Dieterich, K. Classification of arthrogryposis. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Natera-de Benito, D.; Jurgens, J.A.; Yeung, A.; Zaharieva, I.T.; Manzur, A.; DiTroia, S.P.; Di Gioia, S.A.; Pais, L.; Pini, V.; Barry, B.J.; et al. Recessive variants in COL25A1 gene as novel cause of arthrogryposis multiplex congenita with ocular congenital cranial dysinnervation disorder. Hum. Mutat. 2022, 43, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Gelse, K. Collagens—Structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef]
- Myllyharju, J.; Kivirikko, K.I. Collagens and collagen-related diseases. Ann. Med. 2001, 33, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The Collagen Family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef]
- Khoshnoodi, J.; Pedchenko, V.; Hudson, B. Mammalian Collagen IV. Microsc. Res. Tech. 2008, 71, 357–370. [Google Scholar] [CrossRef]
- Hall, J.G.; Kiefer, J. Arthrogryposis as a Syndrome: Gene Ontology Analysis. Mol. Syndromol. 2016, 7, 101–109. [Google Scholar] [CrossRef]
- Foley, A.R.; Mohassel, P.; Donkervoort, S.; Bolduc, V.; Bönnemann, C.G. Collagen VI-Related Dystrophies; GeneReviews®; University of Washington: Seattle, WA, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1503 (accessed on 27 April 2023).
- Di Martino, A.; Cescon, M.; D’Agostino, C.; Schilardi, F.; Sabatelli, P.; Merlini, L.; Faldini, C. Collagen VI in the Musculoskeletal System. Int. J. Mol. Sci. 2023, 24, 5095. [Google Scholar] [CrossRef]
- Jöbsis, G.J.; Boers, J.M.; Barth, P.G.; de Visser, M. Bethlem myopathy: A slowly progressive congenital muscular dystrophy with contractures. Brain 1999, 122, 649–655. [Google Scholar] [CrossRef]
- Allamand, V.; Briñas, L.; Richard, P.; Stojkovic, T.; Quijano-Roy, S.; Bonne, G. ColVI myopathies: Where do we stand, where do we go? Skelet. Muscle 2011, 1, 30. [Google Scholar] [CrossRef]
- Baker, N.L.; Mörgelin, M.; Peat, R.; Goemans, N.; North, K.N.; Bateman, J.F.; Lamandé, S.R. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum. Mol. Genet. 2005, 14, 279–293. [Google Scholar] [CrossRef]
- Lampe, A.K.; Dunn, D.M.; von Niederhausern, A.C.; Hamil, C.; Aoyagi, A.; Laval, S.H.; Marie, S.K.; Chu, M.-L.; Swoboda, K.; Muntoni, F.; et al. Automated genomic sequence analysis of the three collagen VI genes: Applications to Ullrich congenital muscular dystrophy and Bethlem myopathy. J. Med. Genet. 2005, 42, 108–120. [Google Scholar] [CrossRef]
- Camacho Vanegas, O.; Bertini, E.; Zhang, R.Z.; Petrini, S.; Minosse, C.; Sabatelli, P.; Giusti, B.; Chu, M.L.; Pepe, G. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc. Natl. Acad. Sci. USA 2001, 98, 7516–7521. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kawahara, G.; Noguchi, S.; Sugie, K.; Murayama, K.; Nonaka, I.; Hayashi, Y.K.; Nishino, I. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007, 69, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Picillo, E.; Torella, A.; Passamano, L.; Nigro, V.; Politano, L. Autosomal dominant Ullrich congenital muscular dystrophy due to a de novo mutation in COL6A3 gene. A case report. Acta Myol. 2022, 41, 95–98. [Google Scholar] [CrossRef]
- Pan, T.-C.; Zhang, R.-Z.; Arita, M.; Bogdanovich, S.; Adams, S.M.; Gara, S.K.; Wagener, R.; Khurana, T.S.; Birk, D.E.; Chu, M.-L. A mouse model for dominant collagen VI disorders: Heterozygous deletion of Col6a3 Exon 16. J. Biol. Chem. 2014, 289, 10293–10307. [Google Scholar] [CrossRef]
- Bethlem, J.; Wijngaarden, G.K.V. Benign myopathy, with autosomal dominant inheritance: A report on three pedigrees. Brain 1976, 99, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Jöbsis, G.J.; Keizers, H.; Vreijling, J.P.; de Visser, M.; Speer, M.C.; Wolterman, R.A.; Baas, F.; Bolhuis, P.A. Type VI collagen mutations in Bethlem myopathy, an autosomal dominant myopathy with contractures. Nat. Genet. 1996, 14, 113–115. [Google Scholar] [CrossRef]
- Park, H.J.; Choi, Y.C.; Kim, S.M.; Kim, S.H.; Hong, Y.B.; Yoon, B.R.; Chung, K.W.; Choi, B.O. Molecular Genetic Diagnosis of a Bethlem Myopathy Family with an Autosomal-Dominant COL6A1 Mutation, as Evidenced by Exome Sequencing. J. Clin. Neurol. 2015, 11, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Saroja, A.O.; Naik, K.R.; Nalini, A.; Gayathri, N. Bethlem myopathy: An autosomal dominant myopathy with flexion contractures, keloids, and follicular hyperkeratosis. Ann. Indian Acad. Neurol. 2013, 16, 712–715. [Google Scholar] [CrossRef]
- Sasabe, F.; Takase, Y.; Fukusako, T.; Yamamoto, K.; Morimatsu, M. Early-onset benign autosomal dominant limb-girdle myopathy with contractures (Bethlem myopathy) in a Japanese family. Rinsho Shinkeigaku 1992, 32, 138–142. [Google Scholar] [PubMed]
- Caria, F.; Cescon, M.; Gualandi, F.; Pichiecchio, A.; Rossi, R.; Rimessi, P.; Cotti Piccinelli, S.; Gallo Cassarino, S.; Gregorio, I.; Galvagni, A.; et al. Autosomal recessive Bethlem myopathy: A clinical, genetic and functional study. Neuromuscul. Disord. 2019, 29, 657–663. [Google Scholar] [CrossRef]
- Radev, Z.; Hermel, J.-M.; Elipot, Y.; Bretaud, S.; Arnould, S.; Duchateau, P.; Ruggiero, F.; Joly, J.-S.; Sohm, F. A TALEN-Exon Skipping Design for a Bethlem Myopathy Model in Zebrafish. PLoS ONE 2015, 10, e0133986. [Google Scholar] [CrossRef]
- Pan, T.-C.; Zhang, R.-Z.; Sudano, D.G.; Marie, S.K.; Bönnemann, C.G.; Chu, M.-L. New molecular mechanism for Ullrich congenital muscular dystrophy: A heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am. J. Hum. Genet. 2003, 73, 355–369. [Google Scholar] [CrossRef]
- Pepe, G.; Lucarini, L.; Zhang, R.-Z.; Pan, T.-C.; Giusti, B.; Quijano-Roy, S.; Gartioux, C.; Bushby, K.M.D.; Guicheney, P.; Chu, M.-L. COL6A1 genomic deletions in Bethlem myopathy and Ullrich muscular dystrophy. Ann. Neurol. 2006, 59, 190–195. [Google Scholar] [CrossRef]
- Lucioli, S.; Giusti, B.; Mercuri, E.; Vanegas, O.C.; Lucarini, L.; Pietroni, V.; Urtizberea, A.; Ben Yaou, R.; de Visser, M.; van der Kooi, A.J.; et al. Detection of common and private mutations in the COL6A1 gene of patients with Bethlem myopathy. Neurology 2005, 64, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.-R.; Bristow, J. The Ehlers-Danlos syndrome: On beyond collagens. J. Clin. Investig. 2001, 107, 1063–1069. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, R.-Z.; Sabatelli, P.; Chu, M.-L.; Bönnemann, C.G. Muscle interstitial fibroblasts are the main source of collagen VI synthesis in skeletal muscle: Implications for congenital muscular dystrophy types Ullrich and Bethlem. J. Neuropathol. Exp. Neurol. 2008, 67, 144–154. [Google Scholar] [CrossRef]
- Urciuolo, A.; Quarta, M.; Morbidoni, V.; Gattazzo, F.; Molon, S.; Grumati, P.; Montemurro, F.; Tedesco, F.S.; Blaauw, B.; Cossu, G.; et al. Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat. Commun. 2013, 4, 1964. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Braghetta, P.; Zanetti, M.; Piccolo, S.; Volpin, D.; Bressan, G.M. Collagen VI deficiency induces early onset myopathy in the mouse: An animal model for Bethlem myopathy. Hum. Mol. Genet. 1998, 7, 2135–2140. [Google Scholar] [CrossRef] [PubMed]
- Grumati, P.; Coletto, L.; Sabatelli, P.; Cescon, M.; Angelin, A.; Bertaggia, E.; Blaauw, B.; Urciuolo, A.; Tiepolo, T.; Merlini, L.; et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 2010, 16, 1313–1320. [Google Scholar] [CrossRef]
- Irwin, W.A.; Bergamin, N.; Sabatelli, P.; Reggiani, C.; Megighian, A.; Merlini, L.; Braghetta, P.; Columbaro, M.; Volpin, D.; Bressan, G.M.; et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 2003, 35, 367–371. [Google Scholar] [CrossRef]
- Merlini, L.; Angelin, A.; Tiepolo, T.; Braghetta, P.; Sabatelli, P.; Zamparelli, A.; Ferlini, A.; Maraldi, N.M.; Bonaldo, P.; Bernardi, P. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc. Natl. Acad. Sci. USA 2008, 105, 5225–5229. [Google Scholar] [CrossRef]
- Telfer, W.R.; Busta, A.S.; Bonnemann, C.G.; Feldman, E.L.; Dowling, J.J. Zebrafish models of collagen VI-related myopathies. Hum. Mol. Genet. 2010, 19, 2433–2444. [Google Scholar] [CrossRef]
- Zulian, A.; Rizzo, E.; Schiavone, M.; Palma, E.; Tagliavini, F.; Blaauw, B.; Merlini, L.; Maraldi, N.M.; Sabatelli, P.; Braghetta, P.; et al. NIM811, a cyclophilin inhibitor without immunosuppressive activity, is beneficial in collagen VI congenital muscular dystrophy models. Hum. Mol. Genet. 2014, 23, 5353–5363. [Google Scholar] [CrossRef] [PubMed]
- Billich, A.; Hammerschmid, F.; Peichl, P.; Wenger, R.; Zenke, G.; Quesniaux, V.; Rosenwirth, B. Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus (HIV) type 1: Interference with HIV protein-cyclophilin A interactions. J. Virol. 1995, 69, 2451–2461. [Google Scholar] [CrossRef]
- Angelin, A.; Tiepolo, T.; Sabatelli, P.; Grumati, P.; Bergamin, N.; Golfieri, C.; Mattioli, E.; Gualandi, F.; Ferlini, A.; Merlini, L.; et al. Mitochondrial dysfunction in the pathogenesis of Ullrich congenital muscular dystrophy and prospective therapy with cyclosporins. Proc. Natl. Acad. Sci. USA 2007, 104, 991–996. [Google Scholar] [CrossRef]
- Angelin, A.; Bonaldo, P.; Bernardi, P. Altered threshold of the mitochondrial permeability transition pore in Ullrich congenital muscular dystrophy. Biochim. Biophys. Acta 2008, 1777, 893–896. [Google Scholar] [CrossRef]
- Sabatelli, P.; Palma, E.; Angelin, A.; Squarzoni, S.; Urciuolo, A.; Pellegrini, C.; Tiepolo, T.; Bonaldo, P.; Gualandi, F.; Merlini, L.; et al. Critical evaluation of the use of cell cultures for inclusion in clinical trials of patients affected by collagen VI myopathies. J. Cell. Physiol. 2012, 227, 2927–2935. [Google Scholar] [CrossRef]
- Jimenez-Mallebrera, C.; Maioli, M.A.; Kim, J.; Brown, S.C.; Feng, L.; Lampe, A.K.; Bushby, K.; Hicks, D.; Flanigan, K.M.; Bonnemann, C.; et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscul. Disord. NMD 2006, 16, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.J.; Maslen, C.L.; Keene, D.R.; Glanville, R.W. Type VI collagen anchors endothelial basement membranes by interacting with type IV collagen. J. Biol. Chem. 1997, 272, 26522–26529. [Google Scholar] [CrossRef] [PubMed]
- Sabatelli, P.; Bonaldo, P.; Lattanzi, G.; Braghetta, P.; Bergamin, N.; Capanni, C.; Mattioli, E.; Columbaro, M.; Ognibene, A.; Pepe, G.; et al. Collagen VI deficiency affects the organization of fibronectin in the extracellular matrix of cultured fibroblasts. Matrix Biol. J. Int. Soc. Matrix Biol. 2001, 20, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Sardone, F.; Traina, F.; Bondi, A.; Merlini, L.; Santi, S.; Maraldi, N.M.; Faldini, C.; Sabatelli, P. Tendon Extracellular Matrix Alterations in Ullrich Congenital Muscular Dystrophy. Front. Aging Neurosci. 2016, 8, 131. [Google Scholar] [CrossRef]
- Antoniel, M.; Traina, F.; Merlini, L.; Andrenacci, D.; Tigani, D.; Santi, S.; Cenni, V.; Sabatelli, P.; Faldini, C.; Squarzoni, S. Tendon Extracellular Matrix Remodeling and Defective Cell Polarization in the Presence of Collagen VI Mutations. Cells 2020, 9, 409. [Google Scholar] [CrossRef]
- Dziadek, M.; Darling, P.; Bakker, M.; Overall, M.; Zhang, R.Z.; Pan, T.C.; Tillet, E.; Timpl, R.; Chu, M.L. Deposition of collagen VI in the extracellular matrix during mouse embryogenesis correlates with expression of the alpha 3(VI) subunit gene. Exp. Cell Res. 1996, 226, 302–315. [Google Scholar] [CrossRef]
- Shinwari, J.M.A.; Khan, A.; Awad, S.; Shinwari, Z.; Alaiya, A.; Alanazi, M.; Tahir, A.; Poizat, C.; Al Tassan, N. Recessive mutations in COL25A1 are a cause of congenital cranial dysinnervation disorder. Am. J. Hum. Genet. 2015, 96, 147–152. [Google Scholar] [CrossRef]
- Mercer, D.K.; Nicol, P.F.; Kimbembe, C.; Robins, S.P. Identification, expression, and tissue distribution of the three rat lysyl hydroxylase isoforms. Biochem. Biophys. Res. Commun. 2003, 307, 803–809. [Google Scholar] [CrossRef]
- Nuytinck, L.; Freund, M.; Lagae, L.; Pierard, G.E.; Hermanns-Le, T.; De Paepe, A. Classical Ehlers-Danlos Syndrome Caused by a Mutation in Type I Collagen. Am. J. Hum. Genet. 2000, 66, 1398–1402. [Google Scholar] [CrossRef]
- Cosgrove, D.; Liu, S. Collagen IV Diseases: A Focus on the Glomerular Basement Membrane in Alport Syndrome. Matrix Biol. J. Int. Soc. Matrix Biol. 2017, 57–58, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Wakabayashi, T.; Oizumi, H.; Nishio, S.; Sato, T.; Harada, A.; Fujii, D.; Matsuo, Y.; Hashimoto, T.; Iwatsubo, T. CLAC-P/Collagen Type XXV Is Required for the Intramuscular Innervation of Motoneurons during Neuromuscular Development. J. Neurosci. 2014, 34, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.O.; Al-Mesfer, S. Recessive COL25A1 mutations cause isolated congenital ptosis or exotropic Duane syndrome with synergistic divergence. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2015, 19, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Yüksel Ülker, A.; Uludağ Alkaya, D.; Elkanova, L.; Şeker, A.; Akpınar, E.; Akarsu, N.A.; Uyguner, Z.O.; Tüysüz, B. Long-Term Follow-Up Outcomes of 19 Patients with Osteogenesis Imperfecta Type XI and Bruck Syndrome Type I Caused by FKBP10 Variants. Calcif. Tissue Int. 2021, 109, 633–644. [Google Scholar] [CrossRef]
- Otaify, G.A.; Abdel-Hamid, M.S.; Hassib, N.F.; Elhossini, R.M.; Abdel-Ghafar, S.F.; Aglan, M.S. Bruck syndrome in 13 new patients: Identification of five novel FKBP10 and PLOD2 variants and further expansion of the phenotypic spectrum. Am. J. Med. Genet. A. 2022, 188, 1815–1825. [Google Scholar] [CrossRef]
- Schwarze, U.; Cundy, T.; Pyott, S.M.; Christiansen, H.E.; Hegde, M.R.; Bank, R.A.; Pals, G.; Ankala, A.; Conneely, K.; Seaver, L.; et al. Mutations in FKBP10, which result in Bruck syndrome and recessive forms of osteogenesis imperfecta, inhibit the hydroxylation of telopeptide lysines in bone collagen. Hum. Mol. Genet. 2013, 22, 1–17. [Google Scholar] [CrossRef]
- Gjaltema, R.A.F.; van der Stoel, M.M.; Boersema, M.; Bank, R.A. Disentangling mechanisms involved in collagen pyridinoline cross-linking: The immunophilin FKBP65 is critical for dimerization of lysyl hydroxylase 2. Proc. Natl. Acad. Sci. USA 2016, 113, 7142–7147. [Google Scholar] [CrossRef]
- Gistelinck, C.; Weis, M.; Rai, J.; Schwarze, U.; Niyazov, D.; Song, K.M.; Byers, P.H.; Eyre, D.R. Abnormal Bone Collagen Cross-Linking in Osteogenesis Imperfecta/Bruck Syndrome Caused by Compound Heterozygous PLOD2 Mutations. JBMR Plus 2021, 5, e10454. [Google Scholar] [CrossRef]
- Gistelinck, C.; Witten, P.E.; Huysseune, A.; Symoens, S.; Malfait, F.; Larionova, D.; Simoens, P.; Dierick, M.; Van Hoorebeke, L.; De Paepe, A.; et al. Loss of Type I Collagen Telopeptide Lysyl Hydroxylation Causes Musculoskeletal Abnormalities in a Zebrafish Model of Bruck Syndrome. J. Bone Miner. Res. 2016, 31, 1930–1942. [Google Scholar] [CrossRef]
- Eyre, D.R.; Koob, T.J.; Van Ness, K.P. Quantitation of hydroxypyridinium crosslinks in collagen by high-performance liquid chromatography. Anal. Biochem. 1984, 137, 380–388. [Google Scholar] [CrossRef]
- Leal, G.F.; Nishimura, G.; Voss, U.; Bertola, D.R.; Åström, E.; Svensson, J.; Yamamoto, G.L.; Hammarsjö, A.; Horemuzova, E.; Papadogiannakis, N.; et al. Expanding the Clinical Spectrum of Phenotypes Caused by Pathogenic Variants in PLOD2. J. Bone Miner. Res. 2018, 33, 753–760. [Google Scholar] [CrossRef]
- Hershkovitz, D.; Mandel, H.; Ishida-Yamamoto, A.; Chefetz, I.; Hino, B.; Luder, A.; Indelman, M.; Bergman, R.; Sprecher, E. Defective lamellar granule secretion in arthrogryposis, renal dysfunction, and cholestasis syndrome caused by a mutation in VPS33B. Arch. Dermatol. 2008, 144, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Galmes, R.; Gogolina, E.; Straatman-Iwanowska, A.; Reay, K.; Banushi, B.; Bruce, C.K.; Cullinane, A.R.; Romero, R.; Chang, R.; et al. Associations among genotype, clinical phenotype, and intracellular localization of trafficking proteins in ARC syndrome. Hum. Mutat. 2012, 33, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.-L.; Chen, P.; Zhou, Y.-Y.; Jiang, Y.-M.; Gao, Y.; Zhang, H.-Z.; Guan, L.-H.; Wang, C.-H.; Liu, J.-L.; Huang, M.; et al. Hepatic Vps33b deficiency aggravates cholic acid-induced cholestatic liver injury in male mice. Acta Pharmacol. Sin. 2022, 43, 933–940. [Google Scholar] [CrossRef]
- Gruber, R.; Rogerson, C.; Windpassinger, C.; Banushi, B.; Straatman-Iwanowska, A.; Hanley, J.; Forneris, F.; Strohal, R.; Ulz, P.; Crumrine, D.; et al. Autosomal Recessive Keratoderma-Ichthyosis-Deafness (ARKID) Syndrome Is Caused by VPS33B Mutations Affecting Rab Protein Interaction and Collagen Modification. J. Investig. Dermatol. 2017, 137, 845–854. [Google Scholar] [CrossRef]
- Bull, L.N.; Mahmoodi, V.; Baker, A.J.; Jones, R.; Strautnieks, S.S.; Thompson, R.J.; Knisely, A.S. VPS33B mutation with ichthyosis, cholestasis, and renal dysfunction but without arthrogryposis: Incomplete ARC syndrome phenotype. J. Pediatr. 2006, 148, 269–271. [Google Scholar] [CrossRef]
- Qiu, Y.-L.; Liu, T.; Abuduxikuer, K.; Hao, C.-Z.; Gong, J.-Y.; Zhang, M.-H.; Li, L.-T.; Yan, Y.-Y.; Li, J.-Q.; Wang, J.-S. Novel missense mutation in VPS33B is associated with isolated low gamma-glutamyltransferase cholestasis: Attenuated, incomplete phenotype of arthrogryposis, renal dysfunction, and cholestasis syndrome. Hum. Mutat. 2019, 40, 2247–2257. [Google Scholar] [CrossRef] [PubMed]
- Alter, S.; Hotz, A.; Jahn, A.; Di Donato, N.; Schröck, E.; Smitka, M.; von der Hagen, M.; Schallner, J.; Menschikowski, M.; Gillitzer, C.; et al. Novel VPS33B mutation in a patient with autosomal recessive keratoderma-ichthyosis-deafness syndrome. Am. J. Med. Genet. A 2018, 176, 2862–2866. [Google Scholar] [CrossRef]
- Rogerson, C.; Gissen, P. VPS33B and VIPAR are essential for epidermal lamellar body biogenesis and function. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Aflatounian, M.; Smith, H.; Farahani, F.; Tofighi Naeem, A.; Straatman-Iwanowska, A.; Zoghi, S.; Khatri, U.; Tajdini, P.; Fallahi, G.H.; Gissen, P.; et al. Novel VIPAS39 mutation in a syndromic patient with arthrogryposis, renal tubular dysfunction and intrahepatic cholestasis. Eur. J. Med. Genet. 2016, 59, 237–239. [Google Scholar] [CrossRef]
- Rautavuoma, K.; Takaluoma, K.; Sormunen, R.; Myllyharju, J.; Kivirikko, K.I.; Soininen, R. Premature aggregation of type IV collagen and early lethality in lysyl hydroxylase 3 null mice. Proc. Natl. Acad. Sci. USA 2004, 101, 14120–14125. [Google Scholar] [CrossRef] [PubMed]
- Ewans, L.J.; Colley, A.; Gaston-Massuet, C.; Gualtieri, A.; Cowley, M.J.; McCabe, M.J.; Anand, D.; Lachke, S.A.; Scietti, L.; Forneris, F.; et al. Pathogenic variants in PLOD3 result in a Stickler syndrome-like connective tissue disorder with vascular complications. J. Med. Genet. 2019, 56, 629–638. [Google Scholar] [CrossRef]
- Maddirevula, S.; Alzahrani, F.; Al-Owain, M.; Al Muhaizea, M.A.; Kayyali, H.R.; AlHashem, A.; Rahbeeni, Z.; Al-Otaibi, M.; Alzaidan, H.I.; Balobaid, A.; et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet. Med. 2019, 21, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Vahidnezhad, H.; Youssefian, L.; Saeidian, A.H.; Touati, A.; Pajouhanfar, S.; Baghdadi, T.; Shadmehri, A.A.; Giunta, C.; Kraenzlin, M.; Syx, D.; et al. Mutations in PLOD3, encoding lysyl hydroxylase 3, cause a complex connective tissue disorder including recessive dystrophic epidermolysis bullosa-like blistering phenotype with abnormal anchoring fibrils and type VII collagen deficiency. Matrix Biol. J. Int. Soc. Matrix Biol. 2019, 81, 91–106. [Google Scholar] [CrossRef]
- Risteli, M.; Ruotsalainen, H.; Salo, A.M.; Sormunen, R.; Sipilä, L.; Baker, N.L.; Lamandé, S.R.; Vimpari-Kauppinen, L.; Myllylä, R. Reduction of Lysyl Hydroxylase 3 Causes Deleterious Changes in the Deposition and Organization of Extracellular Matrix. J. Biol. Chem. 2009, 284, 28204–28211. [Google Scholar] [CrossRef]
- Wartosch, L.; Günesdogan, U.; Graham, S.C.; Luzio, J.P. Recruitment of VPS33A to HOPS by VPS16 Is Required for Lysosome Fusion with Endosomes and Autophagosomes. Traffic Cph. Den. 2015, 16, 727–742. [Google Scholar] [CrossRef]
- Cullinane, A.R.; Straatman-Iwanowska, A.; Zaucker, A.; Wakabayashi, Y.; Bruce, C.K.; Luo, G.; Rahman, F.; Gürakan, F.; Utine, E.; Ozkan, T.B.; et al. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat. Genet. 2010, 42, 303–312. [Google Scholar] [CrossRef]
- Myllylä, R.; Wang, C.; Heikkinen, J.; Juffer, A.; Lampela, O.; Risteli, M.; Ruotsalainen, H.; Salo, A.; Sipilä, L. Expanding the lysyl hydroxylase toolbox: New insights into the localization and activities of lysyl hydroxylase 3 (LH3). J. Cell. Physiol. 2007, 212, 323–329. [Google Scholar] [CrossRef]
- Ruotsalainen, H.; Sipilä, L.; Vapola, M.; Sormunen, R.; Salo, A.M.; Uitto, L.; Mercer, D.K.; Robins, S.P.; Risteli, M.; Aszodi, A.; et al. Glycosylation catalyzed by lysyl hydroxylase 3 is essential for basement membranes. J. Cell Sci. 2006, 119, 625–635. [Google Scholar] [CrossRef]
- Sipilä, L.; Ruotsalainen, H.; Sormunen, R.; Baker, N.L.; Lamandé, S.R.; Vapola, M.; Wang, C.; Sado, Y.; Aszodi, A.; Myllylä, R. Secretion and assembly of type IV and VI collagens depend on glycosylation of hydroxylysines. J. Biol. Chem. 2007, 282, 33381–33388. [Google Scholar] [CrossRef] [PubMed]
- Sricholpech, M.; Perdivara, I.; Nagaoka, H.; Yokoyama, M.; Tomer, K.B.; Yamauchi, M. Lysyl hydroxylase 3 glucosylates galactosylhydroxylysine residues in type I collagen in osteoblast culture. J. Biol. Chem. 2011, 286, 8846–8856. [Google Scholar] [CrossRef] [PubMed]
- Agakidou, E.; Agakidis, C.; Kambouris, M.; Printza, N.; Farini, M.; Vourda, E.; Gerou, S.; Sarafidis, K. A Novel Mutation of VPS33B Gene Associated with Incomplete Arthrogryposis-Renal Dysfunction-Cholestasis Phenotype. Case Rep. Genet. 2020, 2020, 8872294. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, F.S.; Mancini, G.M.S.; Maugeri, A.; Cobben, J.M. Ehlers Danlos syndrome, kyphoscoliotic type due to Lysyl Hydroxylase 1 deficiency in two children without congenital or early onset kyphoscoliosis. Eur. J. Med. Genet. 2017, 60, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Reilly, D.M.; Lozano, J. Skin collagen through the lifestages: Importance for skin health and beauty. Plast. Aesthet. Res. 2021, 8, 2. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Gene | Protein Encoded | Function of Protein | Clinical Phenotype |
|---|---|---|---|
| PLOD2 | LH2 | Post-translational modification of collagen | Congenital contractures, joint stiffness, osteoporosis, recurrent fractures, short stature, pterygia, kyphosis, idiopathic scoliosis, fragile skin |
| FKBP10 | FKBP65 | Forms a complex with LH2 to allow LH2 dimerization and telopeptide hydroxylase activity | Congenital contractures, joint stiffness, osteoporosis, recurrent fractures, short stature, pterygia, kyphosis, idiopathic scoliosis, aplastic patellae & radius, talipes, reduced tendon reflexes, melanocytic naevus, fragile skin |
| COL6A1 | Collagen VI alpha 1 chain | Structural role in basement membrane, inhibition of apoptosis, regulation of cellular adhesion and cellular proliferation | Congenital contractures, muscle weakness, hyperextensibility of joints, kyphosis, spinal rigidity, decreased foetal movements, diaphragmatic weakness, hip dislocation, torticollis, delayed motor skills, keloid scarring, follicular hyperkeratosis |
| COL6A2 | Collagen VI alpha 2 chain | Structural role in basement membrane, inhibition of apoptosis, regulation of cellular adhesion and cellular proliferation | Congenital contractures, muscle weakness, hyperextensibility of joints, kyphosis, spinal rigidity, decreased foetal movements, diaphragmatic weakness, hip dislocation, torticollis, delayed motor skills, keloid scarring, follicular hyperkeratosis |
| COL6A3 | Collagen VI alpha 3 chain | Structural role in basement membrane, inhibition of apoptosis, regulation of cellular adhesion and cellular proliferation | Congenital contractures, muscle weakness, hyperextensibility of joints, kyphosis, spinal rigidity, decreased foetal movements, diaphragmatic weakness, hip dislocation, torticollis, delayed motor skills, keloid scarring, follicular hyperkeratosis |
| COL25A1 | Collagen XXV | Role in myogenesis during the primary formation of myofibrils by enabling motor innervation | Congenital ptosis, Duane syndrome, congenital contractures |
| VPS33B | VPS33B | LH3-mediated post-translational modification of collagen | Congenital contractures, renal tubular dysfunction, cholestasis, ichthyosis |
| VIPAS39 | VIPAR | LH3-mediated post-translational modification of collagen | Congenital contractures, renal tubular dysfunction, cholestasis, ichthyosis |
| PLOD3 | LH3 | Post-translational modification of collagen | Finger contractures, ocular abnormalities, sensorineural hearing loss, congenital heart defects, facial abnormalities, skin blistering |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picker, S.M.; Parker, G.; Gissen, P. Features of Congenital Arthrogryposis Due to Abnormalities in Collagen Homeostasis, a Scoping Review. Int. J. Mol. Sci. 2023, 24, 13545. https://doi.org/10.3390/ijms241713545
Picker SM, Parker G, Gissen P. Features of Congenital Arthrogryposis Due to Abnormalities in Collagen Homeostasis, a Scoping Review. International Journal of Molecular Sciences. 2023; 24(17):13545. https://doi.org/10.3390/ijms241713545
Chicago/Turabian StylePicker, Sarah MacKenzie, George Parker, and Paul Gissen. 2023. "Features of Congenital Arthrogryposis Due to Abnormalities in Collagen Homeostasis, a Scoping Review" International Journal of Molecular Sciences 24, no. 17: 13545. https://doi.org/10.3390/ijms241713545