
Postprandial Hyperlipidemia: Its Pathophysiology, Diagnosis, Atherogenesis, and Treatments

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Evaluation of Postprandial Hyperlipidemia
2.1. OFLT
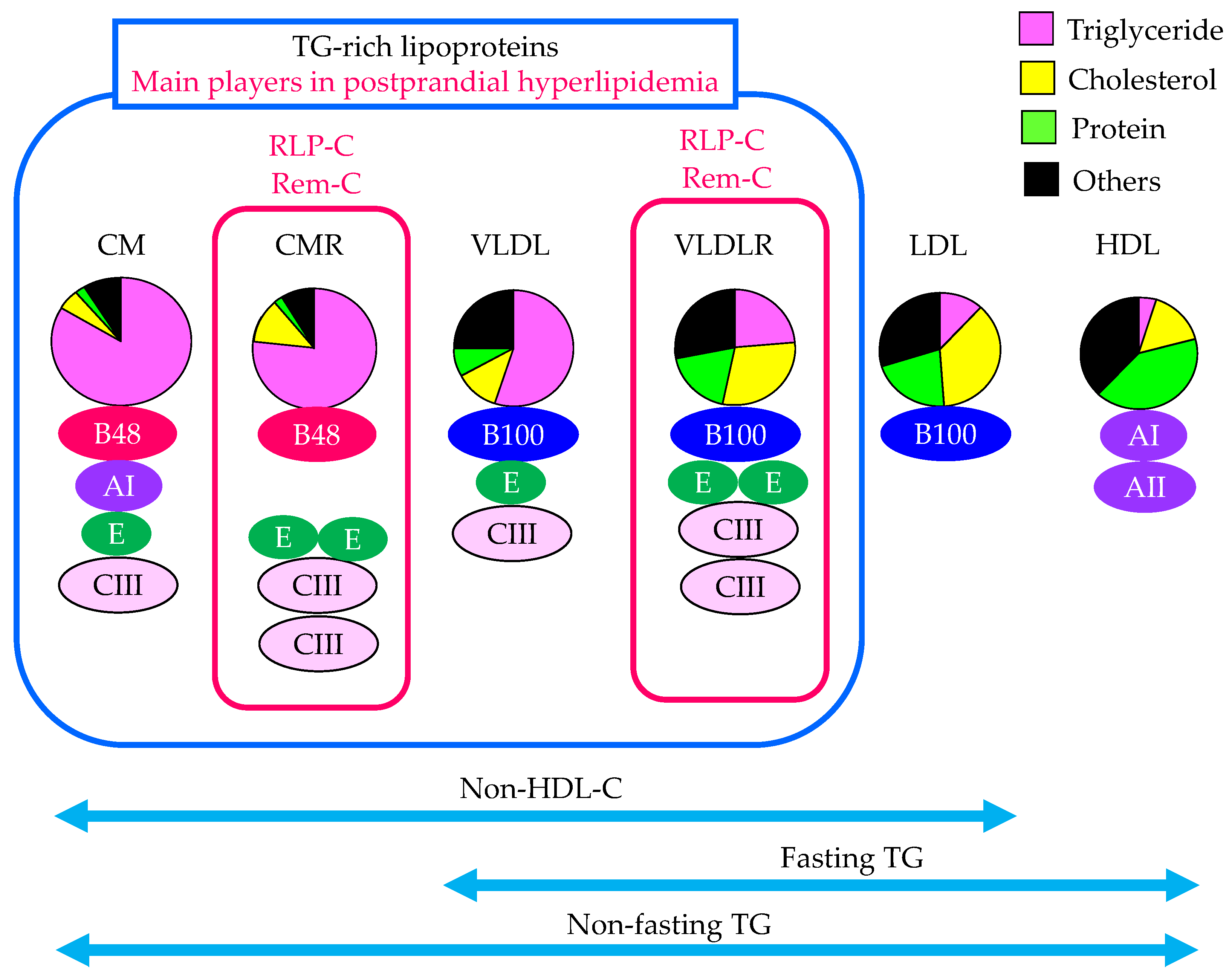
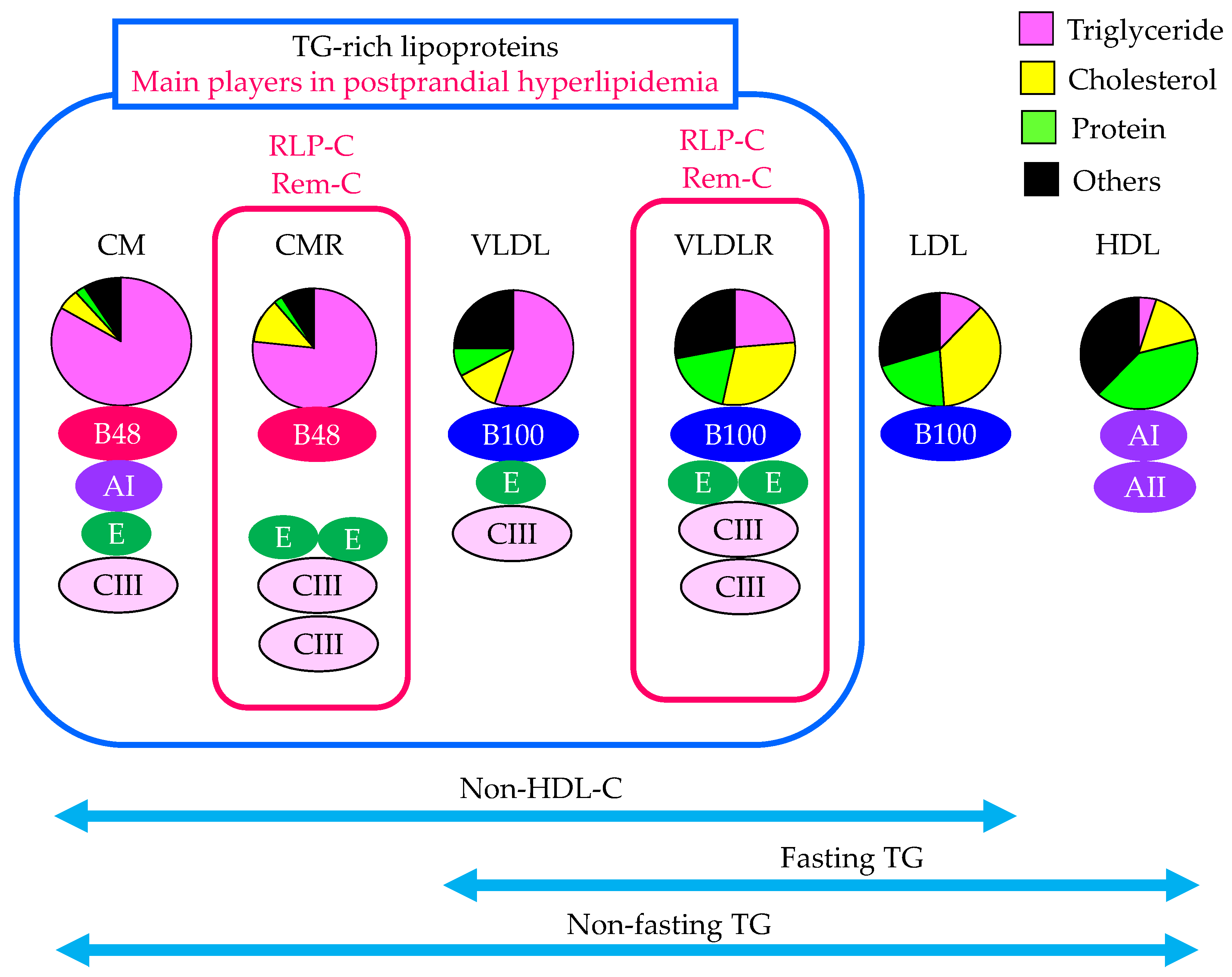
2.2. The Contribution of Remnant Lipoproteins to Postprandial Hyperlipidemia
2.3. The Evaluation of Remnant Lipoproteins in the Clinical Setting
2.3.1. Fasting TG
2.3.2. Non-Fasting TG
2.3.3. Non-HDL-C
2.3.4. Fasting Serum Apo B48
2.3.5. RLP-C
2.3.6. RemL-C
3. Pathological Factors That Induce Postprandial Hyperlipidemia
3.1. Familial Type III Hyperlipoproteinemia
3.2. FCHL
3.3. CKD
3.4. Metabolic Syndrome and Type 2 Diabetes
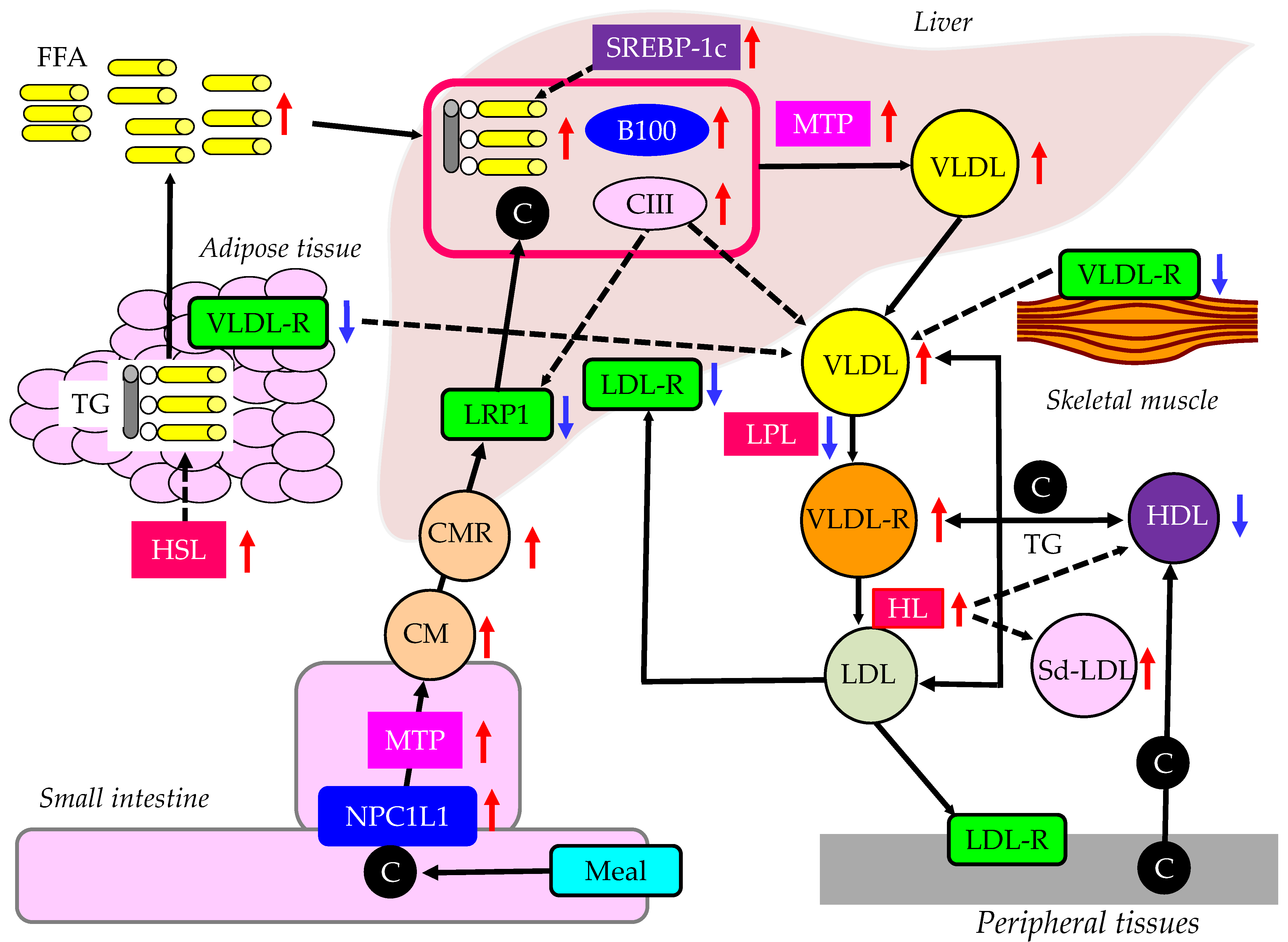
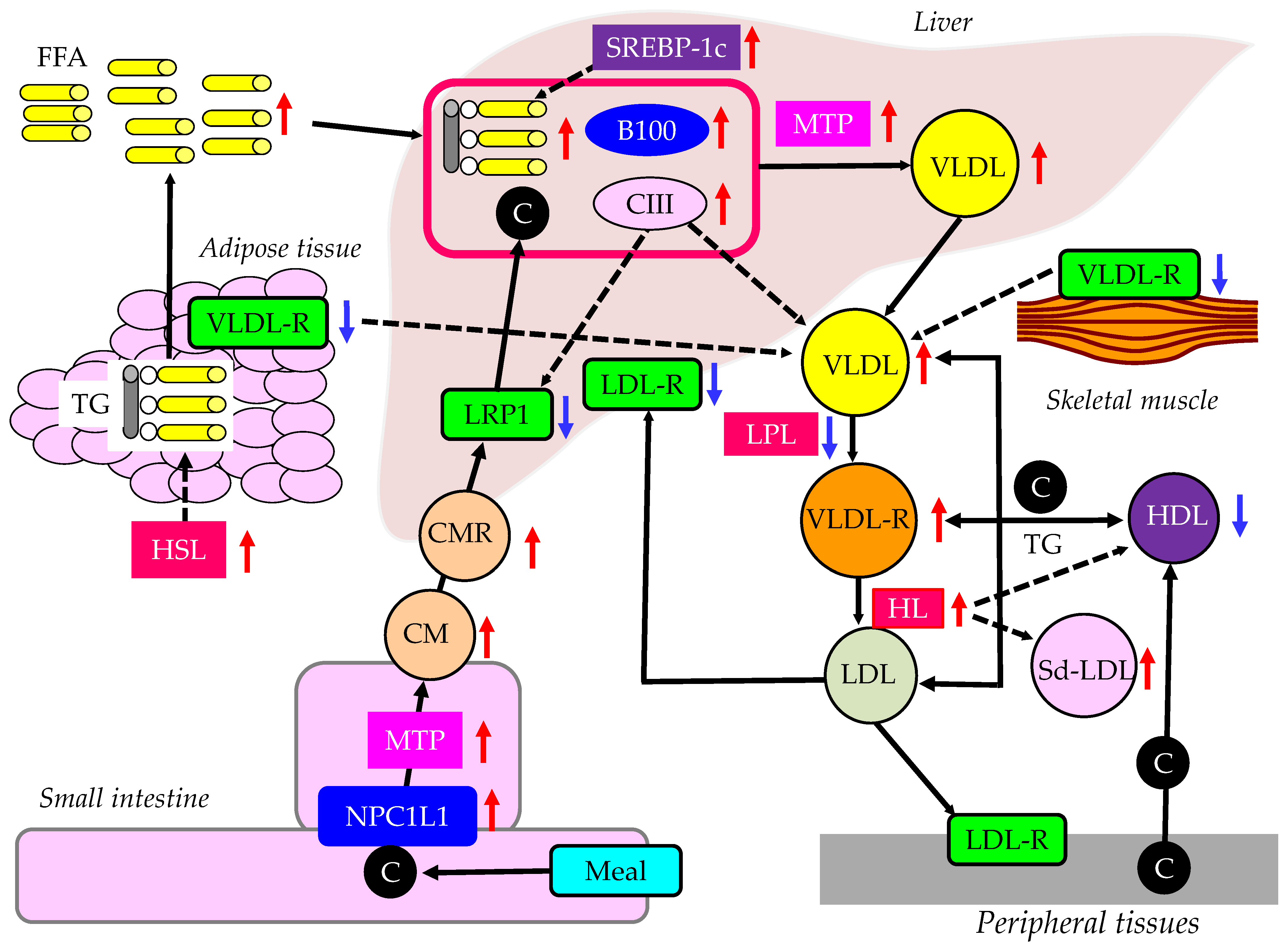
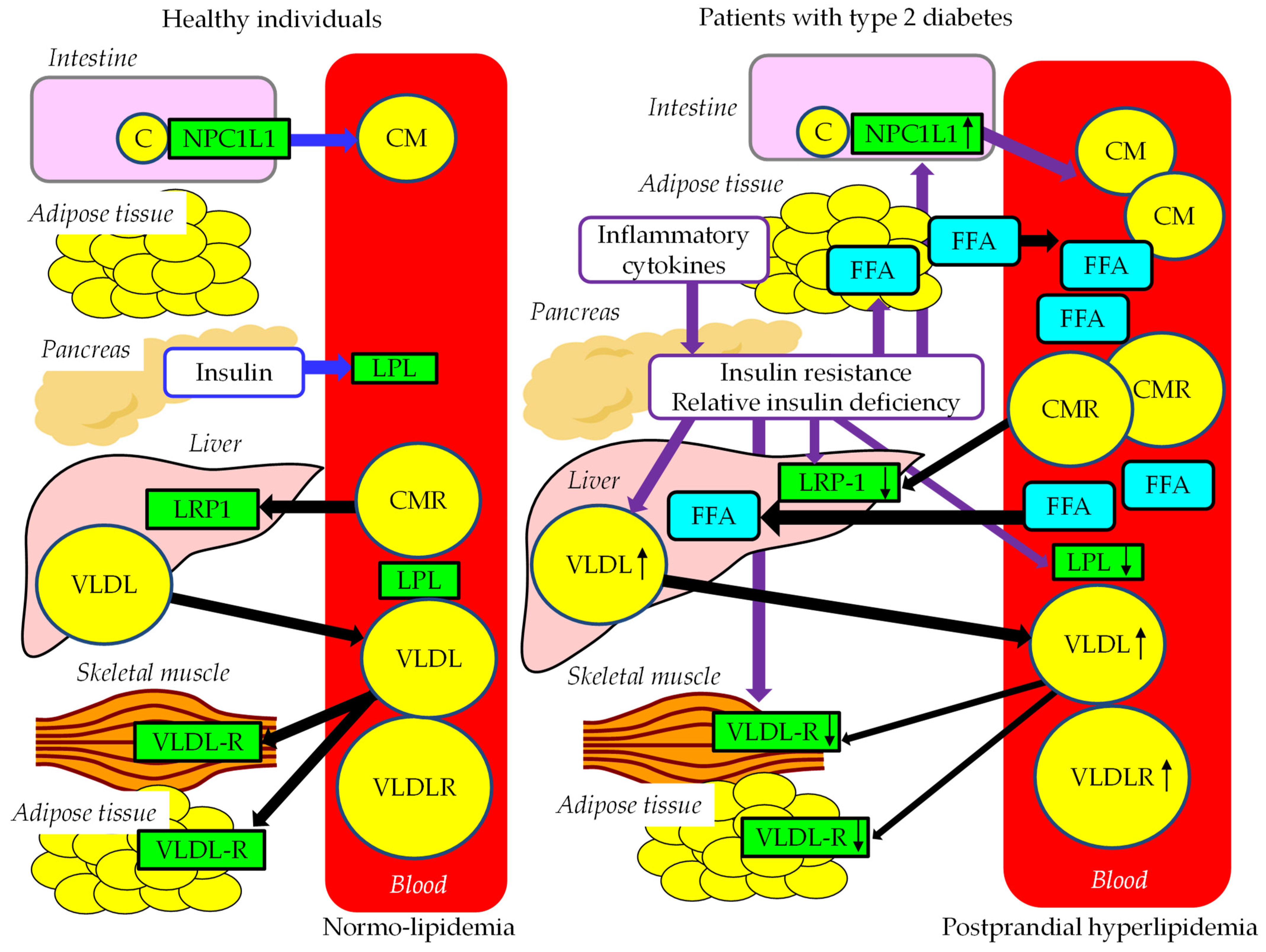
3.4.1. The Effects of Insulin Resistance on TRL Metabolism
3.4.2. Postprandial Hyperlipidemia in Patients with Obesity and/or Type 2 Diabetes
4. Postprandial Hyperglycemia and Postprandial Hyperlipidemia
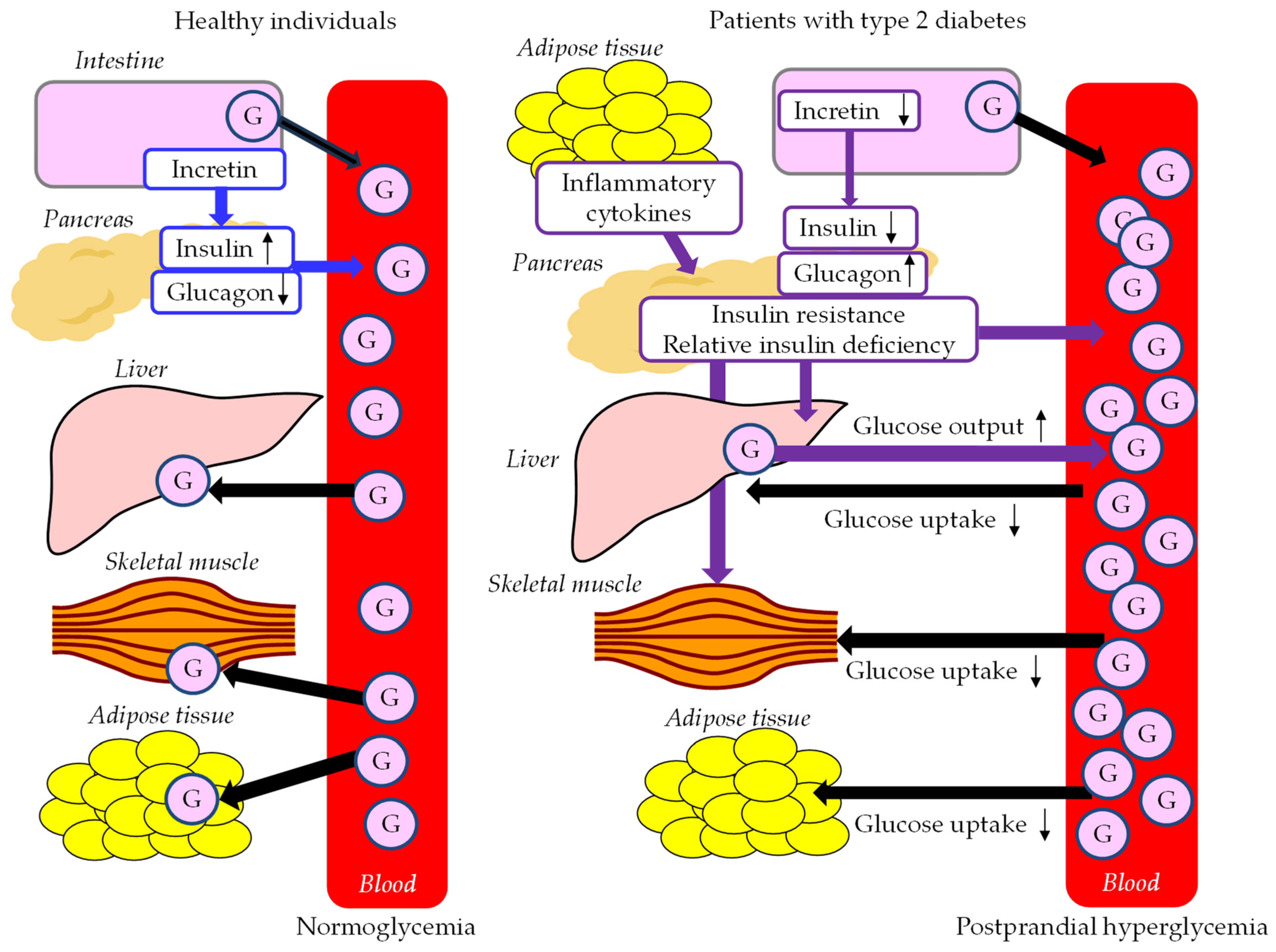
4.1. Abnormal Postprandial Glucose Metabolism in Postprandial Hyperglycemia
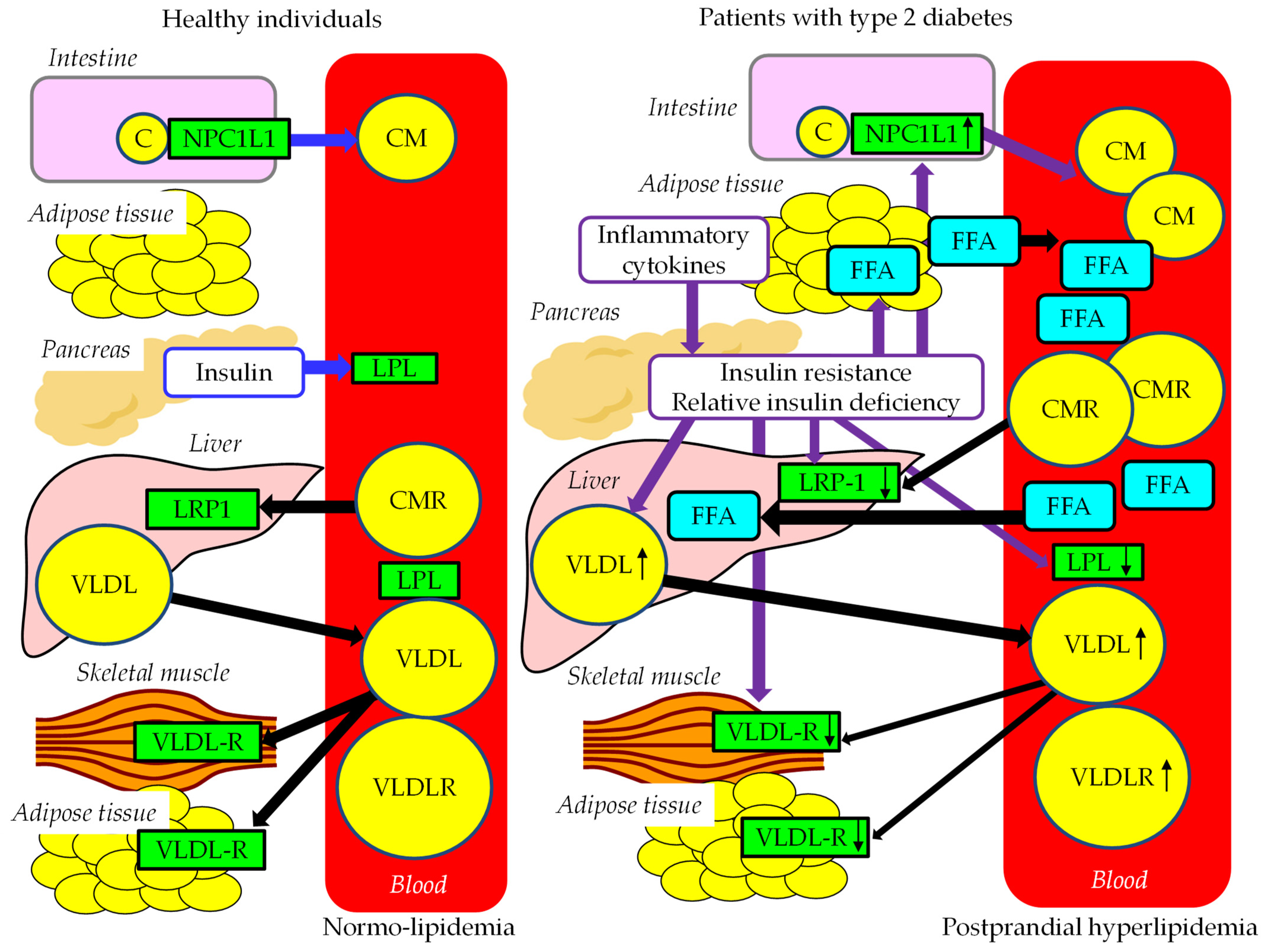
4.2. Abnormal FFA and TRL Metabolism in Postprandial Hyperlipidemia
4.3. The Association between Postprandial Hyperglycemia and Postprandial Hyperlipidemia
4.4. Differences in the Management of Postprandial Hyperglycemia and Postprandial Hyperlipidemia
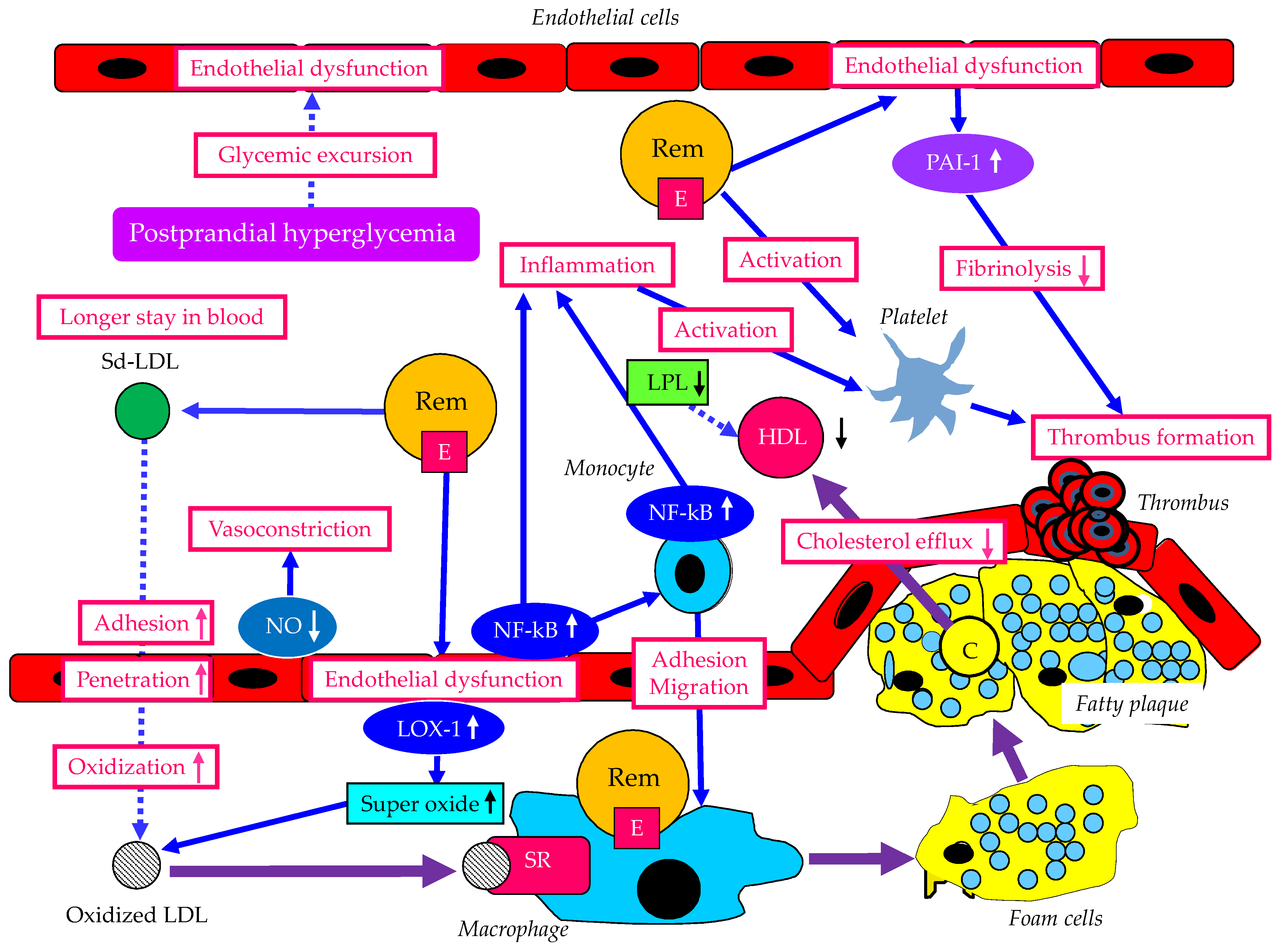
5. The Atherogenic Properties of Remnant Lipoproteins and Metabolic Disorders Associated with Postprandial Hyperlipidemia
6. Treatments for Postprandial Hyperglycemia
6.1. The Modification of Lifestyle, Including Diet and Exercise
6.2. Anti-Hyperlipidemic Drugs
6.2.1. Pemafibrate
6.2.2. Fenofibrate
6.2.3. Bezafibrate
6.2.4. Ezetimibe
6.2.5. Eicosapentaenoic Acid (EPA)
6.3. Anti-Diabetic Drugs
6.3.1. Metformin
6.3.2. Alpha-Glucosidase Inhibitors
6.3.3. Pioglitazone
6.3.4. Dipeptidyl-Peptidase-4 Inhibitors (DPP4is)
6.3.5. Glucagon-like Peptide 1 Analogues (GLP1A)
6.3.6. Sodium Glucose Cotransporter-2 Inhibitors (SGLT2is)
7. The Limitations of the Strengths of Our Review Article
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zilversmit, D.B. Atherogenesis: A postprandial phenomenon. Circulation 1979, 60, 473–485. [Google Scholar] [CrossRef]
- Nikkilä, M.; Solakivi, T.; Lehtimäki, T.; Koivula, T.; Laippala, P.; Aström, B. Postprandial plasma lipoprotein changes in relation to apolipoprotein E phenotypes and low density lipoprotein size in men with and without coronary artery disease. Atherosclerosis 1994, 106, 149–157. [Google Scholar] [CrossRef]
- Patsch, J.R.; Miesenböck, G.; Hopferwieser, T.; Mühlberger, V.; Knapp, E.; Dunn, J.K.; Gotto, A.M., Jr.; Patsch, W. Relation of triglyceride metabolism and coronary artery disease. Studies in the postprandial state. Arterioscler. Thromb. 1992, 12, 1336–1345. [Google Scholar] [CrossRef]
- Durlach, V.; Attia, N.; Zahouani, A.; Leutenegger, M.; Girard-Globa, A. Postprandial cholesteryl ester transfer and high density lipoprotein composition in normotriglyceridemic non-insulin-dependent diabetic patients. Atherosclerosis 1996, 120, 155–165. [Google Scholar] [CrossRef]
- Chen, Y.D.; Skowronski, R.; Coulston, A.M.; Pietarinen, J.; Hollenbeck, C.B.; Reaven, G.M. Effect of acute variations in dietary fat and carbohydrate intake on retinyl ester content of intestinally derived lipoproteins. J. Clin. Endocrinol. Metab. 1992, 74, 28–32. [Google Scholar]
- Zampelas, A.; Peel, A.S.; Gould, B.J.; Wright, J.; Williams, C.M. Polyunsaturated fatty acids of the n-6 and n-3 series: Effects on postprandial lipid and apolipoprotein levels in healthy men. Eur. J. Clin. Nutr. 1994, 48, 842–848. [Google Scholar]
- Nakamura, K.; Miyoshi, T.; Yunoki, K.; Ito, H. Postprandial hyperlipidemia as a potential residual risk factor. J. Cardiol. 2016, 67, 335–339. [Google Scholar] [CrossRef]
- Aldred, H.E.; Perry, I.C.; Hardman, A.E. The effect of a single bout of brisk walking on postprandial lipemia in normolipidemic young adults. Metabolism 1994, 43, 836–841. [Google Scholar] [CrossRef]
- Havel, R.J. Postprandial hyperlipidemia and remnant lipoproteins. Curr. Opin. Lipidol. 1994, 5, 102–109. [Google Scholar] [CrossRef]
- Havel, R.J. Remnant lipoproteins as therapeutic targets. Curr. Opin. Lipidol. 2000, 11, 615–620. [Google Scholar] [CrossRef]
- Nakajima, K.; Saito, T.; Tamura, A.; Suzuki, M.; Nakano, T.; Adachi, M.; Tanaka, A.; Tada, N.; Nakamura, H.; Campos, E.; et al. Cholesterol in remnant-like lipoproteins in human serum using monoclonal anti apo B-100 and anti apo A-I immunoaffinity mixed gels. Clin. Chim. Acta 1993, 223, 53–71. [Google Scholar] [CrossRef]
- Nakajima, K.; Nakano, T.; Tokita, Y.; Nagamine, T.; Inazu, A.; Kobayashi, J.; Mabuchi, H.; Stanhope, K.L.; Havel, P.J.; Okazaki, M.; et al. Postprandial lipoprotein metabolism: VLDL vs chylomicrons. Clin. Chim. Acta 2011, 412, 1306–1318. [Google Scholar] [CrossRef]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Atherogenic Lipoproteins for the Statin Residual Cardiovascular Disease Risk. Int. J. Mol. Sci. 2022, 23, 13499. [Google Scholar] [CrossRef]
- Masuda, D.; Nakagawa-Toyama, Y.; Nakatani, K.; Inagaki, M.; Tsubakio-Yamamoto, K.; Sandoval, J.C.; Ohama, T.; Nishida, M.; Ishigami, M.; Yamashita, S. Ezetimibe improves postprandial hyperlipidaemia in patients with type IIb hyperlipidaemia. Eur. J. Clin. Investig. 2009, 39, 689–698. [Google Scholar] [CrossRef]
- Kahri, J.; Fruchart-Najib, J.; Matikainen, N.; Fruchart, J.C.; Vakkilainen, J.; Taskinen, M.R. The increase of apolipoprotein A-V during postprandial lipemia parallels the response of triglyceride-rich lipoproteins in type 2 diabetes: No relationship between apoA-V and postheparin plasma lipolytic activity. Diabetes Care 2007, 30, 2083–2085. [Google Scholar] [CrossRef]
- Nakajima, K.; Nakano, T.; Tokita, Y.; Nagamine, T.; Yatsuzuka, S.; Shimomura, Y.; Tanaka, A.; Sumino, H.; Nara, M.; Machida, T.; et al. The characteristics of remnant lipoproteins in the fasting and postprandial plasma. Clin. Chim. Acta 2012, 413, 1077–1086. [Google Scholar] [CrossRef]
- Iso, H.; Naito, Y.; Sato, S.; Kitamura, A.; Okamura, T.; Sankai, T.; Shimamoto, T.; Iida, M.; Komachi, Y. Serum triglycerides and risk of coronary heart disease among Japanese men and women. Am. J. Epidemiol. 2001, 153, 490–499. [Google Scholar] [CrossRef]
- Eberly, L.E.; Stamler, J.; Neaton, J.D.; Multiple Risk Factor Intervention Trial Research Group. Relation of triglyceride levels, fasting and nonfasting, to fatal and nonfatal coronary heart disease. Arch. Intern. Med. 2003, 163, 1077–1083. [Google Scholar] [CrossRef]
- White, K.T.; Moorthy, M.V.; Akinkuolie, A.O.; Demler, O.; Ridker, P.M.; Cook, N.R.; Mora, S. Identifying an Optimal Cutpoint for the Diagnosis of Hypertriglyceridemia in the Nonfasting State. Clin. Chem. 2015, 61, 1156–1163. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar]
- Nordestgaard, B.G.; Langsted, A.; Mora, S.; Kolovou, G.; Baum, H.; Bruckert, E.; Watts, G.F.; Sypniewska, G.; Wiklund, O.; Borén, J.; et al. Fasting is not routinely required for determination of a lipid profile: Clinical and laboratory implications including flagging at desirable concentration cut-points-a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur. Heart J. 2016, 37, 1944–1958. [Google Scholar]
- Japan Atherosclerosis Society (JAS). Guidelines for Prevention of Atherosclerotic Cardiovascular Diseases; Japan Atherosclerosis Society (JAS): Tokyo, Japan, 2022. [Google Scholar]
- Masuda, D.; Sakai, N.; Sugimoto, T.; Kitazume-Taneike, R.; Yamashita, T.; Kawase, R.; Nakaoka, H.; Inagaki, M.; Nakatani, K.; Yuasa-Kawase, M.; et al. Fasting serum apolipoprotein B-48 can be a marker of postprandial hyperlipidemia. J. Atheroscler. Thromb. 2011, 18, 1062–1070. [Google Scholar] [CrossRef]
- Campos, E.; Nakajima, K.; Tanaka, A.; Havel, R.J. Properties of an apolipoprotein E-enriched fraction of triglyceride-rich lipoproteins isolated from human blood plasma with a monoclonal antibody to apolipoprotein B-100. J. Lipid Res. 1992, 33, 369–380. [Google Scholar] [CrossRef]
- Vigne, J.L.; Havel, R.J. Metabolism of apolipoprotein A-I of chylomicrons in rats and humans. Can. J. Biochem. 1981, 59, 613–618. [Google Scholar] [CrossRef]
- Marcoux, C.; Tremblay, M.; Nakajima, K.; Davignon, J.; Cohn, J.S. Characterization of remnant-like particles isolated by immunoaffinity gel from the plasma of type III and type IV hyperlipoproteinemic patients. J. Lipid Res. 1999, 40, 636–647. [Google Scholar] [CrossRef]
- Miyauchi, K.; Kayahara, N.; Ishigami, M.; Kuwata, H.; Mori, H.; Sugiuchi, H.; Irie, T.; Tanaka, A.; Yamashita, S.; Yamamura, T. Development of a homogeneous assay to measure remnant lipoprotein cholesterol. Clin. Chem. 2007, 53, 2128–2135. [Google Scholar] [CrossRef]
- Nakada, Y.; Kurosawa, H.; Tohyama, J.; Inoue, Y.; Ikewaki, K. Increased remnant lipoprotein in patients with coronary artery disease--evaluation utilizing a newly developed remnant assay, remnant lipoproteins cholesterol homogenous assay (RemL-C). J. Atheroscler. Thromb. 2007, 14, 56–64. [Google Scholar] [CrossRef]
- Yoshida, H.; Kurosawa, H.; Hirowatari, Y.; Ogura, Y.; Ikewaki, K.; Abe, I.; Saikawa, S.; Domitsu, K.; Ito, K.; Yanai, H.; et al. Characteristic comparison of triglyceride-rich remnant lipoprotein measurement between a new homogenous assay (RemL-C) and a conventional immunoseparation method (RLP-C). Lipids Health Dis. 2008, 7, 18. [Google Scholar] [CrossRef]
- Mahley, R.W.; Huang, Y.; Rall, S.C., Jr. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J. Lipid Res. 1999, 40, 1933–1949. [Google Scholar] [CrossRef]
- Heidemann, B.E.; Koopal, C.; van Lennep, J.E.R.; Stroes, E.S.G.; Riksen, N.P.; Mulder, M.T.; van der Zee, L.C.V.V.; Blackhurst, D.M.; Marais, A.D.; Visseren, F.L.J. Effect of evolocumab on fasting and post fat load lipids and lipoproteins in familial dysbetalipoproteinemia. J. Clin. Lipidol. 2023, 17, 112–123. [Google Scholar] [CrossRef]
- de Beer, F.; Stalenhoef, A.F.; Hoogerbrugge, N.; Kastelein, J.J.; Gevers Leuven, J.A.; van Duijn, C.M.; Havekes, L.M.; Smelt, A.H. Expression of type III hyperlipoproteinemia in apolipoprotein E2 (Arg158 → Cys) homozygotes is associated with hyperinsulinemia. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Schrott, H.G.; Hazzard, W.R.; Bierman, E.L.; Motulsky, A.G. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J. Clin. Investig. 1973, 52, 1544–1568. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, M.C.; de Bruin, T.W.; Kock, L.A.; Kortlandt, W.; Van Linde-Sibenius Trip, M.; Jansen, H.; Erkelens, D.W. Simvastatin improves chylomicron remnant removal in familial combined hyperlipidemia without changing chylomicron conversion. Metabolism 1993, 42, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, M.C.; de Bruin, T.W.; Jansen, H.; Kock, L.A.; Kortlandt, W.; Erkelens, D.W. Impaired chylomicron remnant clearance in familial combined hyperlipidemia. Arterioscler. Thromb. 1993, 13, 804–814. [Google Scholar] [CrossRef]
- Castro Cabezas, M.; Erkelens, D.W.; Kock, L.A.; De Bruin, T.W. Postprandial apolipoprotein B100 and B48 metabolism in familial combined hyperlipidaemia before and after reduction of fasting plasma triglycerides. Eur. J. Clin. Investig. 1994, 24, 669–678. [Google Scholar] [CrossRef]
- Meijssen, S.; Cabezas, M.C.; Twickler, T.B.; Jansen, H.; Erkelens, D.W. In vivo evidence of defective postprandial and postabsorptive free fatty acid metabolism in familial combined hyperlipidemia. J. Lipid Res. 2000, 41, 1096–1102. [Google Scholar] [CrossRef]
- de Graaf, J.; van der Vleuten, G.M.; ter Avest, E.; Dallinga-Thie, G.M.; Stalenhoef, A.F. High plasma level of remnant-like particles cholesterol in familial combined hyperlipidemia. J. Clin. Endocrinol. Metab. 2007, 92, 1269–1275. [Google Scholar] [CrossRef]
- Carr, M.C.; Brunzell, J.D. Abdominal obesity and dyslipidemia in the metabolic syndrome: Importance of type 2 diabetes and familial combined hyperlipidemia in coronary artery disease risk. J. Clin. Endocrinol. Metab. 2004, 89, 2601–2607. [Google Scholar] [CrossRef]
- Brahm, A.J.; Hegele, R.A. Combined hyperlipidemia: Familial but not (usually) monogenic. Curr. Opin. Lipidol. 2016, 27, 131–140. [Google Scholar] [CrossRef]
- Shoji, T.; Emoto, M.; Kawagishi, T.; Kimoto, E.; Yamada, A.; Tabata, T.; Ishimura, E.; Inaba, M.; Okuno, Y.; Nishizawa, Y. Atherogenic lipoprotein changes in diabetic nephropathy. Atherosclerosis 2001, 156, 425–433. [Google Scholar] [CrossRef]
- Pandya, V.; Rao, A.; Chaudhary, K. Lipid abnormalities in kidney disease and management strategies. World J. Nephrol. 2015, 4, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Abe, T.; Matsuo, H.; Egusa, G.; Yamasaki, Y.; Kashihara, N.; Shirai, K.; Kashiwagi, A. Committee of Renal and Peripheral Arteries, Japan Atherosclerosis Society. Chronic kidney disease, dyslipidemia, and atherosclerosis. J. Atheroscler. Thromb. 2012, 19, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Hirowatari, Y.; Yoshida, H.; Fueki, Y.; Ito, M.; Ogura, Y.; Sakurai, N.; Miida, T. Measurement of cholesterol concentrations of major serum lipoprotein classes in haemodialysis patients by anion-exchange chromatography. Ann. Clin. Biochem. 2008, 45, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Drukker, A.; Levy, E.; Bronza, N.; Stankiewicz, H.; Goldstein, R. Impaired intestinal fat absorption in chronic renal failure. Nephron 1982, 30, 154–160. [Google Scholar] [CrossRef]
- Nestel, P.J.; Fidge, N.H.; Tan, M.H. Increased lipoprotein-remnant formation in chronic renal failure. N. Engl. J. Med. 1982, 307, 329–333. [Google Scholar] [CrossRef]
- Yanai, H.; Yoshida, H.; Tomono, Y.; Hirowatari, Y.; Kurosawa, H.; Matsumoto, A.; Tada, N. Effects of diacylglycerol on glucose, lipid metabolism, and plasma serotonin levels in lean Japanese. Obesity 2008, 16, 47–51. [Google Scholar] [CrossRef]
- Ito, K.; Yoshida, H.; Yanai, H.; Kurosawa, H.; Sato, R.; Manita, D.; Hirowatari, Y.; Tada, N. Relevance of intermediate-density lipoprotein cholesterol to Framingham risk score of coronary heart disease in middle-aged men with increased non-HDL cholesterol. Int. J. Cardiol. 2013, 168, 3853–3858. [Google Scholar] [CrossRef]
- Yoshida, H.; Hirowatari, Y.; Kurosawa, H.; Manita, D.; Yanai, H.; Ito, K.; Tada, N. Estimation of lipoprotein profile in patients with type II diabetes and its relevance to remnant lipoprotein cholesterol levels. Atherosclerosis 2012, 222, 541–544. [Google Scholar] [CrossRef]
- Yanai, H.; Hamasaki, H.; Adachi, H.; Moriyama, S.; Hirowatari, Y. Effects of liraglutide, a human glucagon-like peptide-1 analog, on glucose/lipid metabolism, and adipocytokines in patients with type 2 diabetes. J. Endocrinol. Metab. 2011, 1, 149–151. [Google Scholar] [CrossRef]
- Yanai, H.; Hirowatari, Y.; Ito, K.; Kurosawa, H.; Tada, N.; Yoshida, H. Understanding of Diabetic Dyslipidemia by Using the Anion-Exchange High Performance Liquid Chromatography Data. J. Clin. Med. Res. 2016, 8, 424–426. [Google Scholar] [CrossRef]
- Sztalryd, C.; Kraemer, F.B. Regulation of hormone-sensitive lipase in streptozotocin-induced diabetic rats. Metabolism 1995, 44, 1391–1396. [Google Scholar] [CrossRef]
- Fisher, E.A. The degradation of apolipoprotein B100: Multiple opportunities to regulate VLDL triglyceride production by different proteolytic pathways. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2012, 1821, 778–781. [Google Scholar] [CrossRef]
- Taghibiglou, C.; Carpentier, A.; Van Iderstine, S.C.; Chen, B.; Rudy, D.; Aiton, A.; Lewis, G.F.; Adeli, K. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J. Biol. Chem. 2000, 275, 8416–8425. [Google Scholar] [PubMed]
- Avramoglu, R.K.; Basciano, H.; Adeli, K. Lipid and lipoprotein dysregulation in insulin resistant states. Clin. Chim. Acta 2006, 368, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Sakai, J.; Fujino, T.; Hattori, H.; Zenimaru, Y.; Suzuki, J.; Miyamori, I.; Yamamoto, T.T. The very low-density lipoprotein (VLDL) receptor: Characterization and functions as a peripheral lipoprotein receptor. J. Atheroscler. Thromb. 2004, 11, 200–208. [Google Scholar] [CrossRef]
- Yuan, G.; Liu, Y.; Sun, T.; Xu, Y.; Zhang, J.; Yang, Y.; Zhang, M.; Cianflone, K.; Wang, D.W. The therapeutic role of very low-density lipoprotein receptor gene in hyperlipidemia in type 2 diabetic rats. Hum. Gene Ther. 2011, 22, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Nikkila, E.A.; Huttunen, J.K.; Ehnholm, C. Postheparin plasma lipoprotein lipase and hepatic lipase in diabetes mellitus. Relationship to plasma triglyceride metabolism. Diabetes 1977, 26, 11–21. [Google Scholar] [CrossRef] [PubMed]
- de Silva, H.V.; Lauer, S.J.; Wang, J.; Simonet, W.S.; Weisgraber, K.H.; Mahley, R.W.; Taylor, J.M. Overexpression of human apolipoprotein C-III in transgenic mice results in an accumulation of apolipoprotein B48 remnants that is corrected by excess apolipoprotein E. J. Biol. Chem. 1994, 269, 2324–2335. [Google Scholar] [CrossRef]
- Cohn, J.S.; Patterson, B.W.; Uffelman, K.D.; Davignon, J.; Steiner, G. Rate of production of plasma and very-low-density lipoprotein (VLDL) apolipoprotein C-III is strongly related to the concentration and level of production of VLDL triglyceride in male subjects with different body weights and levels of insulin sensitivity. J. Clin. Endocrinol. Metab. 2004, 89, 3949–3955. [Google Scholar] [CrossRef]
- Nikkila, E.A.; Taskinen, M.R.; Kekki, M. Relation of plasma high-density lipoprotein cholesterol to lipoprotein-lipase activity in adipose tissue and skeletal muscle of man. Atherosclerosis 1978, 29, 497–501. [Google Scholar] [CrossRef]
- Baynes, C.; Henderson, A.D.; Anyaoku, V.; Richmond, W.; Hughes, C.L.; Johnston, D.G.; Elkeles, R.S. The role of insulin insensitivity and hepatic lipase in the dyslipidaemia of type 2 diabetes. Diabet. Med. 1991, 8, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.C.; Hokanson, J.E.; Zambon, A.; Deeb, S.S.; Barrett, P.H.; Purnell, J.Q.; Brunzell, J.D. The contribution of intraabdominal fat to gender differences in hepatic lipase activity and low/high density lipoprotein heterogeneity. J. Clin. Endocrinol. Metab. 2001, 86, 2831–2837. [Google Scholar] [CrossRef] [PubMed]
- Allayee, H.; Dominguez, K.M.; Aouizerat, B.E.; Krauss, R.M.; Rotter, J.I.; Lu, J.; Cantor, R.M.; de Bruin, T.W.; Lusis, A.J. Contribution of the hepatic lipase gene to the atherogenic lipoprotein phenotype in familial combined hyperlipidemia. J. Lipid Res. 2000, 41, 245–252. [Google Scholar] [CrossRef]
- Deeb, S.S.; Zambon, A.; Carr, M.C.; Ayyobi, A.F.; Brunzell, J.D. Hepatic lipase and dyslipidemia: Interactions among genetic variants, obesity, gender, and diet. J. Lipid Res. 2003, 44, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Chandra, N.C. A comprehensive account of insulin and LDL receptor activity over the years: A highlight on their signaling and functional role. J. Biochem. Mol. Toxicol. 2021, 35, e22840. [Google Scholar] [CrossRef]
- Lally, S.; Owens, D.; Tomkin, G.H. Genes that affect cholesterol synthesis, cholesterol absorption, and chylomicron assembly: The relationship between the liver and intestine in control and streptozotosin diabetic rats. Metabolism 2007, 56, 430–438. [Google Scholar] [CrossRef]
- Lally, S.; Tan, C.Y.; Owens, D.; Tomkin, G.H. Messenger RNA levels of genes involved in dysregulation of postprandial lipoproteins in type 2 diabetes: The role of Niemann-Pick C1-like 1, ATP-binding cassette, transporters G5 and G8, and of microsomal triglyceride transfer protein. Diabetologia 2006, 49, 1008–1016. [Google Scholar] [CrossRef]
- Kinoshita, M.; Ohnishi, H.; Maeda, T.; Yoshimura, N.; Takeoka, Y.; Yasuda, D.; Kusano, J.; Mashimo, Y.; Saito, S.; Shimamoto, K.; et al. Increased serum apolipoprotein B48 concentration in patients with metabolic syndrome. J. Atheroscler. Thromb. 2009, 16, 517–522. [Google Scholar] [CrossRef]
- Irawati, D.; Mamo, J.C.; Dhaliwal, S.S.; Soares, M.J.; Slivkoff-Clark, K.M.; James, A.P. Plasma triglyceride and high density lipoprotein cholesterol are poor surrogate markers of pro-atherogenic chylomicron remnant homeostasis in subjects with the metabolic syndrome. Lipids Health Dis. 2016, 15, 169. [Google Scholar] [CrossRef]
- Actis Dato, V.; Chiabrando, G.A. The Role of Low-Density Lipoprotein Receptor-Related Protein 1 in Lipid Metabolism, Glucose Homeostasis and Inflammation. Int. J. Mol. Sci. 2018, 19, 1780. [Google Scholar] [CrossRef]
- Laatsch, A.; Merkel, M.; Talmud, P.J.; Grewal, T.; Beisiegel, U.; Heeren, J. Insulin stimulates hepatic low density lipoprotein receptor-related protein 1 (LRP1) to increase postprandial lipoprotein clearance. Atherosclerosis 2009, 204, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.E.; Attie, A.D.; Biddinger, S.B. The regulation of ApoB metabolism by insulin. Trends Endocrinol. Metab. 2013, 24, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Carstensen, M.; Thomsen, C.; Hermansen, K. Incremental area under response curve more accurately describes the triglyceride response to an oral fat load in both healthy and type 2 diabetic subjects. Metabolism 2003, 52, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Umpaichitra, V.; Banerji, M.A.; Castells, S. Postprandial hyperlipidemia after a fat loading test in minority adolescents with type 2 diabetes mellitus and obesity. J. Pediatr. Endocrinol. Metab. 2004, 17, 853–864. [Google Scholar] [CrossRef]
- Watanabe, N.; Taniguchi, T.; Taketoh, H.; Kitagawa, Y.; Namura, H.; Yoneda, N.; Kurimoto, Y.; Yamada, S.; Ishikawa, Y. Elevated remnant-like lipoprotein particles in impaired glucose tolerance and type 2 diabetic patients. Diabetes Care 1999, 22, 152–156. [Google Scholar] [CrossRef]
- Vilsbøll, T.; Holst, J.J. Incretins, insulin secretion and Type 2 diabetes mellitus. Diabetologia 2004, 47, 357–366. [Google Scholar] [CrossRef]
- DeFronzo, R.A. Pathogenesis of type 2 (non-insulin dependent) diabetes mellitus: A balanced overview. Diabetologia 1992, 35, 389–397. [Google Scholar] [CrossRef]
- Kawamori, R. The biology of insulin action in diabetes. In Diabetes in the New Millennium; Turtle, J.R., Kaneko, T., Osato, S., Eds.; Wiley: Hoboken, NJ, USA, 1999; pp. 73–84. [Google Scholar]
- de Vries, M.A.; Alipour, A.; Klop, B.; van de Geijn, G.J.; Janssen, H.W.; Njo, T.L.; van der Meulen, N.; Rietveld, A.P.; Liem, A.H.; Westerman, E.M.; et al. Glucose-dependent leukocyte activation in patients with type 2 diabetes mellitus, familial combined hyperlipidemia and healthy controls. Metabolism 2015, 64, 213–217. [Google Scholar] [CrossRef]
- Vakkilainen, J.; Porkka, K.V.; Nuotio, I.; Pajukanta, P.; Suurinkeroinen, L.; Ylitalo, K.; Viikari, J.S.; Ehnholm, C.; Taskinen, M.R. Glucose intolerance in familial combined hyperlipidaemia. EUFAM study group. Eur. J. Clin. Investig. 1998, 28, 24–32. [Google Scholar] [CrossRef]
- Ascaso, J.F.; Sales, J.; Merchante, A.; Real, J.; Lorente, R.; Martinez-Valls, J.; Carmena, R. Influence of obesity on plasma lipoproteins, glycaemia and insulinaemia in patients with familial combined hyperlipidaemia. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 360–366. [Google Scholar] [CrossRef]
- Jadhakhan, F.; Marshall, T.; Ryan, R.; Gill, P. Risk of chronic kidney disease in young adults with impaired glucose tolerance/impaired fasting glucose: A retrospective cohort study using electronic primary care records. BMC Nephrol. 2018, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Canpolat, N.; Caliskan, S.; Sever, L.; Guzeltas, A.; Kantarci, F.; Candan, C.; Civilibal, M.; Kasapcopur, O.; Arisoy, N. Glucose intolerance: Is it a risk factor for cardiovascular disease in children with chronic kidney disease? Pediatr. Nephrol. 2012, 27, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Ikee, R.; Honda, K.; Ishioka, K.; Oka, M.; Maesato, K.; Moriya, H.; Hidaka, S.; Ohtake, T.; Kobayashi, S. Postprandial hyperglycemia and hyperinsulinemia associated with renal arterio-arteriolosclerosis in chronic kidney disease. Hypertens. Res. 2010, 33, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Ai, M.; Tanaka, A.; Ogita, K.; Sekinc, M.; Numano, F.; Numano, F.; Reaven, G.M. Relationship between plasma insulin concentration and plasma remnant lipoprotein response to an oral fat load in patients with type 2 diabetes. J. Am. Coll. Cardiol. 2001, 38, 1628–1632. [Google Scholar] [CrossRef] [PubMed]
- Genuth, S.; Alberti, K.G.; Bennett, P.; Buse, J.; Defronzo, R.; Kahn, R.; Kitzmiller, J.; Knowler, W.C.; Lebovitz, H.; Lernmark, A.; et al. Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care 2003, 26, 3160–3167. [Google Scholar] [PubMed]
- Sathish, T.; Khunti, K.; Narayan, K.M.V.; Mohan, V.; Davies, M.J.; Yates, T.; Oldenburg, B.; Thankappan, K.R.; Tapp, R.J.; Bajpai, R. Effect of Conventional Lifestyle Interventions on Type 2 Diabetes Incidence by Glucose-Defined Prediabetes Phenotype: An Individual-Participant Data Meta-analysis of Randomized Controlled Trials. Diabetes Care 2023, dc230696. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, Y.; Mu, Y.M.; Huang, Y.; Xuan, J. Network Meta-analysis of the Therapeutic Effects of Hypoglycemic Drugs and Intensive Lifestyle Modification on Impaired Glucose Tolerance. Clin. Ther. 2021, 43, 1524–1556. [Google Scholar] [CrossRef]
- Garrib, A.; Kivuyo, S.; Bates, K.; Ramaiya, K.; Wang, D.; Majaliwa, E.; Simbauranga, R.; Charles, G.; van Widenfelt, E.; Luo, H.; et al. Metformin for the prevention of diabetes among people with HIV and either impaired fasting glucose or impaired glucose tolerance (prediabetes) in Tanzania: A Phase II randomised placebo-controlled trial. Diabetologia 2023, 66, 1882–1896. [Google Scholar] [CrossRef]
- Ipsen, E.Ø.; Madsen, K.S.; Chi, Y.; Pedersen-Bjergaard, U.; Richter, B.; Metzendorf, M.I.; Hemmingsen, B. Pioglitazone for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2020, 11, CD013516. [Google Scholar]
- Costantino, S.; Paneni, F.; Battista, R.; Castello, L.; Capretti, G.; Chiandotto, S.; Tanese, L.; Russo, G.; Pitocco, D.; Lanza, G.A.; et al. Impact of Glycemic Variability on Chromatin Remodeling, Oxidative Stress, and Endothelial Dysfunction in Patients with Type 2 Diabetes and with Target HbA1c Levels. Diabetes 2017, 66, 2472–2482. [Google Scholar] [CrossRef]
- Akasaka, T.; Sueta, D.; Tabata, N.; Takashio, S.; Yamamoto, E.; Izumiya, Y.; Tsujita, K.; Kojima, S.; Kaikita, K.; Matsui, K.; et al. Effects of the Mean Amplitude of Glycemic Excursions and Vascular Endothelial Dysfunction on Cardiovascular Events in Nondiabetic Patients with Coronary Artery Disease. J. Am. Heart Assoc. 2017, 6, e004841. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.G.; Zhang, Y.Q.; Zhao, D.K.; Wu, J.X.; Zhao, J.; Jiao, X.M.; Chen, B.; Lv, X.F. Relationship between blood glucose fluctuation and macrovascular endothelial dysfunction in type 2 diabetic patients with coronary heart disease. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3593–3600. [Google Scholar] [PubMed]
- O’Keefe, J.H.; Gheewala, N.M.; O’Keefe, J.O. Dietary strategies for improving post-prandial glucose, lipids, inflammation, and cardiovascular health. J. Am. Coll. Cardiol. 2008, 51, 249–255. [Google Scholar] [CrossRef]
- Yamashita, S.; Arai, H.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; K-877 Study Group. Effects of pemafibrate (K-877) on cholesterol efflux capacity and postprandial hyperlipidemia in patients with atherogenic dyslipidemia. J. Clin. Lipidol. 2018, 12, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Katsuyama, H.; Hakoshima, M. Effects of a Novel Selective Peroxisome Proliferator-Activated Receptor α Modulator, Pemafibrate, on Metabolic Parameters: A Retrospective Longitudinal Study. Biomedicines 2022, 10, 401. [Google Scholar] [CrossRef]
- Föger, B.; Drexel, H.; Hopferwieser, T.; Miesenböck, G.; Ritsch, A.; Lechleitner, M.; Tröbinger, G.; Patsch, J.R. Fenofibrate improves postprandial chylomicron clearance in II B hyperlipoproteinemia. Clin. Investig. 1994, 72, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Genest, J., Jr.; Nguyen, N.H.; Theroux, P.; Davignon, J.; Cohn, J.S. Effect of micronized fenofibrate on plasma lipoprotein levels and hemostatic parameters of hypertriglyceridemic patients with low levels of high-density lipoprotein cholesterol in the fed and fasted state. J. Cardiovasc. Pharmacol. 2000, 35, 164–172. [Google Scholar] [CrossRef]
- Cavallero, E.; Dachet, C.; Assadolahi, F.; Martin, C.; Navarro, N.; Ansquer, J.C.; Corda, C.; Foucher, C.; Juhan-Vague, I.; Jacotot, B. Micronized fenofibrate normalizes the enhanced lipidemic response to a fat load in patients with type 2 diabetes and optimal glucose control. Atherosclerosis 2003, 166, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Wolff, D.A.; Huskin, A.L.; Helenowski, I.B.; Rademaker, A.W. Fenofibrate therapy ameliorates fasting and postprandial lipoproteinemia, oxidative stress, and the inflammatory response in subjects with hypertriglyceridemia and the metabolic syndrome. Diabetes Care 2007, 30, 1945–1951. [Google Scholar] [CrossRef]
- Ohno, Y.; Miyoshi, T.; Noda, Y.; Oe, H.; Toh, N.; Nakamura, K.; Kohno, K.; Morita, H.; Ito, H. Bezafibrate improves postprandial hypertriglyceridemia and associated endothelial dysfunction in patients with metabolic syndrome: A randomized crossover study. Cardiovasc. Diabetol. 2014, 13, 71. [Google Scholar] [CrossRef]
- Nakamura, A.; Sato, K.; Kanazawa, M.; Kondo, M.; Endo, H.; Takahashi, T.; Nozaki, E. Impact of decreased insulin resistance by ezetimibe on postprandial lipid profiles and endothelial functions in obese, non-diabetic-metabolic syndrome patients with coronary artery disease. Heart Vessel. 2019, 34, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Tsubata, H.; Hashimoto, N.; Takabe, M.; Miyata, T.; Aoki, K.; Yamashita, S.; Oishi, S.; Osue, T.; Yokoi, K.; et al. Effects of 6-month eicosapentaenoic acid treatment on postprandial hyperglycemia, hyperlipidemia, insulin secretion ability, and concomitant endothelial dysfunction among newly-diagnosed impaired glucose metabolism patients with coronary artery disease. An open label, single blinded, prospective randomized controlled trial. Cardiovasc. Diabetol. 2016, 15, 121. [Google Scholar] [PubMed]
- Weintraub, M.S.; Charach, G.; Grosskopf, I. Effects of fibric acid derivatives and metformin on postprandial lipemia. Atherosclerosis 1998, 141, S71–S75. [Google Scholar] [CrossRef]
- Kado, S.; Murakami, T.; Aoki, A.; Nagase, T.; Katsura, Y.; Noritake, M.; Matsuoka, T.; Nagata, N. Effect of acarbose on postprandial lipid metabolism in type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 1998, 41, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, F.; Lamendola, C.; Leary, E.T.; Reaven, G.M. Pioglitazone decreases postprandial accumulation of remnant lipoproteins in insulin-resistant smokers. Diabetes Obes. Metab. 2009, 11, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Itoh, Y.; Obata, T.; Tajima, N. Effects of pioglitazone vs glibenclamide on postprandial increases in glucose and triglyceride levels and on oxidative stress in Japanese patients with type 2 diabetes. Endocrine 2006, 29, 143–148. [Google Scholar] [CrossRef]
- Noda, Y.; Miyoshi, T.; Oe, H.; Ohno, Y.; Nakamura, K.; Toh, N.; Kohno, K.; Morita, H.; Kusano, K.; Ito, H. Alogliptin ameliorates postprandial lipemia and postprandial endothelial dysfunction in non-diabetic subjects: A preliminary report. Cardiovasc. Diabetol. 2013, 12, 8. [Google Scholar] [CrossRef]
- Derosa, G.; Bonaventura, A.; Bianchi, L.; Romano, D.; Fogari, E.; D’Angelo, A.; Maffioli, P. Vildagliptin compared to glimepiride on post-prandial lipemia and on insulin resistance in type 2 diabetic patients. Metabolism 2014, 63, 957–967. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Björnson, E.; Matikainen, N.; Söderlund, S.; Pietiläinen, K.H.; Ainola, M.; Hakkarainen, A.; Lundbom, N.; Fuchs, J.; Thorsell, A.; et al. Effects of liraglutide on the metabolism of triglyceride-rich lipoproteins in type 2 diabetes. Diabetes Obes. Metab. 2021, 23, 1191–1201. [Google Scholar] [CrossRef]
- Hermansen, K.; Bækdal, T.A.; Düring, M.; Pietraszek, A.; Mortensen, L.S.; Jørgensen, H.; Flint, A. Liraglutide suppresses postprandial triglyceride and apolipoprotein B48 elevations after a fat-rich meal in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled, cross-over trial. Diabetes Obes. Metab. 2013, 15, 1040–1048. [Google Scholar] [CrossRef]
- Bunck, M.C.; Cornér, A.; Eliasson, B.; Heine, R.J.; Shaginian, R.M.; Wu, Y.; Yan, P.; Smith, U.; Yki-Järvinen, H.; Diamant, M.; et al. One-year treatment with exenatide vs. insulin glargine: Effects on postprandial glycemia, lipid profiles, and oxidative stress. Atherosclerosis 2010, 212, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, H.; Hakoshima, M.; Umeyama, S.; Iida, S.; Adachi, H.; Yanai, H. Real-World Efficacy of Glucagon-like Peptide-1 (GLP-1) Receptor Agonist, Dulaglutide, on Metabolic Parameters in Japanese Patients with Type 2 Diabetes: A Retrospective Longitudinal Study. Biomedicines 2023, 11, 869. [Google Scholar] [CrossRef]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Katsuyama, H. A Significant Effect of Oral Semaglutide on Cardiovascular Risk Factors in Patients with Type 2 Diabetes. Cardiol. Res. 2022, 13, 303–308. [Google Scholar] [CrossRef]
- Kakuda, H.; Kobayashi, J.; Sakurai, M.; Kakuda, M.; Takekoshi, N. The Effect of Tofogliflozin Treatment on Postprandial Glucose and Lipid Metabolism in Japanese Men with Type 2 Diabetes: A Pilot Study. J. Clin. Med. Res. 2017, 9, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Burggraaf, B.; Pouw, N.M.C.; Fernández Arroyo, S.; van Vark-van der Zee, L.C.; van de Geijn, G.M.; Birnie, E.; Huisbrink, J.; van der Zwan, E.M.; de Herder, W.W.; Mulder, M.T.; et al. Effects of dapagliflozin on postprandial lipid metabolism in type 2 diabetes mellitus. Eur. J. Endocrinol. 2022, 186, 597–605. [Google Scholar] [CrossRef]
- Hattori, S. Empagliflozin decreases remnant-like particle cholesterol in type 2 diabetes patients with insulin resistance. J. Diabetes Investig. 2018, 9, 870–874. [Google Scholar] [CrossRef]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Kawaguchi, A.; Waragai, Y.; Harigae, T.; Masui, Y.; Kakuta, K.; Hamasaki, H.; Katsuyama, H.; et al. Effects of Six Kinds of Sodium-Glucose Cotransporter 2 Inhibitors on Metabolic Parameters, and Summarized Effect and Its Correlations with Baseline Data. J. Clin. Med. Res. 2017, 9, 605–612. [Google Scholar] [CrossRef]
- Yanai, H.; Hakoshima, M.; Adachi, H.; Katsuyama, H. Multi-Organ Protective Effects of Sodium Glucose Cotransporter 2 Inhibitors. Int. J. Mol. Sci. 2021, 22, 4416. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Postprandial Hyperlipidemia: Its Pathophysiology, Diagnosis, Atherogenesis, and Treatments. Int. J. Mol. Sci. 2023, 24, 13942. https://doi.org/10.3390/ijms241813942
Yanai H, Adachi H, Hakoshima M, Katsuyama H. Postprandial Hyperlipidemia: Its Pathophysiology, Diagnosis, Atherogenesis, and Treatments. International Journal of Molecular Sciences. 2023; 24(18):13942. https://doi.org/10.3390/ijms241813942
Chicago/Turabian StyleYanai, Hidekatsu, Hiroki Adachi, Mariko Hakoshima, and Hisayuki Katsuyama. 2023. "Postprandial Hyperlipidemia: Its Pathophysiology, Diagnosis, Atherogenesis, and Treatments" International Journal of Molecular Sciences 24, no. 18: 13942. https://doi.org/10.3390/ijms241813942
APA StyleYanai, H., Adachi, H., Hakoshima, M., & Katsuyama, H. (2023). Postprandial Hyperlipidemia: Its Pathophysiology, Diagnosis, Atherogenesis, and Treatments. International Journal of Molecular Sciences, 24(18), 13942. https://doi.org/10.3390/ijms241813942