
Anti-HIV Potential of Beesioside I Derivatives as Maturation Inhibitors: Synthesis, 3D-QSAR, Molecular Docking and Molecular Dynamics Simulations
 , , and
, , and
Abstract
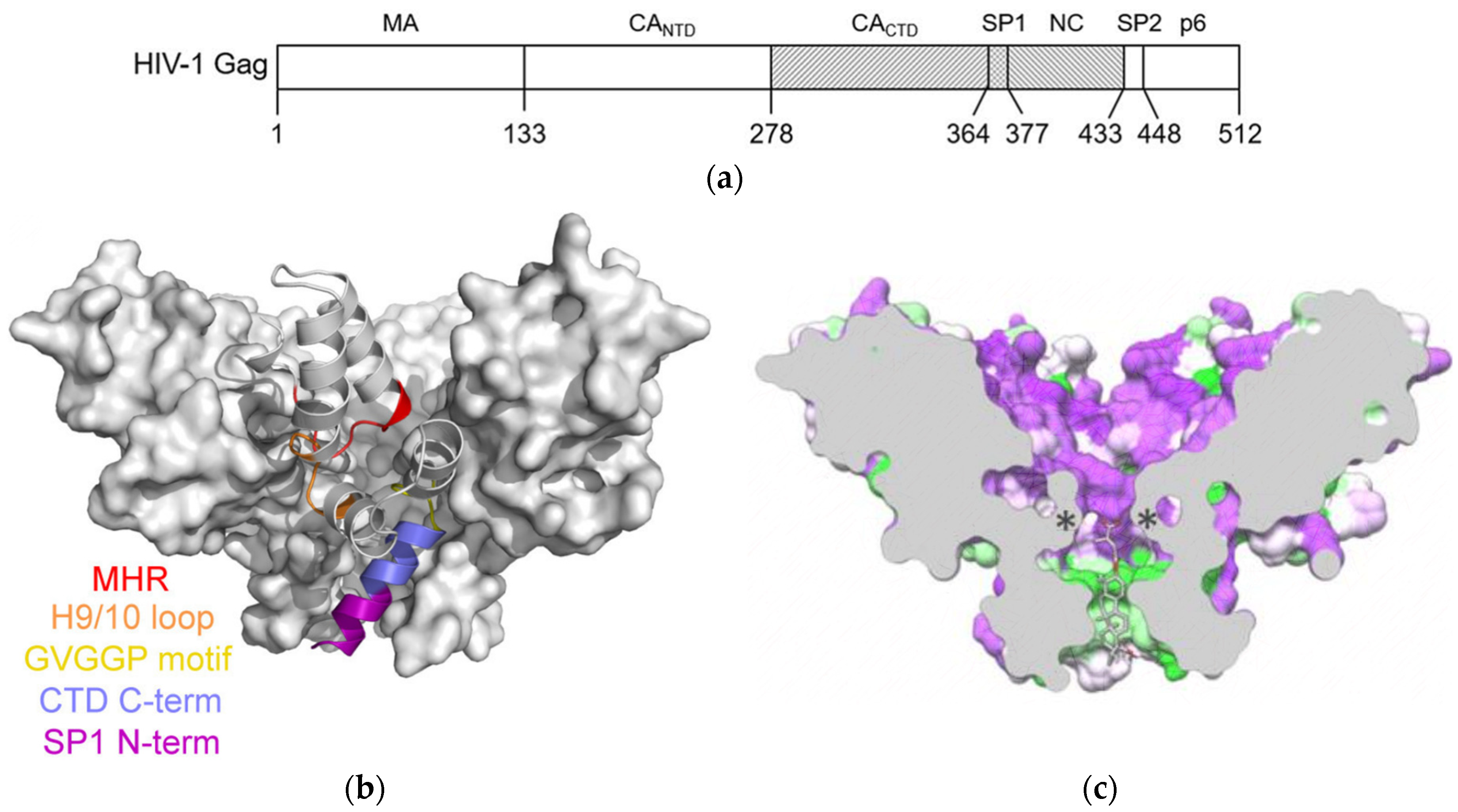
:1. Introduction
2. Results and Discussion
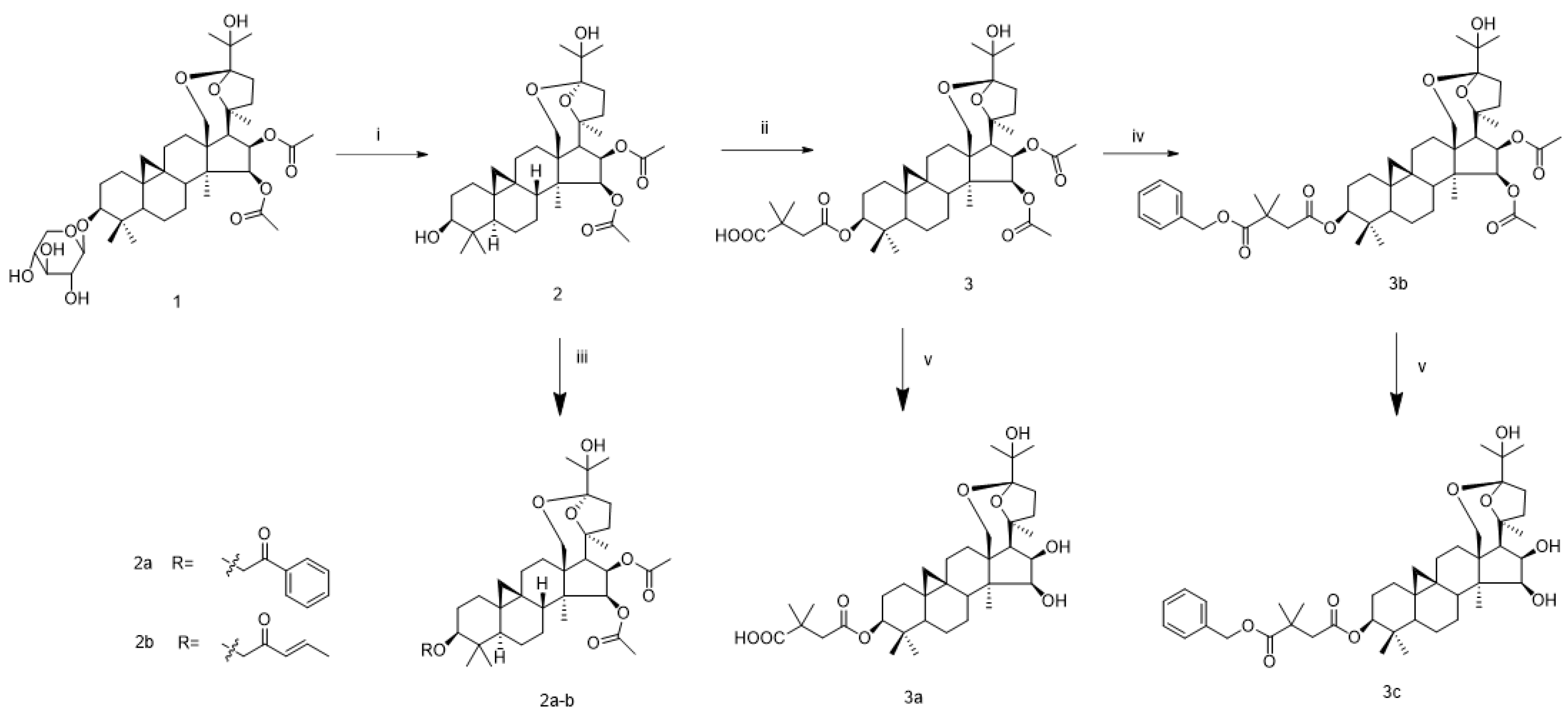
2.1. Chemistry
2.2. Evaluation of Anti-HIV Activity
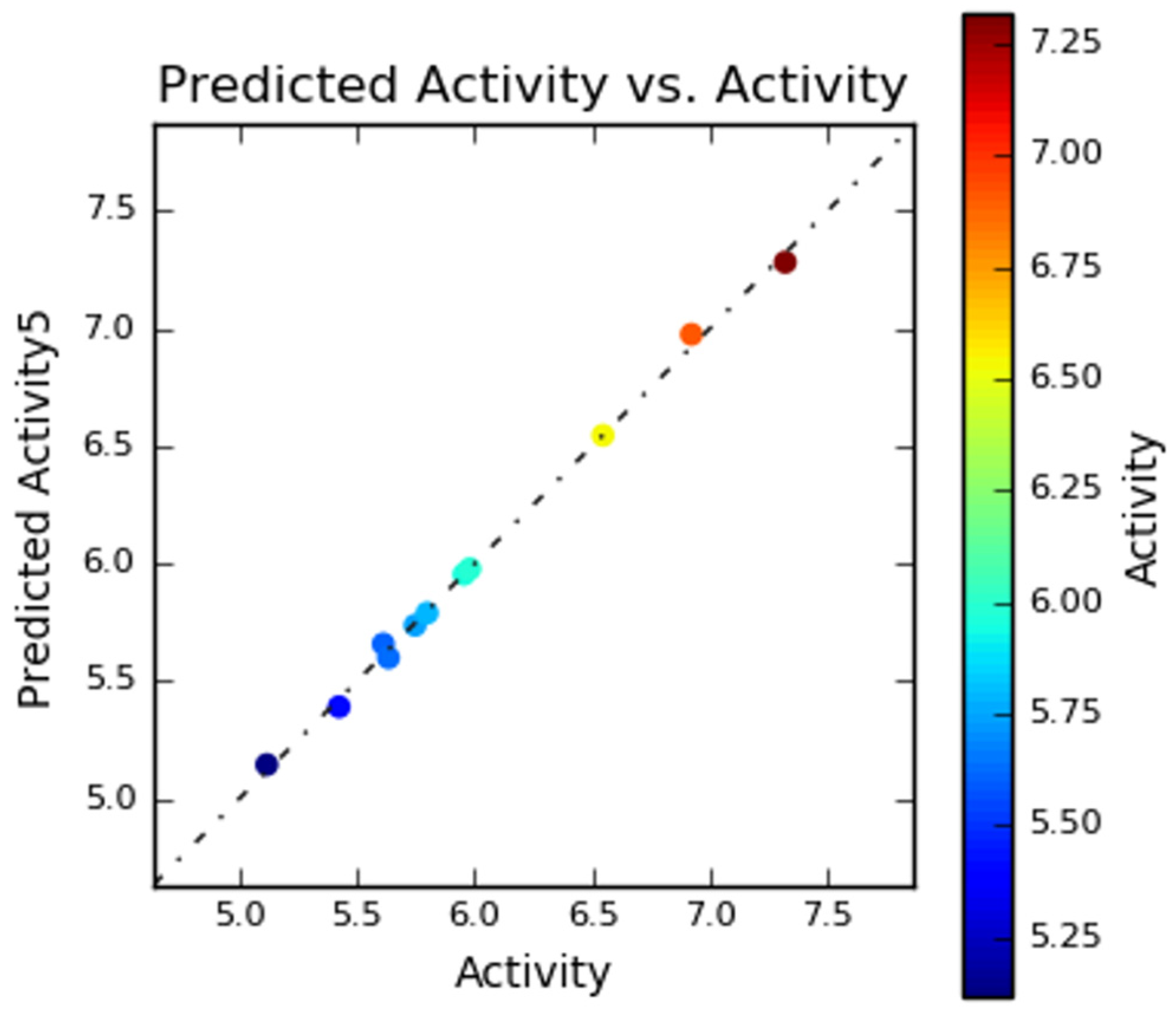
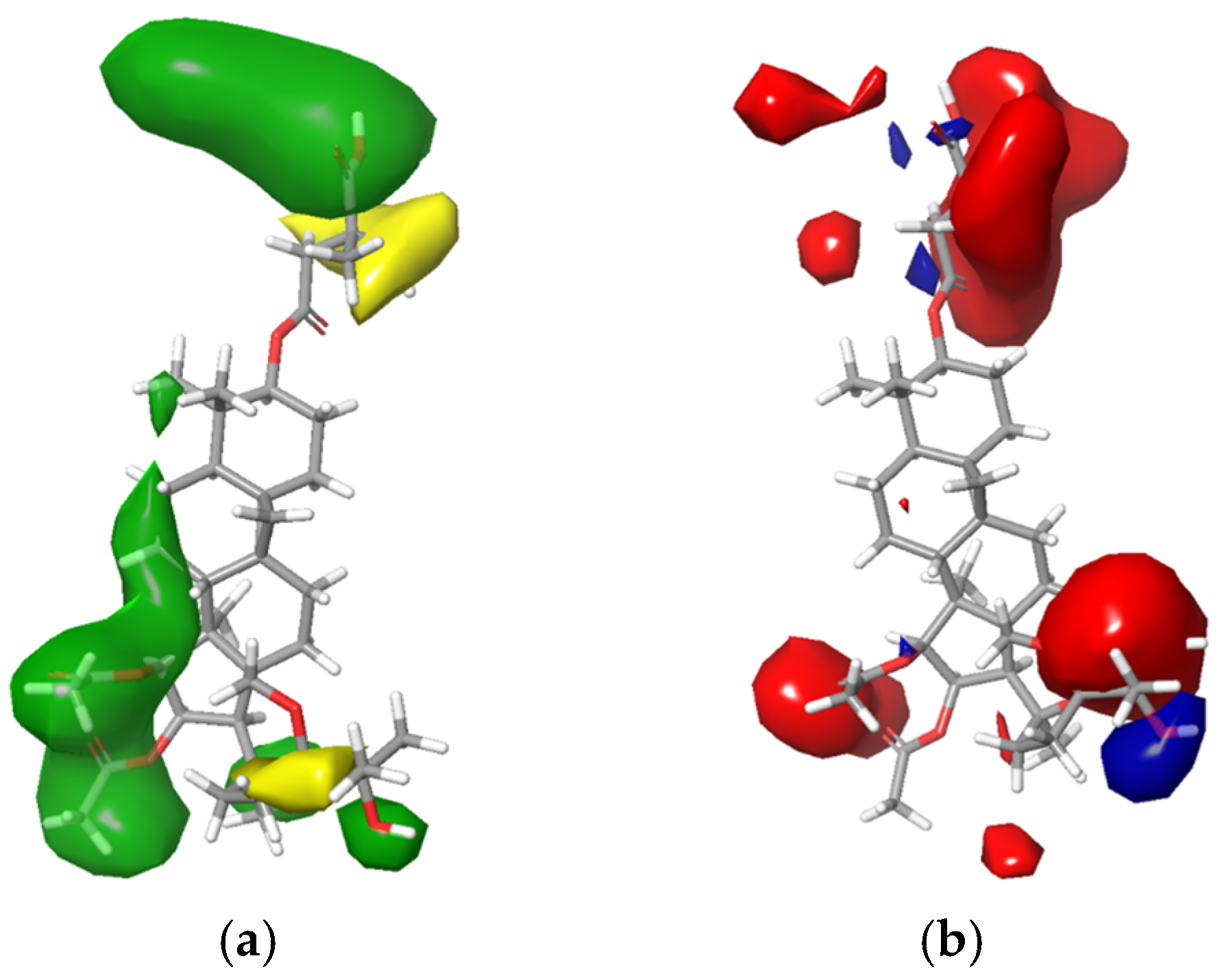
2.3. 3D-QSAR Study
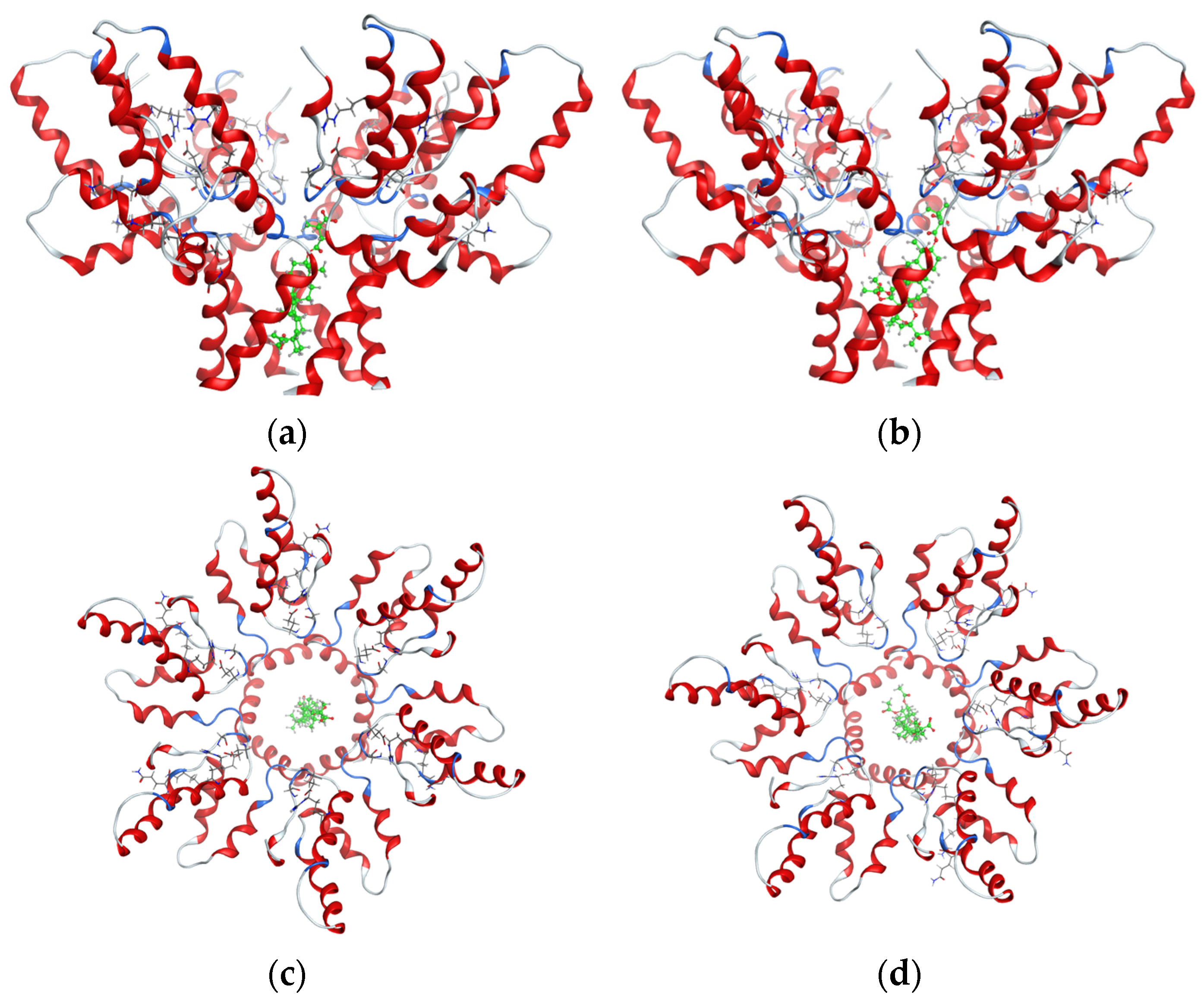
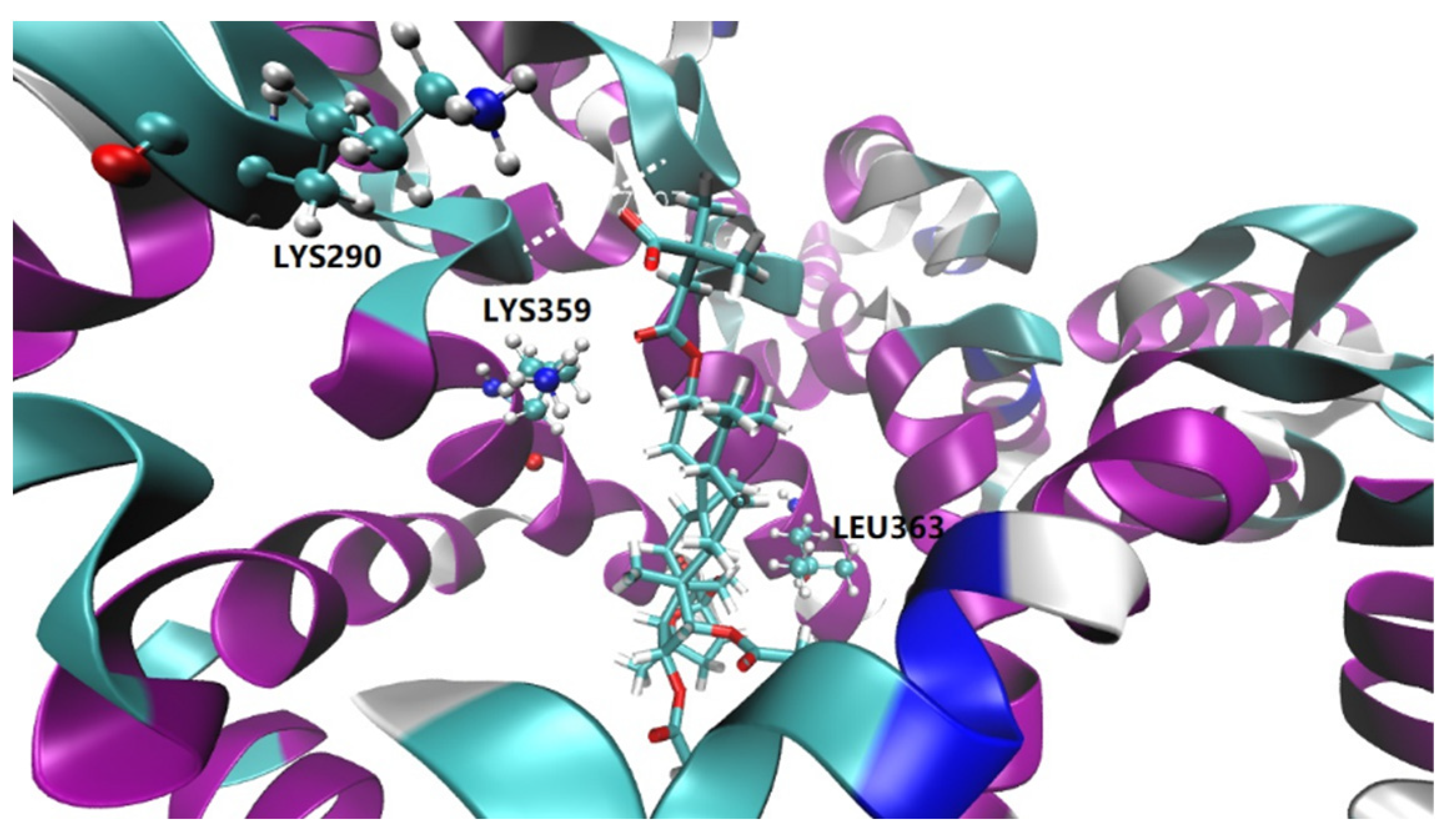
2.4. Molecular Docking Study
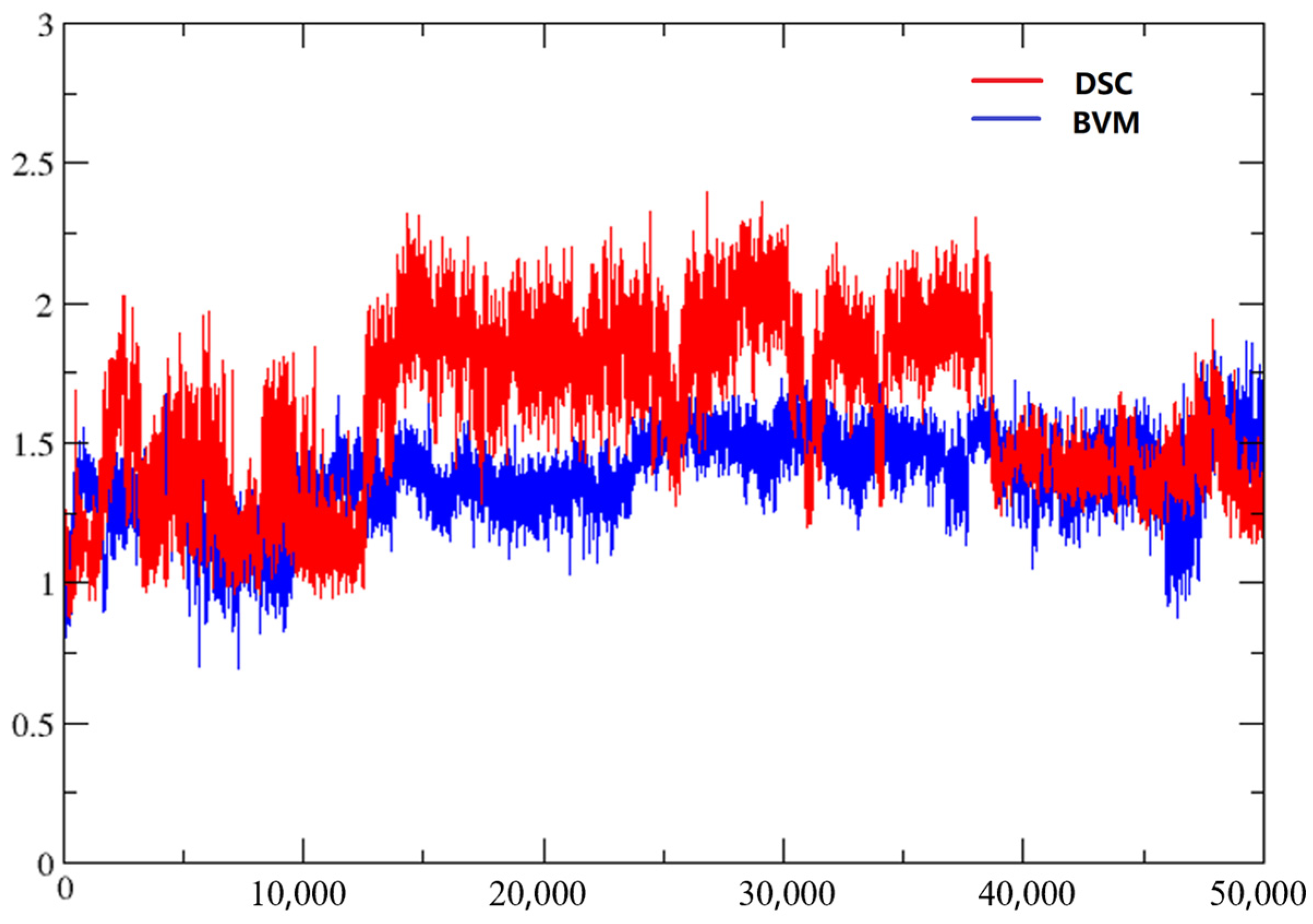
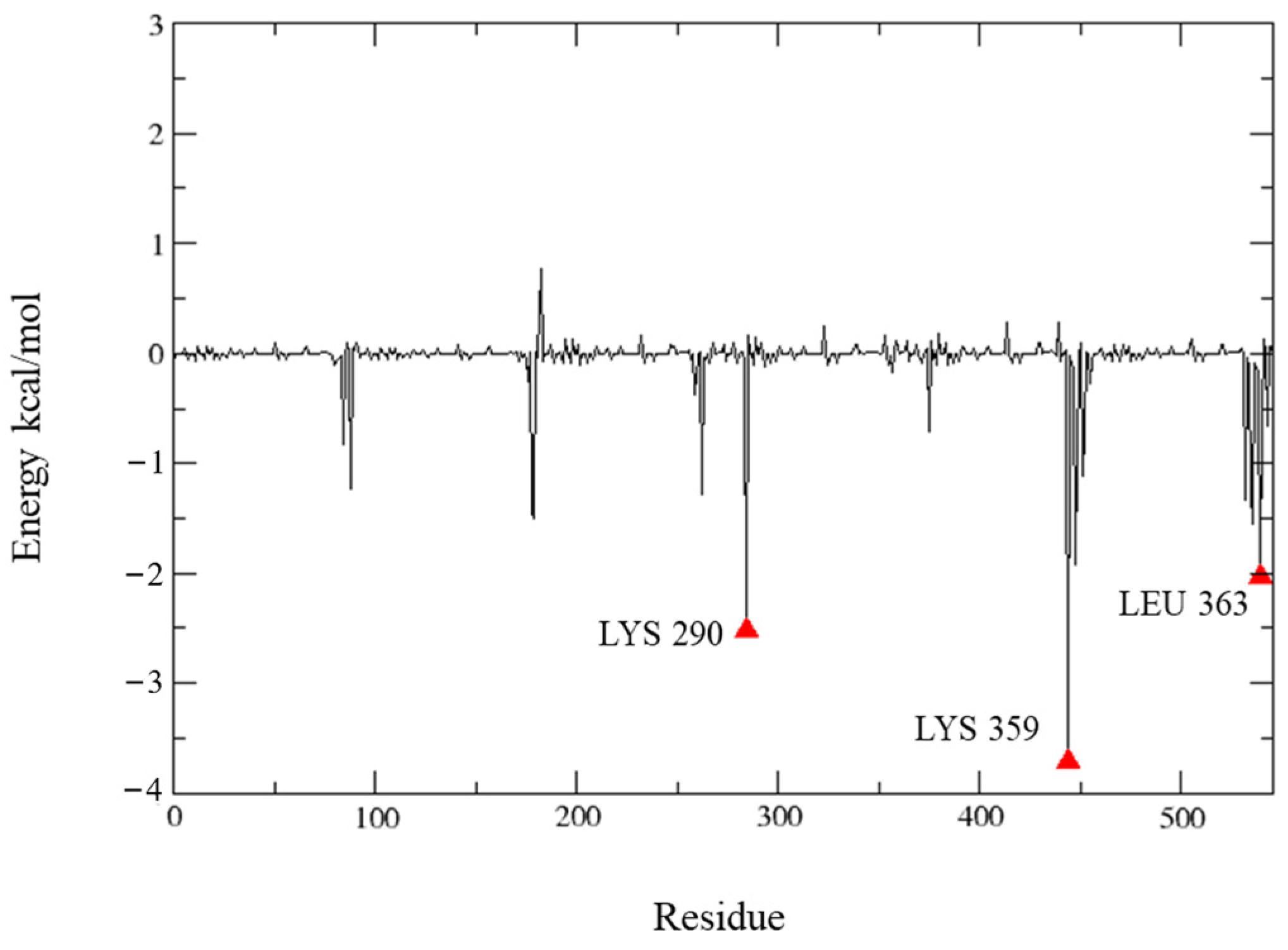
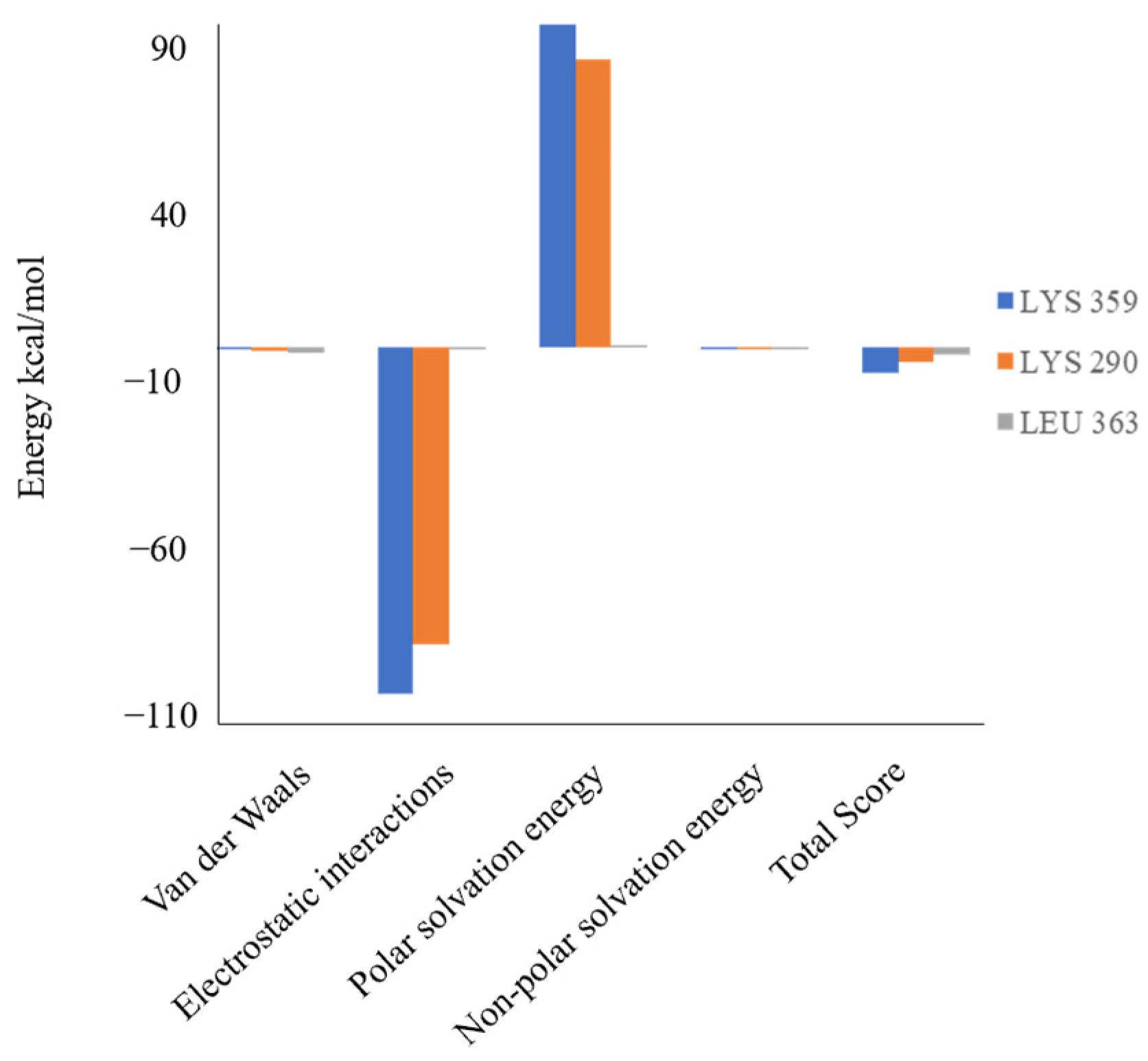
2.5. Molecular Dynamics
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure for Synthesis of Compounds 2a–b
3.2.1. (20S,24S)-15β,16β-Diacetoxy-18,24; 20,24-diepoxy-9,19-cyclolanostane-25-hydroxy-3I-yl benzoate (2a)
3.2.2. (20S,24S)-15β,16β-Diacetoxy-18,24; 20,24-diepoxy-9,19-cyclolanostane-25-hydroxy-3β-yl Crotonate (2b)
3.3. General Procedure for Synthesis of Compounds 3a–c
3.3.1. (20S,24S)-18,24;20,24-Diepoxy-9,19-cyclolanostane-3β,15β,16β,25-tetraol-3-O-3′,3′-dimethylsuccinate (3a)
3.3.2. (20S,24S)-15β,16β-Diacetoxy-18,24;20,24-diepoxy-9,19-cyclolanostane-3β,25-diol-3-O-4′-(benzyloxy) carbonyl-3′,3′-dimethylsuccinate (3b)
3.3.3. (20S,24S)-18,24; 20,24-Diepoxy-9,19-cyclolanostane-3β,15β,16β,25-tetraol-3-O-4′-(benzyloxy) carbonyl-3′,3′-dimethylsuccinate (3c)
3.4. Biological Experiments
3.4.1. Anti-HIV Assay
3.4.2. Cytotoxicity Assay
3.5. 3D-QSAR Study
3.6. Molecular Docking
3.7. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dang, Z.; Ho, P.; Zhu, L.; Qian, K.D.; Lee, K.H.; Huang, L.; Chen, C.H. New betulinic acid derivatives for bevirimat-resistant human immunodeficiency virus type-1. J. Med. Chem. 2013, 56, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, C.H.; Morris-Natschke, S.L.; Lee, K.H. Design, synthesis, and structure activity relationship analysis of new betulinic acid derivatives as potent HIV inhibitors. Eur. J. Med. Chem. 2021, 215, 113287. [Google Scholar] [CrossRef]
- Huang, Q.X.; Chen, H.F.; Luo, X.R.; Zhang, Y.X.; Yao, X.; Zheng, X. Structure and anti-HIV activity of betulinic acid analogues. Curr. Med. Sci. 2018, 38, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 maturation: Lessons learned from inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef]
- Popović-Djordjević, J.; Quispe, C.; Giordo, R.; Kostić, A.; Stanković, J.S.K.; Fokou, P.V.T.; Carbone, K.; Martorell, M.; Kumar, M.; Pintus, G.; et al. Natural products and synthetic analogues against HIV: A perspective to develop new potential anti-HIV drugs. Eur. J. Med. Chem. 2022, 233, 114217. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Waki, K.; Durell, S.R.; Soheilian, F.; Nagashima, K.; Butler, S.L.; Freed, E.O. Structural and functional insights into the HIV-1 maturation inhibitor binding pocket. PLoS Pathog. 2012, 8, e1002997. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.W.; Coric, P.; Bouaziz, S. 1H, 13C and 15N backbone resonance assignment of HIV-1 Gag (276–432) encompassing the C-terminal domain of the capsid protein, the spacer peptide 1 and the nucleocapsid protein. Biomol. NMR Assign. 2021, 15, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Dick, R.A.; Zadrozny, K.K.; Xu, C.Y.; Schur, F.K.M.; Lyddon, T.D.; Ricana, C.L.; Wagner, J.M.; Perilla, J.R.; Ganser-Pornillos, B.K.; Johnson, M.C.; et al. Inositol phosphates are assembly co-factors for HIV-1. Nature 2018, 560, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Schur, F.K.M.; Obr, M.; Hagen, W.J.H.; Wan, W.; Jakobi, A.J.; Kirkpatrick, J.M.; Sachse, C.; Kräeusslich, H.G.; Briggs, J.A.G. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science 2016, 353, 506–508. [Google Scholar] [CrossRef]
- Bayro, M.J.; Ganser-Pornillos, B.K.; Zadrozny, K.K.; Yeager, M.; Tycko, R. Helical conformation in the CA-SP1 junction of the immature HIV-1 lattice determined from solid-state NMR of virus-like particles. J. Am. Chem. Soc. 2016, 138, 12029–12032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, J.M.; Zadrozny, K.K.; Chrustowicz, J.; Purdy, M.D.; Yeager, M.; Ganser-Pornillos, B.K.; Pornillos, O. Crystal structure of an HIV assembly and maturation switch. Elife 2016, 5, e17063. [Google Scholar] [CrossRef] [PubMed]
- Pak, A.J.; Purdy, M.D.; Yeager, M.; Voth, G.A. Preservation of HIV-1 Gag helical bundle symmetry by bevirimat is central to maturation inhibition. J. Am. Chem. Soc. 2021, 143, 19137–19148. [Google Scholar] [CrossRef]
- Lingappa, J.R.; Reed, J.C.; Tanaka, M.; Chutiraka, K.; Robinson, B.A. How HIV-1 Gag assembles in cells: Putting together pieces of the puzzle. Virus Res. 2014, 193, 89–107. [Google Scholar] [CrossRef] [Green Version]
- Purdy, M.D.; Shi, D.; Chrustowicz, J.; Hattne, J.; Gonen, T.; Yeager, M. MicroED structures of HIV-1 Gag CTD-SP1 reveal binding interactions with the maturation inhibitor bevirimat. Proc. Natl. Acad. Sci. USA 2018, 115, 13258–13263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, B.; Guba, W.; Hert, J.; Banner, D.; Bissantz, C.; Ceccarelli, S.; Haap, W.; Koerner, M.; Kuglstatter, A.; Lerner, C.; et al. A real-world perspective on molecular design. J. Med. Chem. 2016, 59, 4087–4102. [Google Scholar] [CrossRef]
- Aminpour, M.; Montemagno, C.; Tuszynski, J.A. An overview of molecular modeling for drug discovery with specific illustrative examples of applications. Molecules 2019, 24, 1693. [Google Scholar] [CrossRef] [Green Version]
- Haghshenas, H.; Kaviani, B.; Firouzeh, M.; Tavakol, H. Developing a variation of 3D-QSAR/MD method in drug design. J. Comput. Chem. 2021, 42, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef] [PubMed]
- Filipe, H.A.L.; Loura, L.M.S. Molecular dynamics simulations: Advances and applications. Molecules 2022, 27, 2105. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Alanko, I.; Bhadane, R.; Bonvin, A.M.J.J.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S.; et al. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 2021, 9, 71. [Google Scholar] [CrossRef]
- Liu, X.W.; Shi, D.F.; Zhou, S.Y.; Liu, H.L.; Liu, H.X.; Yao, X.J. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Dis. 2018, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.; Allen, J.E.; Yang, Y.; Bennett, W.F.D.; Gokhale, M.; Moshiri, N.; Rosing, T.S. Accelerators for classical molecular dynamics simulations of biomolecules. J. Chem. Theory Comput. 2022, 18, 4047–4069. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.N.; Wang, Z.Z.; Sun, X.D.; Yang, J.; Ma, C.Y.; Li, W.; Liu, H.M. 3D-QSAR (CoMFA, CoMSIA), molecular docking and molecular dynamics simulations study of 6-aryl-5-cyano-pyrimidine derivatives to explore the structure requirements of LSD1 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3521–3528. [Google Scholar] [CrossRef]
- Panwar, U.; Singh, S.K. Atom-based 3D-QSAR, molecular docking, DFT, and simulation studies of acylhydrazone, hydrazine, and diazene derivatives as IN-LEDGF/p75 inhibitors. Struct. Chem. 2021, 32, 337–352. [Google Scholar] [CrossRef]
- Si, L.L.; Meng, K.; Tian, Z.; Sun, J.Q.; Li, H.Q.; Zhang, Z.W.; Soloveva, V.; Li, H.W.; Fu, G.; Xia, Q.; et al. Triterpenoids manipulate a broad range of virus-host fusion via wrapping the HR2 domain prevalent in viral envelopes. Sci Adv. 2018, 4, eaau8408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.W.; Sun, J.Q.; Xiao, S.L.; Zhang, L.H.; Zhou, D.M. Triterpenoid-mediated inhibition of virus-host interaction: Is now the time for discovering viral entry/release inhibitors from nature? J. Med. Chem. 2020, 63, 15371–15388. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.F.; Morris-Natschke, S.L.; Xu, X.D.; Yang, M.H.; Cheng, Y.Y.; Yu, S.S.; Lee, K.H. Recent advances in natural anti-HIV triterpenoids and analogs. Med. Res. Rev. 2020, 40, 2339–2385. [Google Scholar] [CrossRef]
- Wu, H.F.; Ma, G.X.; Yang, Q.W.; Zhu, Y.D.; Huang, L.; Tian, Y.; Yang, X.M.; Zhang, M.H.; Chen, C.H.; Morris-Natschke, S.L.; et al. Discovery and synthesis of novel beesioside I derivatives with potent anti-HIV activity. Eur. J. Med. Chem. 2019, 166, 159–166. [Google Scholar] [CrossRef]
- Neyret, A.; Gay, B.; Cransac, A.; Briant, L.; Coric, P.; Turcaud, S.; Laugaa, P.; Bouaziz, S.; Chazal, N. Insight into the mechanism of action of EP-39, a bevirimat derivative that inhibits HIV-1 maturation. Antivir. Res. 2019, 164, 162–175. [Google Scholar] [CrossRef]
- Dang, Z.; Zhu, L.; Lai, W.; Bogerd, H.; Lee, K.H.; Huang, L.; Chen, C.H. Aloperine and its derivatives as a new class of HIV-1 entry inhibitors. ACS Med. Chem. Lett. 2016, 7, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machireddy, B.; Kalra, G.; Jonnalagadda, S.; Ramanujachary, K.; Wu, C. Probing the binding pathway of BRACO19 to a parallel-stranded human telomeric G-quadruplex using molecular dynamics binding simulation with AMBER DNA OL15 and ligand GAFF2 force fields. J. Chem. Inf. Model. 2017, 57, 2846–2864. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham III, T.E. Parallelization of CPPTRAJ enables large scale analysis of molecular dynamics trajectory data. J. Comput. Chem. 2018, 39, 2110–2117. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Vora, J.; Patel, D.; Sinha, S.; Jha, P.C.; Shrivastava, N. Identification of natural inhibitors against prime targets of SARS-CoV-2 using molecular docking, molecular dynamics simulation and MM-PBSA approaches. J. Biomol. Struct. Dyn. 2022, 40, 3296–3311. [Google Scholar] [CrossRef]
- Han, D.; Su, M.; Tan, J.; Li, C.; Zhang, X.; Wang, C. Structure-activity relationship and binding mode studies for a series of diketo-acids as HIV integrase inhibitors by 3D-QSAR, molecular docking and molecular dynamics simulations. RSC Adv. 2016, 6, 27594–27606. [Google Scholar] [CrossRef]
- Fu, H.; Chen, H.; Blazhynska, M.; de Lacam, E.G.C.; Szczepaniak, F.; Pavlova, A.; Shao, X.; Gumbart, J.C.; Dehez, F.; Roux, B.; et al. Accurate determination of protein: Ligand standard binding free energies from molecular dynamics simulations. Nat. Protoc. 2022, 17, 1114–1141. [Google Scholar] [CrossRef]
- Liu, X.; Peng, L.; Zhou, Y.; Zhang, Y.; Zhang, J.Z.H. Computational alanine scanning with interaction entropy for protein-ligand binding free energies. J. Chem. Theory Comput. 2018, 14, 1772–1780. [Google Scholar] [CrossRef] [PubMed]
- Neto, R.A.M.; Santos, C.B.R.; Henriques, S.V.C.; Machado, L.O.; Cruz, J.N.; da Silva, C.H.T.P.; Federico, L.B.; de Oliveira, E.H.C.; de Souza, M.P.C.; da Silva, P.N.B.; et al. Novel chalcones derivatives with potential antineoplastic activity investigated by docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2022, 40, 2204–2216. [Google Scholar] [CrossRef]
- Ozdemir, E.S.; Gursoy, A.; Keskin, O. Analysis of single amino acid variations in singlet hot spots of protein-protein interfaces. Bioinformatics 2018, 34, i795–i801. [Google Scholar] [CrossRef]
- Kalescky, R.; Zhou, H.; Liu, J.; Tao, P. Rigid residue scan simulations systematically reveal residue entropic roles in protein allostery. PLoS Comput. Biol. 2016, 12, e1004893. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | EC50 (μM) NL4-3 b | CC50 (μM) MT4 c | TI e |
|---|---|---|---|
| 2 a | >30 | >30 | - |
| 2 b | *-d | *-d | - |
| 3 a | 1.79 ± 0.22 | >30 | >16.8 |
| 3 b | 0.28 ± 0.07 | 22.1 ± 2.7 | 78.9 |
| 3 c | 1.52 ± 0.14 | 13.2 ±1.2 | 8.7 |
| AZT | 0.024 ± 0.0045 | >0.3 | >12 |
 | ||||||
| NO. | R1 | R2 | R3 | EC50 (μM) | PEC50 | Predicte |
| 1 a,*,# | 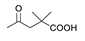 | Ac | Ac | 0.025 | 7.602 | 6.328 |
| 2 a |  | Ac | Ac | 0.048 | 7.319 | 7.280 |
| 3 a | 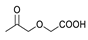 | Ac | Ac | 0.120 | 6.921 | 6.974 |
| 4 a | 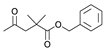 | Ac | Ac | 0.285 | 6.545 | 6.545 |
| 5 b | Xyl | H | OH | 1.047 | 5.98 | 5.978 |
| 6 a | 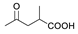 | Ac | Ac | 1.110 | 5.955 | 5.954 |
| 7 a,* | 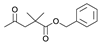 | H | H | 1.524 | 5.817 | 5.643 |
| 8 a | 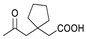 | Ac | Ac | 1.590 | 5.799 | 5.790 |
| 9 a |  | H | H | 1.788 | 5.748 | 5.738 |
| 10 a | Xyl | Ac | Ac | 2.320 | 5.635 | 5.600 |
| 11 a | Ac | Ac | Ac | 2.440 | 5.613 | 5.659 |
| 12 b | Xyl | Glc | H | 3.760 | 5.425 | 5.392 |
| 13 a,* | Xyl | H | Ac | 4.850 | 5.314 | 6.275 |
| 14 a | Xyl | H | H | 7.620 | 5.118 | 5.146 |
| VDW b | EE c | Polar d | Apolar e | Total f | ||
|---|---|---|---|---|---|---|
| MM/GBSA | BVM a | –48.63 | –221.12 | 235.86 | –4.24 | –38.13 |
| DSC | –55.52 | –245.89 | 258.52 | –5.37 | –48.27 | |
| MM/PBSA | BVM a | –47.37 | –235.42 | 246.55 | –6.54 | –42.79 |
| DSC | –56.29 | –246.11 | 262.24 | –8.01 | –48.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Ma, Y.; Li, X.; Morris-Natschke, S.L.; Sun, Z.; Sun, Z.; Ma, G.; Dong, Z.; Zhao, X.; Yang, M.; et al. Anti-HIV Potential of Beesioside I Derivatives as Maturation Inhibitors: Synthesis, 3D-QSAR, Molecular Docking and Molecular Dynamics Simulations. Int. J. Mol. Sci. 2023, 24, 1430. https://doi.org/10.3390/ijms24021430
Zhao Z, Ma Y, Li X, Morris-Natschke SL, Sun Z, Sun Z, Ma G, Dong Z, Zhao X, Yang M, et al. Anti-HIV Potential of Beesioside I Derivatives as Maturation Inhibitors: Synthesis, 3D-QSAR, Molecular Docking and Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2023; 24(2):1430. https://doi.org/10.3390/ijms24021430
Chicago/Turabian StyleZhao, Zixuan, Yinghong Ma, Xiangyuan Li, Susan L. Morris-Natschke, Zhaocui Sun, Zhonghao Sun, Guoxu Ma, Zhengqi Dong, Xiaohong Zhao, Meihua Yang, and et al. 2023. "Anti-HIV Potential of Beesioside I Derivatives as Maturation Inhibitors: Synthesis, 3D-QSAR, Molecular Docking and Molecular Dynamics Simulations" International Journal of Molecular Sciences 24, no. 2: 1430. https://doi.org/10.3390/ijms24021430