Abstract
Apolipoprotein(a) (apo(a)) is the protein component that defines lipoprotein(a) (Lp(a)) particles and is encoded by the LPA gene. The apo(a) is extremely heterogeneous in size due to the copy number variations in the kringle-IV type 2 (KIV2) domains. In this review, we aim to discuss the role of genetics in establishing Lp(a) as a risk factor for coronary heart disease (CHD) by examining a series of molecular biology techniques aimed at identifying the best strategy for a possible application in clinical research and practice, according to the current gold standard.
1. Introduction
During the past decades, the attention on Lipoprotein(a) (Lp(a)) has exponentially grown. Lp(a) is a form of a low-density lipoprotein (LDL) and is an established and genetically determined risk factor for atherosclerosis, coronary artery disease (CAD), stroke, and aortic stenosis [1,2]. Moreover, high plasma levels of Lp(a) nearly double the risk of developing peripheral artery disease [3]. Mendelian studies also associate a high Lp(a) with a mildly though significantly higher risk of developing atrial fibrillation. The pathophysiological link between these two conditions, however, has yet to be clarified [4].
A recent comprehensive meta-analysis of 75 epidemiological studies involving 957,253 participants concluded that the hazard ratios (HRs) and 95% confidence intervals (95%CI) for the top versus the bottom tertiles of Lp(a) levels and the risk of all-cause mortality is 1.09 (95%CI: 1.01 to 1.18) in the general population and 1.18 (95%CI: 1.04 to 1.34) in patients with cardiovascular diseases (CVDs). As expected, the HRs for CVD mortality is 1.33 (95%CI: 1.11 to 1.58) in the general population and 1.25 (95%CI: 1.10 to 1.43) in patients with CVDs, reaching exceptionally high values in patients with type 2 diabetes (2.53 (95%CI: 1.13 to 5.64)). The same analysis highlighted a linear dose–response relationship between Lp(a) and an increased risk of CVD, with a 31% and 15% greater risk of CVD death in the general population and in patients with CVD, respectively, for each 50 mg/dL rise in Lp(a) plasma levels [5].
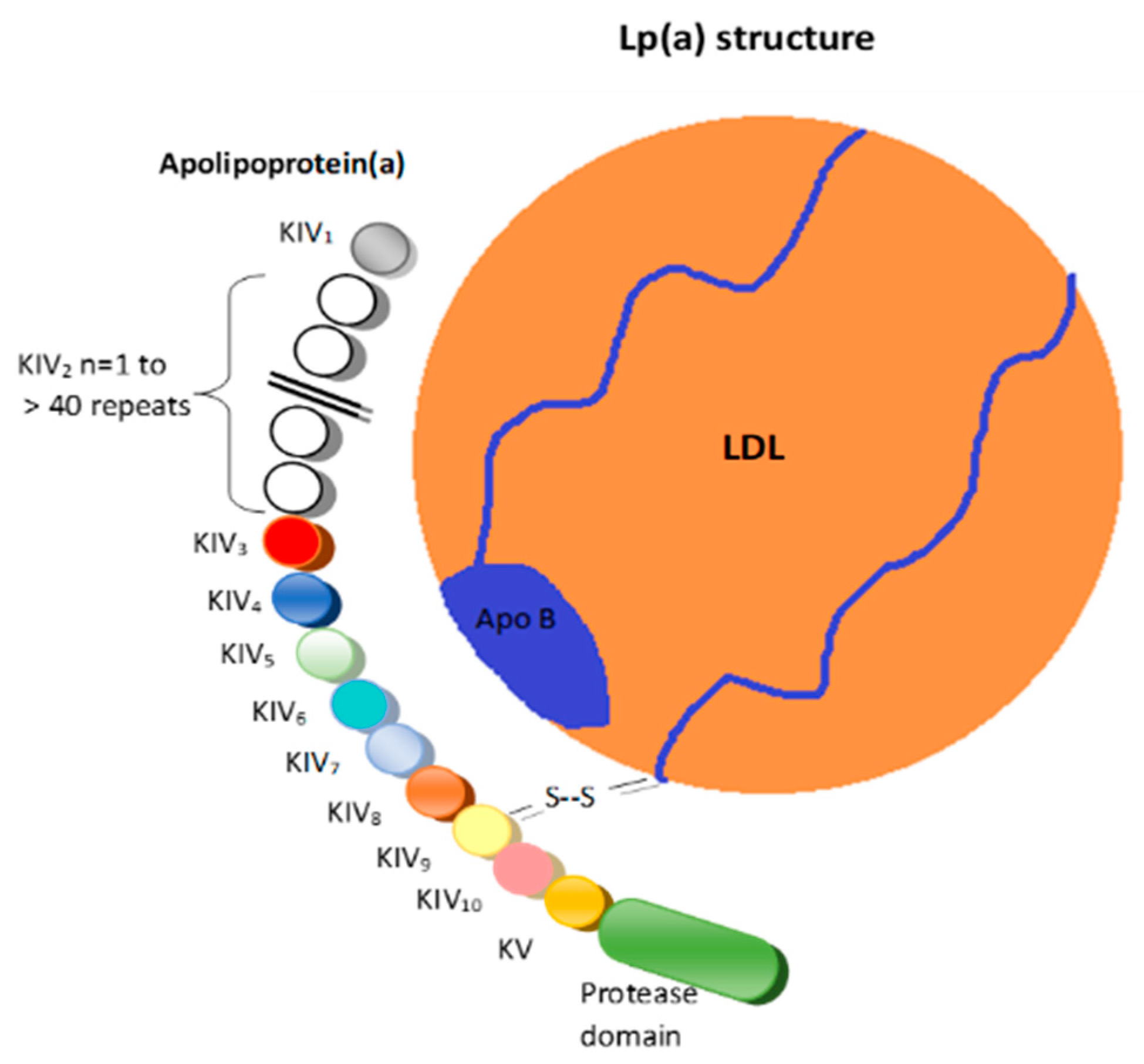
Structurally, Lp(a) is a variant of LDL with an apolipoprotein(a) (apo(a)) that is bound to the apolipoprotein B100 (Figure 1) [6].
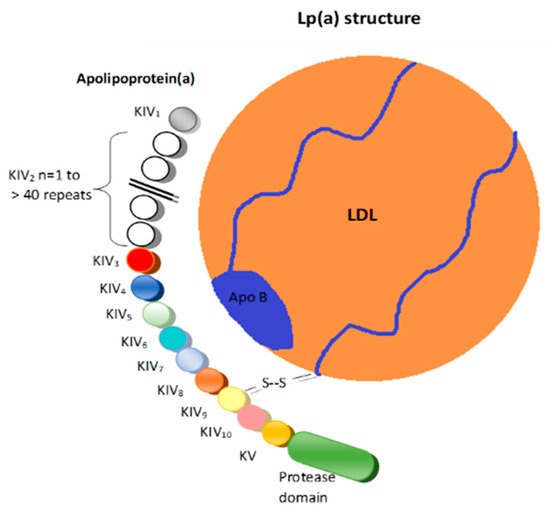
Figure 1.
Lipoprotein(a). LDL = Low-density lipoprotein; K = Kringle; Apo B = Apolipoprotein B 100; S-S = Disulfide bridge.
These two structures are linked by a disulfide bridge and are assembled in the cell membranes of hepatocytes [7].
The plasma levels of Lp(a) are largely mediated by the LPA gene locus on chromosome 6q22-23, with small or negligible effects resulting from diet [8,9]. The levels of Lp(a) in plasma are determined by the rate of entry of these particles into the circulation (i.e., the production rate) and the efficiency of their removal (i.e., fractional clearance rate) [10].
There is an inverse relationship between the plasma concentrations of Lp(a) and the isoform of apo(a) [11]. The variation in the apo(a) isoform is induced by the different numbers of kringle IV type-2 (KIV-2) repeats in the LPA gene, which leads to variable levels of Lp(a) in the general population [12]. Individuals with fewer kringle repeats have smaller Lp(a) particles but higher serum levels of Lp(a). In addition, the larger the isoforms of apo(a), the greater the accumulation of its intracellular precursor in the endoplasmic reticulum. The short alleles of the KIV-2 copy number variation (CNV) have been shown to be associated with an increased risk of coronary heart disease (CHD) in almost all populations [13].
Recently, the circulating levels of individual apo(a) isoform sizes have been shown to be correlated with the fractionated clearance rate and the production rate of Lp(a). Some studies showed that the isoform size is tightly associated with the Lp(a) production rate (i.e., smaller isoforms with fewer KIV-2 repeats had higher rates of Lp(a) production) [14,15], while other studies showed that the fractionated clearance rate and the production rate of Lp(a) are both affected by the isoform size [16,17].
Lp(a) concentrations in the blood vary by more than a thousand-fold between individuals, ranging from less than 0.1 to more than 300 mg/dL, depending on the size of apo(a) that is encoded by the LPA gene [18]. The plasma concentrations of Lp(a) show considerable variability not only among individuals within a population far exceeding those of other plasma lipoproteins, but they also vary across different human populations [19]. The KIV-2 copy number ranges from 1 to >40, and the CNV of KIV-2 shows a >95% heterozygosity in most populations [20]. Screening patients for elevated Lp(a) is strongly encouraged as an effective tool to identify individuals requiring more aggressive lipid-lowering therapy to reduce the CVD risk [21,22,23,24]. Lp(a) levels above 50 mg/dL are correlated with an increased risk for the development of CVD.
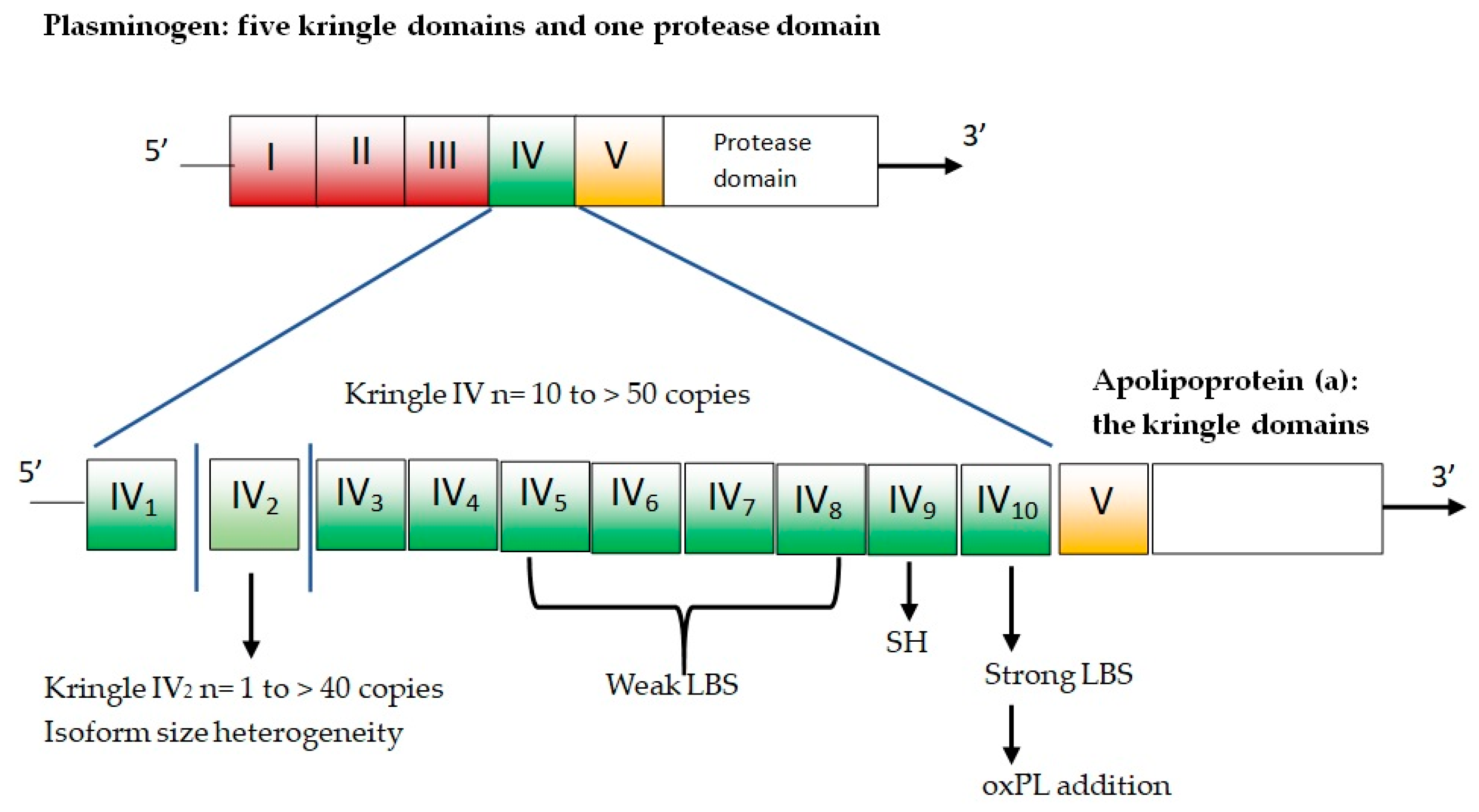
Apo(a) is composed of several kringles—which are three-loop structural domains—and a mutated protease domain with no proteolytic activity. The human LPA gene is evolved via duplication from the PLG gene [25]. The PLG gene is characterized by five different kringle domains and one protease domain (Figure 2).
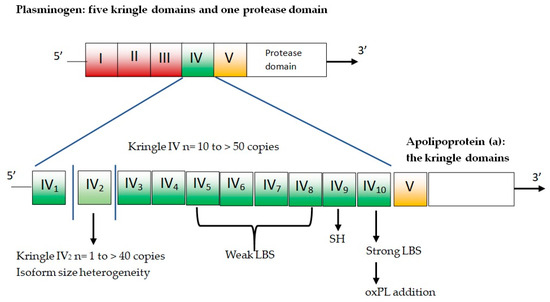
Figure 2.
Plasminogen and apolipoprotein(a). LBS = lysine binding sites; oxPL = oxidized phospholipids; SH = free cysteine residue contained in KIV9 that mediates covalent coupling to apoB-100 in Lp(a).
The five plasminogen kringle domains are numbered from 1 to 5 (i.e., KI, KII, KIII, KIV, and KV). In the LPA gene, KI, KII, and KIII are lost by deletion, while KIV sustains mutations and differentiation, resulting in 10 slightly different KIV domains (KIV types 1–10). There is only one copy for each of these domains, except for KIV-2, which can present up to forty copies and represents the variable part of apo(a) [26]. Homozygous individuals express only one isoform, while heterozygotes have two distinct particle types in plasma. The remainder of apo(a) is composed of KV and the mutated protease domain and has a higher number of KIV-2 copies, and larger apo(a) isoform results are associated with lower plasma concentrations of Lp(a) [6].
High levels of Lp(a) may induce a prothrombotic/antifibrinolytic effect because apo(a) resembles both plasminogen and plasmin but does not have fibrinolytic activity. In 20% of children with early arterial ischemic stroke, high levels of Lp(a) were detected [27]. Higher Lp(a) levels have also been detected in patients affected by retinal thrombosis [28]. In particular, the apo(a) KIV-2 copy number variation is associated with a venous thromboembolism risk [29]. The prothrombotic effect of Lp(a) could also contribute to the higher incidence of microvascular diabetic complications, such as nephropathy [30] and feet ulcers [31]. However, it is not yet clear if specific apo(a) isoforms or the Lp(a) mass is directly associated with a higher risk of type 2 diabetes complications. On the other side, high Lp(a) levels are associated with a risk of ischemic stroke [32]. The reason for this observation has also yet to be further explained, but could be related to the individual high structural variability of Lp(a). In addition, high levels of Lp(a) may accelerate atherosclerosis because this particle is rich in cholesterol [33]. The data from pathophysiology, epidemiology and genetics support the causal role of Lp(a) in CVD and aortic valve calcification and stenosis. In fact, meta-analyses of epidemiological studies confirm that its plasma level is a strong predictor of calcium deposition, both in coronary arteries [34] and the aortic valve [35].
Small isoforms of Lp(a) are linked to elevated serum Lp(a) levels, and thus, to an increased risk of coronary artery disease. Thus, screening for genetic factors of Lp(a) is expected to have added value and proves to be cost-effective in primary prevention, whereas the monitoring of serum Lp(a) levels could improve the prediction of the clinical risk of atherosclerotic damage in patients with ASCVD [36,37].
In this context, the aim of our critical review is to summarize the available genotyping and phenotyping techniques that are potentially able to distinguish subjects at different cardiovascular risks despite having similarly high Lp(a) plasma levels.
2. Techniques to Assess the Size of KIV-2 CNV
Several laboratory techniques can be employed to assess the size of the KIV-2 CNV. In this review article, we will explore the pulsed-field gel electrophoresis (PFGE)/Southern blot, the quantitative polymerase chain reaction (qPCR), the fiber fluorescence in situ hybridization (fiber-FISH), and the Western blot (using apo(a) specific antibodies).
2.1. Genotyping Techniques
The KIV-2 CNV size polymorphisms can be analyzed to characterize the genetic architecture of Lp(a) [38]. Identifying single nucleotide polymorphisms (SNPs) in the LPA gene has a prognostic significance. The most recent evidence from population studies support the role of some SNIPs (rs6415084 and rs12194138) in predicting future ASCV events independently of the concentration of Lp(a) in the blood [39].
Among the experimental procedures based on deoxyribonucleic acid (DNA) analysis, the largely used PFGE/Southern blot, qPCR, and fiber-FISH will be reviewed thereafter.
2.1.1. Pulsed-Field Gel Electrophoresis (PFGE)/Southern Blot
Variations in the number of LPA KIV2 repeats can be estimated via immunoblotting electrophoresis or the pulsed-field electrophoresis of unamplified genomic DNA. Immunoblotting electrophoresis is used to detect apo(a) protein isoforms [40], whereas the pulsed-field electrophoresis of unamplified genomic DNA is used to detect variations in the LPA gene [41]. These experimental methods are both technically challenging, laborious, and time-consuming. In addition, they require high-quality genetic material and larger amounts of starting DNA than the Southern blot.
APO(a) polymorphisms can be examined via PFGE DNA by means of the use of various restriction enzymes (SwaI, KpnI, KspI, SfiI, and NotI) and an apo(a) kringle-IV-specific probe [42]. According to previous evidence, these enzymes ensure similar results by detecting the same polymorphism in the KIV repeat domain of apo(a) [40]. A PFGE analysis using the KpnI restriction enzyme was found to be able to identify 26 different alleles in unrelated individuals (with sizes ranging from 32 kilobases (kb) to 189 kb, progressively increasing by increments of 5.6 kb corresponding to one KIV unit), with a perfect match between the size of apo(a) DNA phenotypes and the size of apo(a) isoforms in plasma [40]. Unfortunately, PFGE is time-consuming and labor-intensive and can only be performed in reference laboratories with skillful technicians [43]. Furthermore, by measuring the DNA fragments in kb, the molecular weights of the genomic products cannot be derived directly, but alleles are derived from multiples of 5.6 kb. Finally, the post-translational processing of APO(a) cannot be detected.
2.1.2. Quantitative Polymerase Chain Reaction (qPCR)
The qPCR is a faster and more sensitive and reliable assay to detect the number of KIV2 repeats in LPA [44].
One major limitation of the qPCR approach is that it measures the total number of KIV2 repeats instead of the number of KIV2 repeats in each single allele. It follows that qPCR would not always be able to differentiate two different individuals. For instance, an individual who inherited 10 KIV2 repeats from a parent and 20 KIV2 repeats from another parent will exhibit a different biochemical phenotype than another individual who inherited 15 KIV2 repeats from either parent. For the same reason, an individual carrying a null allele would appear to be homozygous for the active allele when using immunoblotting, but qPCR would correctly identify the person as having many KIV2 repeats. Despite these limitations, qPCR is a fast and convenient method to identify the relative total number of KIV2 repeats from genomic DNA samples that have been stored. Moreover, the number of KIV2 repeats in LPA—as assessed using qPCR—has been shown to be correlated with both the size of apo(a) isoforms, as determined via immunoblotting, and the plasma concentration of Lp(a) [45].
2.1.3. Fiber Fluorescence In Situ Hybridization (Fiber-FISH)
The fiber-FISH method is the most precise technique available to determine the size of the KIV-2 CNV as it allows one to count, under a fluorescence microscope, the number of KIV-2 copies on individual alleles [46]. Some unavoidable technical limitations (e.g., the conditions of sample collection, preservation, and storage before analysis) make this method not feasible to handle large sample sizes, although it has historically been used to define standards to be applied in large-scale epidemiologic studies [23,24].
The FISH technique uses high-resolution FITC-labeled 4 kb probes that are intron sequences cloned from KIV-2 repeats [47]. In this way, good linear hybridization signals are obtained on heterozygotes, where there are two different sizes corresponding to different KIV-2 repeat numbers of the respective LPA alleles [48]. A prerequisite for the accurate counting of KIV-2 repeats is to avoid irregular (discontinuous) hybridization signals. For this purpose, to improve the analysis and obtain more reliable results, the conventional agarose plug method for isolating high-molecular-length DNA can be modified. The number of KIV-2 repeats in each allele can then be accurately determined by counting the FISH signals that appear in a “dotted line” rather than a “beadsona string” pattern under a fluorescence microscope [49].
2.2. Phenotyping Techniques
Techniques that allow for protein separation such as Western blotting can be used to assess the apo(a) isoform size and associate the plasma concentrations of Lp(a) with specific alleles of LPA.
Western Blot Using Antibodies for apo(a) (Immunoblotting)
Sodium dodecyl sulfate (SDS) agarose gel electrophoresis followed by immunoblotting can be used to assess the apo(a) isoform size and associate the relative proportion of the total Lp(a) plasma concentration to a specific LPA allele [49]. This semi-quantitative analysis via densitometry is the most common tool used to assess the specific allele levels of Lp(a) [48]. However, neither the identification of the unexpressed alleles nor the evaluation of samples with a single band in Western blots is feasible without prior knowledge of the actual size of the KIV-2 CNV of both alleles, which could be null or homozygous in size. For this reason, the most reliable assignment between the allele and Lp(a) concentration is ensured by a combination of KIV-2 CNV genotyping and phenotyping by apo(a) [50].
SDS-PAGE (PolyAcrylamide Gel Electrophoresis) with polyacrylamide gel at a low percentage (4%) separates the proteins primarily by mass [51]. However, the high content of carbohydrates in the apo(a) makes it difficult to obtain an accurate molecular weight estimation in SDS-PAGE gels [52]. As shown by Kamboh et al., SDS-agarose is a more efficient method to assess the apo(a) isoform size, also in consideration of the high molecular weight of apo(a) [53]. As an effect, larger proteins are separated more easily in a gel that has a lower percentage of acrylamide because the holes in the web are larger. In addition, the increased migration distance between bands running close together even allows for the detection of very small differences (1 mm resolution limit) between them [54].
3. Discussion
Even if the epidemiological and pathophysiological link between the Lp(a) plasma levels and CVD has been suspected some decades ago, the role of high Lp(a) levels as an independent CV risk factor has been underestimated in clinical practice for a long time because of the lack of therapeutic options that are able to significantly reduce them [55]. Diet and physical exercise have nearly no impact on the Lp(a) plasma levels. L-carnitine and coenzyme Q10 are the only nutraceuticals and dietary supplements that mildly improve this parameter, even though their effect is quite irrelevant from clinical and prognostic points of view [56]. Statins have a neutral effect on the Lp(a) plasma levels, balancing the negative effect of Lp(a) on the CV risk when the plasma Lp(a) is less than 50 mg/dL, but not for higher concentrations [57]. Nicotinic acid is the only drug that is able to reduce the Lp(a) by 20-30%, but it is not always well tolerated. Moreover, its use in the long term has never been associated with a significant reduction in CV events [58], even if estimates from genetic studies suggest that Lp(a) decreases might yield proportional reductions in the coronary risk by ≈2% overall and 6% in the top quintile by Lp(a) levels. Additionally, it must be recognized that the effect of nicotinic acid on Lp(a) plasma levels is proportional to the Lp(a) isoform size [59], creating the need for a more refined investigation method of Lp(a) to predict the drug effects. Mipomersen—an antisense oligonucleotide (ASO) directed against apolipoprotein-B 100 (apo B-100) mRNA in the liver—is also able to reduce the Lp(a) levels, but there is serious concern regarding its liver safety [60]. The cholesteryl ester transfer protein (CETP) inhibitors increase the potentially protective high-density lipoprotein (HDL) fraction and reduce the atherogenic non-HDL particles, such as Lp(a) [61]. For instance, according to the findings of a comprehensive meta-analysis of 10 randomized clinical studies (34781 patients overall), anacetrapib significantly lowers the blood concentrations of Lp(a) by a weighted mean difference (WMD) of −13.35 (95%CI: −18.31 to −8.39) [62]. Another CETP inhibitor, evacetrapib, at a dosage of 500 mg as a monotherapy or in combination with statin, has been shown to reduce Lp(a) by 30–40% over a period of 12 weeks [63]. Of course, a CV outcome trial is needed to translate these effects into a reduction in CV events.
Plasma proprotein convertase subtilisin/kexin type 9 (PCSK9) seems to be related to the Lp(a) plasma levels [64]. Hereinafter, the PCSK9 inhibitors and small interfering RNA (siRNA) targeting PCSK9 have been shown to significantly—though mildly—reduce the Lp(a) plasma levels [65,66]. A recent network meta-analysis of 41 randomized controlled trials with 17,601 participants—involving 23 unduplicated interventions—concluded that overall, all the available PCSK9 inhibitors are able to significantly reduce Lp(a) plasma levels (by up to 25.1%), where a biweekly dose of either 140 mg Evolocumab or 150 mg Alirocumab is the best treatment option [67]. Inclisiran was also shown to reduce Lp(a) by an average of 20.9% (95%CI: 25.8% to 15.9%) [68]. However, this effect—though positive—is rarely able to significantly impact the management of patients with high to very high Lp(a) plasma levels. For this reason, to date, in patients with severe and progressive CVD and very high levels of Lp(a), the most effective way to significantly reduce Lp(a) concentrations is apheresis, which is an invasive, expensive, impractical, and not widely available procedure [69]. In a study carried out in patients receiving PCSK9 inhibitors, the size of the apo(a) was shown to be an independent determinant of the treatment response, with each additional kringle domain being associated with a 3% additional reduction in Lp(a) [70]. Currently, emerging lipid-lowering drugs—namely small interfering RNA (siRNA) agents (olpasiran and SLN360) and the second generation ASO pelacarsen—are being developed to interfere with Lp(a) synthesis in the liver by blocking the translation of apo(a) mRNA in apo(a) [71]. These new drugs in development are very promising and overall safe, even if their cost-effectiveness will be carefully evaluated as a part of streamlining the health investments in CV prevention.
According to the most recent evidence, the relative expression of apo(a) isoforms does not change after the Lp(a) levels are lowered using ASO apo(a) treatment [72]. Moreover, the latter results suggest that apo(a) ASO treatment does not preferentially affect one isoform size over the other [10]. However, the knowledge of the available laboratory techniques could steer future research and hopefully help to identify individuals with Lp(a) with a greater atherogenic potential, for whom a reduction in Lp(a) levels with olpasiran or pelacarsen could be more advised and cost-effective.
Notwithstanding that the Lp(a) plasma level is per se an independent predictor of the individual CV risk, Lp(a)-associated CV risk in two individuals with similar Lp(a) levels and apo(a) isoform sizes may differ depending on the relative apo(a) allele expression and/or dominance pattern [73]. In particular, in recent years, two common splice site mutations (G4925A and G4733A with 22% and 38% carrier frequencies, respectively) in the LPA KIV-2 repeat region were shown to have a pronounced Lp(a) decreasing effect, with a concomitant lower risk of coronary artery disease in the carriers [74,75,76]. Although most genetic association studies investigating the potential discordance between the apo(a) isoform size and Lp(a) levels on the artery disease risk provided evidence for a causal role of Lp(a) levels in artery disease independent of the apo(a) isoform size, a Mendelian randomization analysis provided evidence that a variant linked to a smaller apo(a) isoform size, though not to Lp(a) levels, is associated with artery disease [77]. This latter finding makes it even more relevant to balance the different ethnicities in phase 3 clinical trials investigating Lp(a)-lowering drugs, since SNPs and the KIV-2 copy number together explain a greater proportion of the variation in the plasma Lp(a) concentrations in European Caucasians than in Chinese and South Asian individuals [78].
Some cost-effective and practical methods aimed at estimating the Lp(a) mass are not accurate. For instance, the vertical auto profile (VAP) poorly estimates the Lp(a) mass, while it more likely reflects the content of high-density lipoprotein cholesterol (HDL-C) in the overlapping density spectrum of Lp(a) and HDL. For this reason, patients with prior VAP-Lp(a) measurements may have a misclassification of Lp(a)-related risk [79]. Additionally, the assessment of the KIV-2 CNV size could furnish more reliable information on the Lp(a) mass and the related health risks.
Currently, the application of several laboratory techniques can be employed to assess the size of the KIV-2 CNV. However, each method has advantages and disadvantages (Table 1).

Table 1.
Main features of the laboratory techniques that can be employed to assess the size of KIV-2 CNV.
The PFGE and fiber-FISH techniques require large amounts of stored, well-preserved, non-fragmented DNA, so that their use is limited by sample collection conditions and technical requirements. Western blotting has been widely used to determine the apo(a) isoform size and associated Lp(a) concentrations, and therefore provides phenotypic information. Compared with PFGE, this method has advantages and drawbacks. Alleles producing apo(a) are recognized, and the amount of Lp(a) contributed by each allele can be determined in heterozygotes with two expressed alleles and exhibiting double-band phenotypes. In these individuals, the CNV genotype can also be directly deduced from the phenotype. However, the frequencies of double-band phenotypes reported in the literature vary considerably, ranging from 30% to 90% [80]. The reasons for this are twofold. Firstly, there are technical limitations. The number of double-band phenotypes depends on the sensitivity of the blotting system and the resolution of the gel system, and both vary among the studies. Moreover, the genetic architecture of the Lp(a)/apo(a) trait in the population is bound to be taken into consideration [80,81]. In fact, populations differ in allele-associated Lp(a) concentrations [82]. Hence, in populations where long alleles are associated with higher concentrations (e.g., populations from Africa), more double-band phenotypes are expected to be detected. Assuming a detection efficiency of 100%, the “true” number of double-band phenotypes to be expected in a population will depend on the number of loss-of-function alleles (null alleles) in that population, which is presently unknown. Furthermore, it is presently difficult to recognize truncated isoforms or splice variants at the protein level without any prior knowledge from molecular analysis. On the other hand, PFGE allows for the determination of the CNV genotype without providing any phenotypic information. In this regard, it should be noted that Simò et al. showed that the correlation of the plasma Lp(a) concentration is stronger with phenotype measures than with genotype measures, even though the application of both methods simultaneously provides the most comprehensive information [83]. It should also be acknowledged that PFGE genotyping needs large amounts of buffy coat, which are not commonly available in population studies [84]. On the contrary, the qPCR technique allows for the rapid assessment of the apo(a) size from DNA, providing an additional genomic variable to assess the genetic determinants of the plasma Lp(a) concentration in epidemiological studies [45,85]. Moreover, compared to PFGE, PCR-based methods are easy to perform and have high levels of specimen typeability and reproducibility.
Recently, a new DRAGEN KIV-2 CN caller—which utilizes short reads—was demonstrated to have high accuracy compared to optical mapping across 166 whole genome sequencing, and can further phase ∼50% of the samples. The authors compared the KIV-2 CN numbers to 24 previously postulated KIV-2-relevant SNVs, finally showing that many are ineffective predictors of KIV-2 copy numbers [86].
A recent study carried out on 1.020 individuals including 252 clinically diagnosed hypercholesterolemia patients from the Familial Hypercholesterolemia (FH) Register Austria showed that the integration of polygenic scores with some LPA gene variants increases the proportion of individuals with a clearly defined disease by nearly 30% compared to standard scores [87]. Moreover, a genome-wide association study (GWAS) identified within the LPA/PLG locus seven SNPs associated with CHD events in individuals undergoing statin treatment. Each copy of the risk allele G at the lead variant, rs10455872, was associated with a 58% increased risk of CHD events, with this association also being present in individuals with LDL-C ≤ 70 mg/dL [88]. All of the previous considerations have to be put in the context of the single country’s public health policies and priorities. Of course, an advanced investigation of the Lp(a) isoforms and genetics make sense, whereas CVD prevention is a national public health priority, quantitative Lp(a) screening is already widely available, and the access to second-level lipid-lowering treatments exists and is supported by the public health system and/or insurances. In the United States, for instance, universal Lp(a) screening in the youth has been estimated to be feasible and cost-effective [89]. In these circumstances, the simultaneous application of methods for both the phenotyping and genotyping aspects [46] in association with the CV risk—especially for the most studied techniques including Western blotting with SDS agarose [90,91] and qPCR [29,92,93,94]—provide more complete information for the personalized management of individuals with an increased CV risk. Of course, identifying individuals with high Lp(a) levels and a higher CV risk could improve the cost-effectiveness of the novel or up-coming Lp(a)-lowering drugs. Additionally, currently, the application of these methods should be limited to the study of specific family clusters and/or of individuals with recurrent CV events without other CV risk factors expect for hyperlipoproteinemia(a). Further research is needed to improve the available phenotyping and genotyping techniques to validate their prognostic value and increase their availability, reducing their cost.
Author Contributions
Conceptualization, A.F.G.C. and F.F.; methodology, A.F.G.C. and V.D.M.; data curation, V.D.M.; writing—original draft preparation, A.F.G.C., V.D.M. and F.F.; writing—review and editing, M.G., A.A. and E.I.; visualization, V.D.M. and M.G.; supervision, A.F.G.C. and F.F.; project administration, A.F.G.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Koschinsky, M.L.; Kronenberg, F. The long journey of lipoprotein(a) from cardiovascular curiosity to therapeutic target. Atherosclerosis 2022, 349, 1–6. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Kamstrup, P.R. Lipoprotein(a) and cardiovascular and valvular diseases: A genetic epidemiological perspective. Atherosclerosis 2022, 349, 7–16. [Google Scholar] [CrossRef]
- Wang, H.; Wu, P.; Jiang, D.; Zhang, H.; Zhang, J.; Zong, Y.; Han, Y. Relationship between serum homocysteine, fibrinogen, lipoprotein-a level, and peripheral arterial disease: A dose-response meta-analysis. Eur. J. Med. Res. 2022, 27, 261. [Google Scholar] [CrossRef]
- Singh, S.; Baars, D.P.; Desai, R.; Singh, D.; Pinto-Sietsma, S.J. Association between Lipoprotein(a) and risk of atrial fibrillation: A Systematic Review and Meta-analysis of Mendelian Randomization Studies. Curr. Probl. Cardiol. 2023, 49, 102024. [Google Scholar] [CrossRef] [PubMed]
- Amiri, M.; Raeisi-Dehkordi, H.; Verkaar, A.J.C.F.; Wu, Y.; van Westing, A.C.; Berk, K.A.; Bramer, W.M.; Aune, D.; Voortman, T. Circulating lipoprotein (a) and all-cause and cause-specific mortality: A systematic review and dose-response meta-analysis. Eur. J. Epidemiol. 2023, 38, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Chemello, K.; Chan, D.C.; Lambert, G.; Watts, G.F. Recent advances in demystifying the metabolism of lipoprotein(a). Atherosclerosis 2022, 349, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Marcovina, S.M.; Morrisett, J.D. Structure and metabolism of lipoprotein(a). Curr. Opin. Lipidol. 1995, 6, 136–145. [Google Scholar] [CrossRef]
- Coassin, S.; Kronenberg, F. Lipoprotein(a) beyond the kringle IV repeat polymorphism: The complexity of genetic variation in the LPA gene. Atherosclerosis 2022, 349, 17–35. [Google Scholar] [CrossRef]
- Fogacci, F.; Cicero, A.F.G.; D’Addato, S.; Giovannini, M.; Borghi, C.; Brisighella Heart Study Group. Effect of spontaneous changes in dietary components and lipoprotein(a) levels: Data from the Brisighella Heart Study. Atherosclerosis 2017, 262, 202–204. [Google Scholar] [CrossRef]
- Matveyenko, A.; Matienzo, N.; Ginsberg, H.; Nandakumar, R.; Seid, H.; Ramakrishnan, R.; Holleran, S.; Thomas, T.; Reyes-Soffer, G. Relationship of apolipoprotein(a) isoform size with clearance and production of lipoprotein(a) in a diverse cohort. J. Lipid. Res. 2023, 64, 100336. [Google Scholar] [CrossRef]
- Cicero, A.F.G.; Fogacci, F.; Derosa, G.; D’Angelo, A.; Ventura, F.; Rizzoli, E.; D’Addato, S.; Borghi, C.; On Behalf of The Brisighella Heart Study Group. Lipoprotein(a) Serum Levels Predict Pulse Wave Velocity in Subjects in Primary Prevention for Cardiovascular Disease with Large Apo(a) Isoforms: Data from the Brisighella Heart Study. Biomedicines 2022, 10, 656. [Google Scholar] [CrossRef] [PubMed]
- Grüneis, R.; Lamina, C.; Di Maio, S.; Schönherr, S.; Zoescher, P.; Forer, L.; Streiter, G.; Peters, A.; Gieger, C.; Köttgen, A.; et al. The effect of LPA Thr3888Pro on lipoprotein(a) and coronary artery disease is modified by the LPA KIV-2 variant 4925G>A. Atherosclerosis 2022, 349, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Sandholzer, C.; Saha, N.; Kark, J.D.; Rees, A.; Jaross, W.; Dieplinger, H.; Hoppichler, F.; Boerwinkle, E.; Utermann, G. Apo(a) isoforms predict risk for coronary heart disease. A study in six populations. Arterioscler. Thromb. 1992, 12, 1214–1226. [Google Scholar] [CrossRef]
- Rader, D.J.; Cain, W.; Ikewaki, K.; Talley, G.; Zech, L.A.; Usher, D.; Brewer, H.B., Jr. The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J. Clin. Investig. 1994, 93, 2758–2763. [Google Scholar] [CrossRef]
- Ma, L.; Chan, D.C.; Ooi, E.M.M.; Marcovina, S.M.; Barrett, P.H.R.; Watts, G.F. Apolipoprotein(a) Kinetics in Statin-Treated Patients with Elevated Plasma Lipoprotein(a) Concentration. J. Clin. Endocrinol. Metab. 2019, 104, 6247–6255. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Watts, G.F.; Coll, B.; Wasserman, S.M.; Marcovina, S.M.; Barrett, P.H.R. Lipoprotein(a) Particle Production as a Determinant of Plasma Lipoprotein(a) Concentration Across Varying Apolipoprotein(a) Isoform Sizes and Background Cholesterol-Lowering Therapy. J. Am. Heart Assoc. 2019, 8, e011781. [Google Scholar] [CrossRef]
- Jenner, J.L.; Seman, L.J.; Millar, J.S.; Lamon-Fava, S.; Welty, F.K.; Dolnikowski, G.G.; Marcovina, S.M.; Lichtenstein, A.H.; Barrett, P.H.; deLuca, C.; et al. The metabolism of apolipoproteins (a) and B-100 within plasma lipoprotein (a) in human beings. Metabolism. 2005, 54, 361–369. [Google Scholar] [CrossRef]
- Kronenberg, F. Lipoprotein(a). In Prevention and Treatment of Atherosclerosis. Handbook of Experimental Pharmacology; von Eckardstein, A., Binder, C.J., Eds.; Springer: Cham, Switzerland, 2021; Volume 270, pp. 201–232. [Google Scholar] [CrossRef]
- Mehta, A.; Jain, V.; Saeed, A.; Saseen, J.J.; Gulati, M.; Ballantyne, C.M.; Virani, S.S. Lipoprotein(a) and ethnicities. Atherosclerosis 2022, 349, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Kraft, H.G.; Lingenhel, A.; Pang, R.W.; Delport, R.; Trommsdorff, M.; Vermaak, H.; Janus, E.D.; Utermann, G. Frequency distributions of apolipoprotein(a) kringle IV repeat alleles and their effects on lipoprotein(a) levels in Caucasian, Asian, and African populations: The distribution of null alleles is non-random. Eur. J. Hum. Genet. 1996, 4, 74–87. [Google Scholar] [CrossRef]
- Fogacci, F.; Cicero, A.F.; D’Addato, S.; D’Agostini, L.; Rosticci, M.; Giovannini, M.; Bertagnin, E.; Borghi, C.; Brisighella Heart Study Group. Serum lipoprotein(a) level as long-term predictor of cardiovascular mortality in a large sample of subjects in primary cardiovascular prevention: Data from the Brisighella Heart Study. Eur. J. Intern. Med. 2017, 37, 49–55. [Google Scholar] [CrossRef]
- Erqou, S.; Thompson, A.; Di Angelantonio, E.; Saleheen, D.; Kaptoge, S.; Marcovina, S.; Danesh, J. Apolipoprotein(a) isoforms and the risk of vascular disease: Systematic review of 40 studies involving 58,000 participants. J. Am. Coll. Cardiol. 2010, 55, 2160–2167. [Google Scholar] [CrossRef]
- Kraft, H.G.; Lingenhel, A.; Köchl, S.; Hoppichler, F.; Kronenberg, F.; Abe, A.; Mühlberger, V.; Schönitzer, D.; Utermann, G. Apolipoprotein(a) kringle IV repeat number predicts risk for coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.H.; Fortmann, S.P.; Marcovina, S.M. A prospective case-control study of lipoprotein(a) levels and apo(a) size and risk of coronary heart disease in Stanford Five-City Project participants. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Mueller, P.A.; Yerkes, E.; Bergstrom, P.; Rosario, S.; Hay, J.; Pamir, N. A method for lipoprotein (a) Isolation from a small volume of plasma with applications for clinical research. Sci. Rep. 2022, 12, 9138. [Google Scholar] [CrossRef] [PubMed]
- Neidhoefer, C. Structural Analysis of Apolipoprotein(a) Kringle IV Type 2 A Genetical, Key Tandem-Repeat Factor in Cardiovascular Diseases. Ph.D. Thesis, University of Pavia, Pavia, Italy, 2018. [Google Scholar] [CrossRef]
- Maher, K.; Persa, L.; Barry, D.; Lee-Eng, J.; Dichek, H.; Joshi, S.; Amlie-Lefond, C. Thrombophilia screening in the routine clinical care of children with arterial ischemic stroke. Pediatr. Blood. Cancer. 2023, 70, e30381. [Google Scholar] [CrossRef] [PubMed]
- Paciullo, F.; Giannandrea, D.; Virgili, G.; Cagini, C.; Gresele, P. Role of Increased Lipoprotein (a) in Retinal Vein Occlusion: A Systematic Review and Meta-analysis. TH Open 2021, 5, e295–e302. [Google Scholar] [CrossRef]
- Sticchi, E.; Magi, A.; Kamstrup, P.R.; Marcucci, R.; Prisco, D.; Martinelli, I.; Mannucci, P.M.; Abbate, R.; Giusti, B. Apolipoprotein(a) Kringle-IV Type 2 Copy Number Variation Is Associated with Venous Thromboembolism. PLoS ONE 2016, 11, e0149427. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, Z.; Yan, Z. Association Between Lipoprotein (A) and Diabetic Nephropathy in Patients with Type 2 Diabetes Mellitus: A Meta-Analysis. Front. Endocrinol. 2021, 12, 633529. [Google Scholar] [CrossRef]
- Ulloque-Badaracco, J.R.; Mosquera-Rojas, M.D.; Hernandez-Bustamante, E.A.; Alarcón-Braga, E.A.; Ulloque-Badaracco, R.R.; Al-Kassab-Córdova, A.; Herrera-Añazco, P.; Benites-Zapata, V.A.; Hernandez, A.V. Association between Lipid Profile and Apolipoproteins with Risk of Diabetic Foot Ulcer: A Systematic Review and Meta-Analysis. Int. J. Clin. Pract. 2022, 2022, 5450173. [Google Scholar] [CrossRef]
- Tsimikas, S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Kausik, R.; Borén, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur. Heart 2010, 23, 2844–2853. [Google Scholar] [CrossRef]
- Vazirian, F.; Sadeghi, M.; Kelesidis, T.; Budoff, M.J.; Zandi, Z.; Samadi, S.; Mohammadpour, A.H. Predictive value of lipoprotein(a) in coronary artery calcification among asymptomatic cardiovascular disease subjects: A systematic review and meta-analysis. Nutr. Metab. Cardiovasc. Dis. 2023, in press. [Google Scholar] [CrossRef]
- Motawea, K.R.; Elhalag, R.H.; Aboelenein, M.; Ibrahim, N.; Swed, S.; Fathy, H.; Awad, D.M.; Belal, M.M.; Talaat, N.E.; Rozan, S.S.; et al. Association of Aortic Valve Calcification and High Levels of Lipoprotein (a): Systematic Review and Meta-Analysis. Curr. Probl. Cardiol. 2023, 48, 101746. [Google Scholar] [CrossRef] [PubMed]
- Tsarouhas, K.; Hoursalas, A.; Vardavas, A.I.; Tsitsimpikou, C. Lipoprotein a: An emerging risk identifier and evolving clinical target. Public Health Toxicol. 2022, 2, 2. [Google Scholar] [CrossRef]
- Kamstrup, P.R. Lipoprotein(a) and Cardiovascular Disease. Clin. Chem. 2021, 67, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Zysow, B.R.; Lindahl, G.E.; Wade, D.P.; Knight, B.L.; Lawn, R.M. C/T polymorphism in the 5’ untranslated region of the apolipoprotein(a) gene introduces an upstream ATG and reduces in vitro translation. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 58–64. [Google Scholar] [CrossRef]
- Gu, J.X.; Huang, J.; Li, S.S.; Zhou, L.H.; Yang, M.; Li, Y.; Zhang, A.M.; Yin, Y.; Zhang, N.; Jia, M.; et al. Elevated lipoprotein(a) and genetic polymorphisms in the LPA gene may predict cardiovascular events. Sci. Rep. 2022, 12, 3588. [Google Scholar] [CrossRef] [PubMed]
- Kraft, H.G.; Köchl, S.; Menzel, H.J.; Sandholzer, C.; Utermann, G. The apolipoprotein (a) gene: A transcribed hypervariable locus controlling plasma lipoprotein (a) concentration. Hum. Genet. 1992, 90, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Anglés-Cano, E.; Loyau, S.; Cardoso-Saldaña, G.; Couderc, R.; Gillery, P. A novel kringle-4 number-based recombinant apo[a] standard for human apo[a] phenotyping. J. Lipid. Res. 1999, 40, 354–359. [Google Scholar] [CrossRef]
- Lackner, C.; Boerwinkle, E.; Leffert, C.C.; Rahmig, T.; Hobbs, H.H. Molecular basis of apolipoprotein (a) isoform size hetero-geneity as revealed by pulsed-field gel electrophoresis. J. Clin. Investig. 1991, 87, 2153–2161. [Google Scholar] [CrossRef]
- Chiou, C.S.; Wei, H.L.; Yang, L.C. Comparison of pulsed-field gel electrophoresis and coagulase gene restriction profile analysis techniques in the molecular typing of Staphylococcus aureus. J. Clin. Microbiol. 2000, 38, 2186–2190. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Gong, F.; Yu, X.; Zhang, T. A novel deletion mutation in the LPA gene in a middle-aged woman with ischaemic stroke. BMC Med. Genom. 2021, 14, 132. [Google Scholar] [CrossRef] [PubMed]
- Lanktree, M.B.; Rajakumar, C.; Brunt, J.H.; Koschinsky, M.L.; Connelly, P.W.; Hegele, R.A. Determination of lipoprotein(a) krin-gle repeat number from genomic DNA: Copy number variation genotyping using qPCR. J. Lipid. Res. 2009, 50, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein (a). J. Lipid. Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef] [PubMed]
- Erdel, M.; Hubalek, M.; Lingenhel, A.; Kofler, K.; Duba, H.-C.; Utermann, G. Counting the repetitive kringle-IV repeats in the gene encoding human apolipoprotein(a) by fibre-FISH. Nat. Genet. 1999, 21, 357–358. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Anuurad, E.; Zhang, W.; Tran, T.; Berglund, L. Lipoprotein(a): Genotype-phenotype relationship and impact on atherogenic risk. Metab. Syndr. Relat. Disord. 2011, 9, 411–418. [Google Scholar] [CrossRef]
- Pradhan, S.; Apaydin, S.; Bucevičius, J.; Gerasimaitė, R.; Kostiuk, G.; Lukinavičius, G. Sequence-specific DNA labelling for fluorescence microscopy. Biosens. Bioelectron 2023, 230, 115256. [Google Scholar] [CrossRef]
- Zekavat, S.M.; Ruotsalainen, S.; Handsaker, R.E.; Alver, M.; Bloom, J.; Poterba, T.; Seed, C.; Ernst, J.; Chaffin, M.; Engreitz, J.; et al. Deep coverage whole genome sequences and plasma lipoprotein(a) in individuals of European and African ancestries. Nat. Commun. 2018, 9, 2606. [Google Scholar] [CrossRef]
- Mazur, P.; Dumnicka, P.; Tisończyk, J.; Ząbek-Adamska, A.; Drożdż, R. SDS Electrophoresis on Gradient Polyacrylamide Gels as a Semiquantitative Tool for the Evaluation of Proteinuria. Diagnostics 2023, 13, 1513. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Beisiegel, U.; Henne-Bruns, D.; Eaton, D.L.; Lawn, R.M. Apolipoprotein(a) size heterogeneity is related to variable number of repeat sequences in its mRNA. Biochemistry 1990, 29, 640–644. [Google Scholar] [CrossRef]
- Kamboh, M.I.; Ferrell, R.E.; Kottke, B.A. Expressed hypervariable polymorphism of apolipoprotein (a). Am. J. Hum. Genet. 1991, 49, 1063–1074. [Google Scholar] [PubMed]
- Geroldi, D.; Bellotti, V.; Buscaglia, P.; Bonetti, G.; Gazzaruso, C.; Caprioli, A.; Fratino, P. Characterization of apo(a) polymorphism by a modified immunoblotting technique in an Italian population sample. Clin. Chim. Acta 1993, 221, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Koschinsky, M.L.; Stroes, E.S.G.; Kronenberg, F. Daring to dream: Targeting lipoprotein(a) as a causal and risk-enhancing factor. Pharmacol. Res. 2023, 194, 106843. [Google Scholar] [CrossRef]
- Santos, H.O.; Kones, R.; Rumana, U.; Earnest, C.P.; Izidoro, L.F.M.; Macedo, R.C.O. Lipoprotein(a): Current Evidence for a Physiologic Role and the Effects of Nutraceutical Strategies. Clin. Ther. 2019, 41, 1780–1797. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef]
- Schandelmaier, S.; Briel, M.; Saccilotto, R.; Olu, K.K.; Arpagaus, A.; Hemkens, L.G.; Nordmann, A.J. Niacin for primary and secondary prevention of cardiovascular events. Cochrane Database Syst. Rev. 2017, 6, CD009744. [Google Scholar] [CrossRef]
- Parish, S.; Hopewell, J.C.; Hill, M.R.; Marcovina, S.; Valdes-Marquez, E.; Haynes, R.; Offer, A.; Pedersen, T.R.; Baigent, C.; Collins, R.; et al. Impact of Apolipoprotein(a) Isoform Size on Lipoprotein(a) Lowering in the HPS2-THRIVE Study. Circ. Genom. Precis. Med. 2018, 11, e001696. [Google Scholar] [CrossRef] [PubMed]
- Fogacci, F.; Ferri, N.; Toth, P.P.; Ruscica, M.; Corsini, A.; Cicero, A.F.G. Efficacy and Safety of Mipomersen: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Drugs 2019, 79, 751–766. [Google Scholar] [CrossRef]
- Gencer, B.; Mach, F. Potential of Lipoprotein(a)-Lowering Strategies in Treating Coronary Artery Disease. Drugs 2020, 80, 229–239. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, Q.; Wang, Y.; Gao, P.; Chen, D. The effect and safety of anacetrapib in the treatment of dyslipidemia: A systematic review and meta-analysis. Postgrad Med. 2018, 130, 129–136. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ruotolo, G.; Brewer, H.B.; Wang, M.D.; Liu, L.; Willey, M.B.; Deeg, M.A.; Krueger, K.A.; Nissen, S.E. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J. Clin. Lipidol. 2016, 10, 519–527.e4. [Google Scholar] [CrossRef] [PubMed]
- Afanasieva, O.I.; Ezhov, M.V.; Razova, O.A.; Afanasieva, M.I.; Utkina, E.A.; Pokrovsky, S.N. Apolipoprotein(a) phenotype determines the correlations of lipoprotein(a) and proprotein convertase subtilisin/kexin type 9 levels in patients with potential familial hypercholesterolemia. Atherosclerosis 2018, 277, 477–482. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F. The Promise of PCSK9 and Lipoprotein(a) as Targets for Gene Silencing Therapies. Clin. Ther. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, Y.; Pantea Stoian, A.; Cicero, A.F.G.; Fogacci, F.; Nikolic, D.; Sachinidis, A.; Rizvi, A.A.; Janez, A.; Rizzo, M. Inclisiran: A small interfering RNA strategy targeting PCSK9 to treat hypercholesterolemia. Expert Opin. Drug Saf. 2022, 21, 9–20. [Google Scholar] [CrossRef]
- Yu, Z.; Hu, L.; Sun, C.; Wang, Z.; Zhang, X.; Wu, M.; Liu, L. Effect of Different Types and Dosages of Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors on Lipoprotein(a) Levels: A Network Meta-analysis. J. Cardiovasc. Pharmacol. 2023, 81, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.G.; Fogacci, F.; Zambon, A.; Toth, P.P.; Borghi, C. Efficacy and safety of inclisiran a newly approved FDA drug: A systematic review and pooled analysis of available clinical studies. Am. Heart J. 2022, 13, 100127. [Google Scholar] [CrossRef]
- Hanssen, R.; Gouni-Berthold, I. Lipoprotein(a) Management: Pharmacological and Apheretic Treatment. Curr. Med. Chem. 2017, 24, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, V.; Chemello, K.; Hollstein, T.; Hong-Fong, C.C.; Schumann, F.; Grenkowitz, T.; Nativel, B.; Coassin, S.; Croyal, M.; Kassner, U.; et al. The size of apolipoprotein (a) is an independent determinant of the reduction in lipoprotein (a) induced by PCSK9 inhibitors. Cardiovasc Res. 2022, 118, 2103–2111. [Google Scholar] [CrossRef]
- Alhomoud, I.S.; Talasaz, A.; Mehta, A.; Kelly, M.S.; Sisson, E.M.; Bucheit, J.D.; Brown, R.; Dixon, D.L. Role of lipoprotein(a) in atherosclerotic cardiovascular disease: A review of current and emerging therapies. Pharmacotherapy 2023, in press. [CrossRef]
- Karwatowska-Prokopczuk, E.; Clouet-Foraison, N.; Xia, S.; Viney, N.J.; Witztum, J.L.; Marcovina, S.M.; Tsimikas, S. Prevalence and influence of LPA gene variants and isoform size on the Lp(a)-lowering effect of pelacarsen. Atherosclerosis 2021, 324, 102–108. [Google Scholar] [CrossRef]
- Kotani, K.; Serban, M.C.; Penson, P.; Lippi, G.; Banach, M. Evidence-based assessment of lipoprotein(a) as a risk biomarker for cardiovascular diseases—Some answers and still many questions. Crit. Rev. Clin. Lab. Sci. 2016, 53, 370–378. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1732–1741. [Google Scholar] [CrossRef]
- Coassin, S.; Erhart, G.; Weissensteiner, H.; Eca Guimarães de Araújo, M.; Lamina, C.; Schönherr, S.; Forer, L.; Haun, M.; Losso, J.L.; Köttgen, A.; et al. A novel but frequent variant in LPA KIV-2 is associated with a pronounced Lp(a) and cardiovascular risk reduction. Eur Heart J. 2017, 38, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Schachtl-Riess, J.F.; Kheirkhah, A.; Grüneis, R.; Di Maio, S.; Schoenherr, S.; Streiter, G.; Losso, J.L.; Paulweber, B.; Eckardt, K.U.; Köttgen, A.; et al. Frequent LPA KIV-2 Variants Lower Lipoprotein(a) Concentrations and Protect Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2021, 78, 437–449. [Google Scholar] [CrossRef]
- Saleheen, D.; Haycock, P.C.; Zhao, W.; Rasheed, A.; Taleb, A.; Imran, A.; Abbas, S.; Majeed, F.; Akhtar, S.; Qamar, N.; et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: A mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2017, 5, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Lanktree, M.B.; Anand, S.S.; Yusuf, S.; Hegele, R.A.; SHARE Investigators. Comprehensive analysis of genomic variation in the LPA locus and its relationship to plasma lipoprotein(a) in South Asians, Chinese, and European Caucasians. Circ. Cardiovasc. Genet. 2010, 3, 39–46. [Google Scholar] [CrossRef]
- Yeang, C.; Clopton, P.C.; Tsimikas, S. Lipoprotein(a)-cholesterol levels estimated by vertical auto profile correlate poorly with Lp(a) mass in hyperlipidemic subjects: Implications for clinical practice interpretation of Lp(a)-mediated risk. J. Clin. Lipidol. 2016, 10, 1389–1396. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Anuurad, E.; Berglund, L. Lipoprotein (a): Impact by ethnicity and environmental and medical conditions. J. Lipid Res. 2016, 57, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Langsted, A. Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J. Lipid Res. 2016, 57, 1953–1975. [Google Scholar] [CrossRef]
- Barlera, S.; Specchia, C.; Farrall, M.; Chiodini, B.D.; Franzosi, M.G.; Rust, S.; Green, F.; Nicolis, E.B.; Peden, J.; Assmann, G.; et al. Multiple QTL influence the serum Lp(a) concentration: A genome-wide linkage screen in the PROCARDIS study. Eur. J. Hum. Genet. 2007, 15, 221–227. [Google Scholar] [CrossRef]
- Simó, J.M.; Camps, J.; Martín, S.; Pedro-Botet, J.; Ferré, N.; Gómez, F.; Joven, J. Differences between genotyping and phenotyp-ing methods for assessing apolipoprotein(a) size polymorphisms. Clin. Chem. Lab. Med. 2003, 41, 1340–1344. [Google Scholar] [CrossRef]
- Di Maio, S.; Grüneis, R.; Streiter, G.; Lamina, C.; Maglione, M.; Schoenherr, S.; Öfner, D.; Thorand, B.; Peters, A.; Eckardt, K.U.; et al. Investigation of a nonsense mutation located in the complex KIV-2 copy number variation region of apolipoprotein(a) in 10,910 individuals. Genome Med. 2020, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Cheng, Y.C.; Chen, K.; Wang, H.; Gerhard, G.S.; Still, C.D.; Chu, X.; Yang, R.; Parihar, A.; O’Connell, J.R.; et al. Evidence for several independent genetic variants affecting lipoprotein (a) cholesterol levels. Hum. Mol. Genet. 2015, 24, 2390–2400. [Google Scholar] [CrossRef]
- Behera, S.; Belyeu, J.R.; Chen, X.; Paulin, L.F.; Nguyen, N.Q.H.; Newman, E.; Mahmoud, M.; Menon, V.K.; Qi, Q.; Joshi, P.; et al. Identification of allele-specific KIV-2 repeats and impact on Lp(a) measurements for cardiovascular disease risk. bioRxiv 2023. [Google Scholar] [CrossRef]
- Schwaninger, G.; Forer, L.; Ebenbichler, C.; Dieplinger, H.; Kronenberg, F.; Zschocke, J.; Witsch-Baumgartner, M. Filling the gap: Genetic risk assessment in hypercholesterolemia using LDL-C and LPA genetic scores. Clin. Genet. 2023, 104, 334–343. [Google Scholar] [CrossRef]
- Wei, W.Q.; Li, X.; Feng, Q.; Kubo, M.; Kullo, I.J.; Peissig, P.L.; Karlson, E.W.; Jarvik, G.P.; Lee, M.T.M.; Shang, N.; et al. LPA Variants Are Associated with Residual Cardiovascular Risk in Patients Receiving Statins. Circulation 2018, 138, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Alankar, A.; Brar, P.C.; Kohn, B. Lipoprotein(a): A Case for Universal Screening in Youth. Curr. Atheroscler. Rep. 2023, 25, 487–493. [Google Scholar] [CrossRef]
- Ronald, J.; Rajagopalan, R.; Cerrato, F.; Nord, A.S.; Hatsukami, T.; Kohler, T.; Marcovina, S.; Heagerty, P.; Jarviket, G.P. Genetic variation in LPAL2, LPA, and PLG predicts plasma lipoprotein(a) level and carotid artery disease risk. Stroke 2011, 42, 2–9. [Google Scholar] [CrossRef]
- Noureen, A.; Fresser, F.; Utermann, G.; Schmidt, K. Sequence variation within the KIV-2 copy number polymorphism of the human LPA gene in African, Asian, and European populations. PLoS ONE 2015, 10, e0121582. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein(a) levels and improved cardiovascular risk pre-diction. J. Am. Coll. Cardiol. 2013, 61, 1146–1156. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J. Am. Coll. Cardiol. 2014, 63, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Puckey, L.H.; Lawn, R.M.; Knight, B.L. Polymorphisms in the apolipoprotein(a) gene and their relationship to allele size and plasma lipoprotein(a) concentration. Hum. Mol. Genet. 1997, 6, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).