
Investigating the Molecular Mechanisms Underlying Early Response to Inflammation and Helicobacter pylori Infection in Human Gastric Epithelial Cells
 , ,
, ,  , , , ,
, , , ,  and
and
Abstract
:1. Introduction
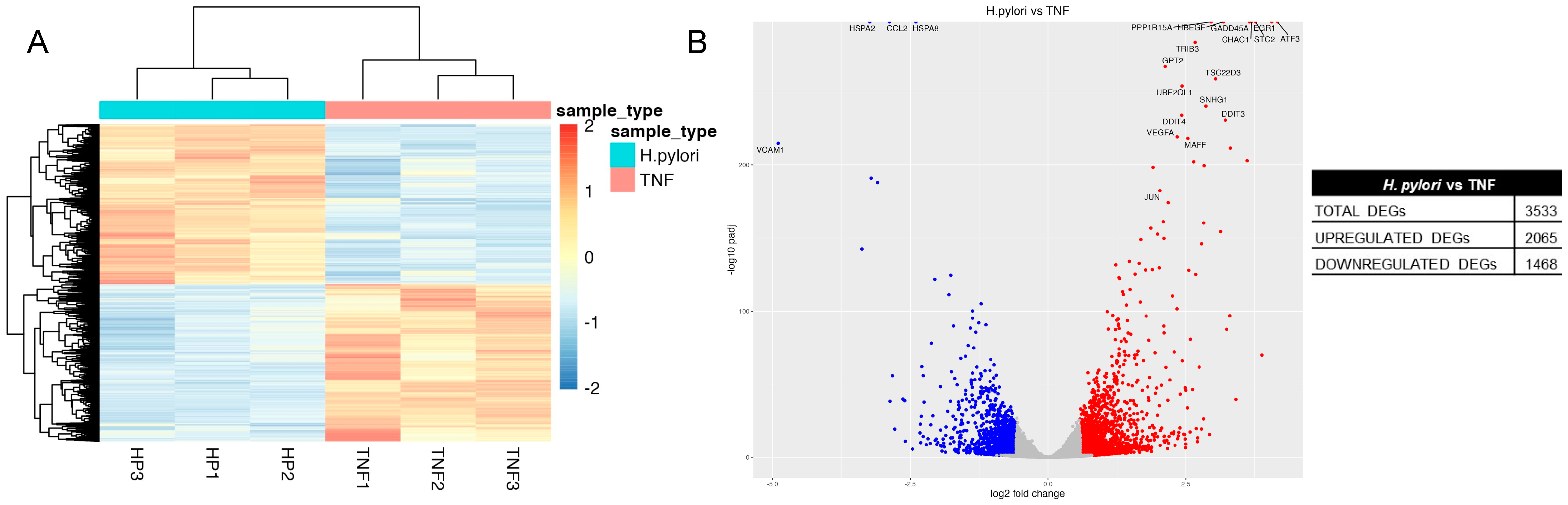
2. Results
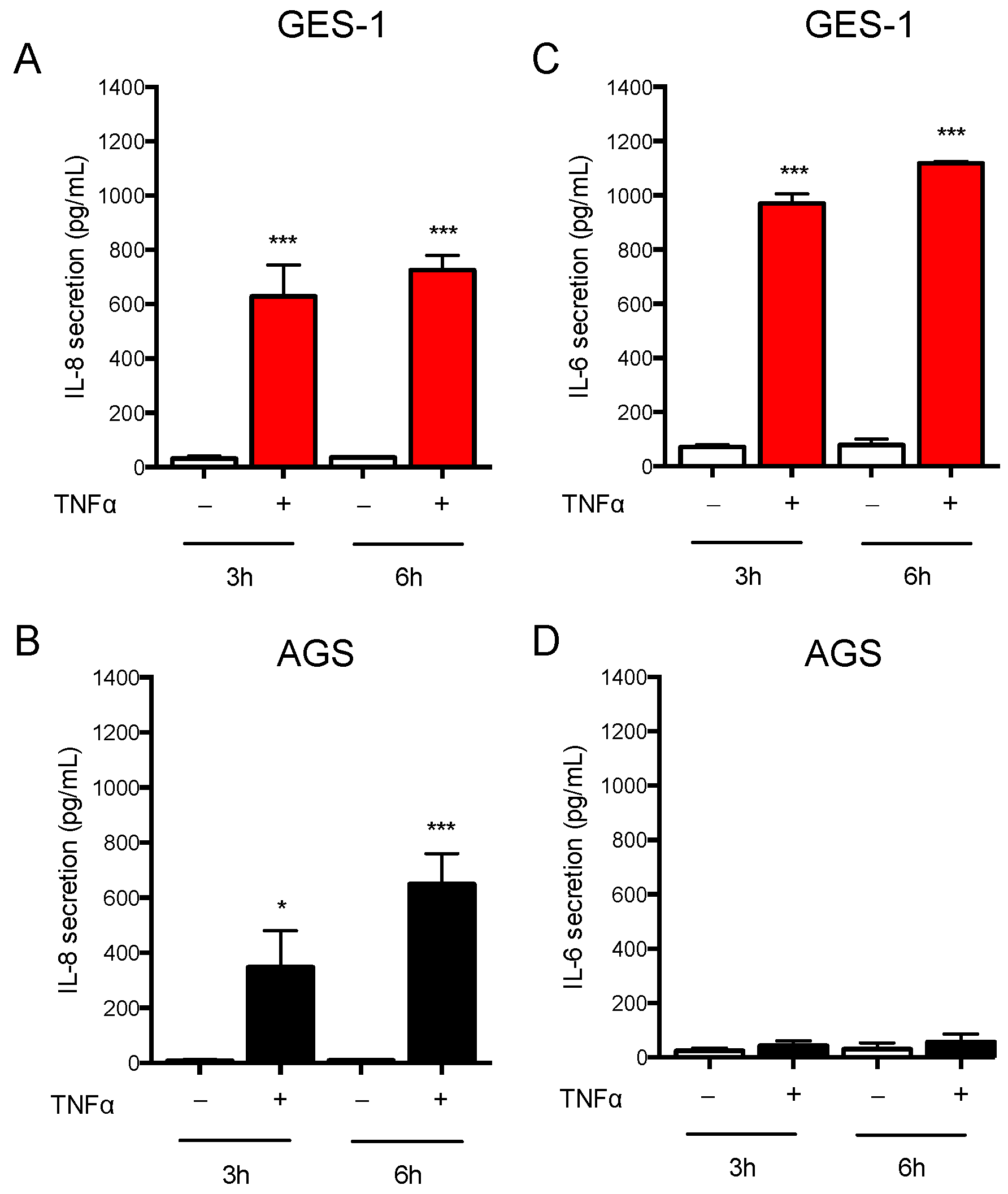
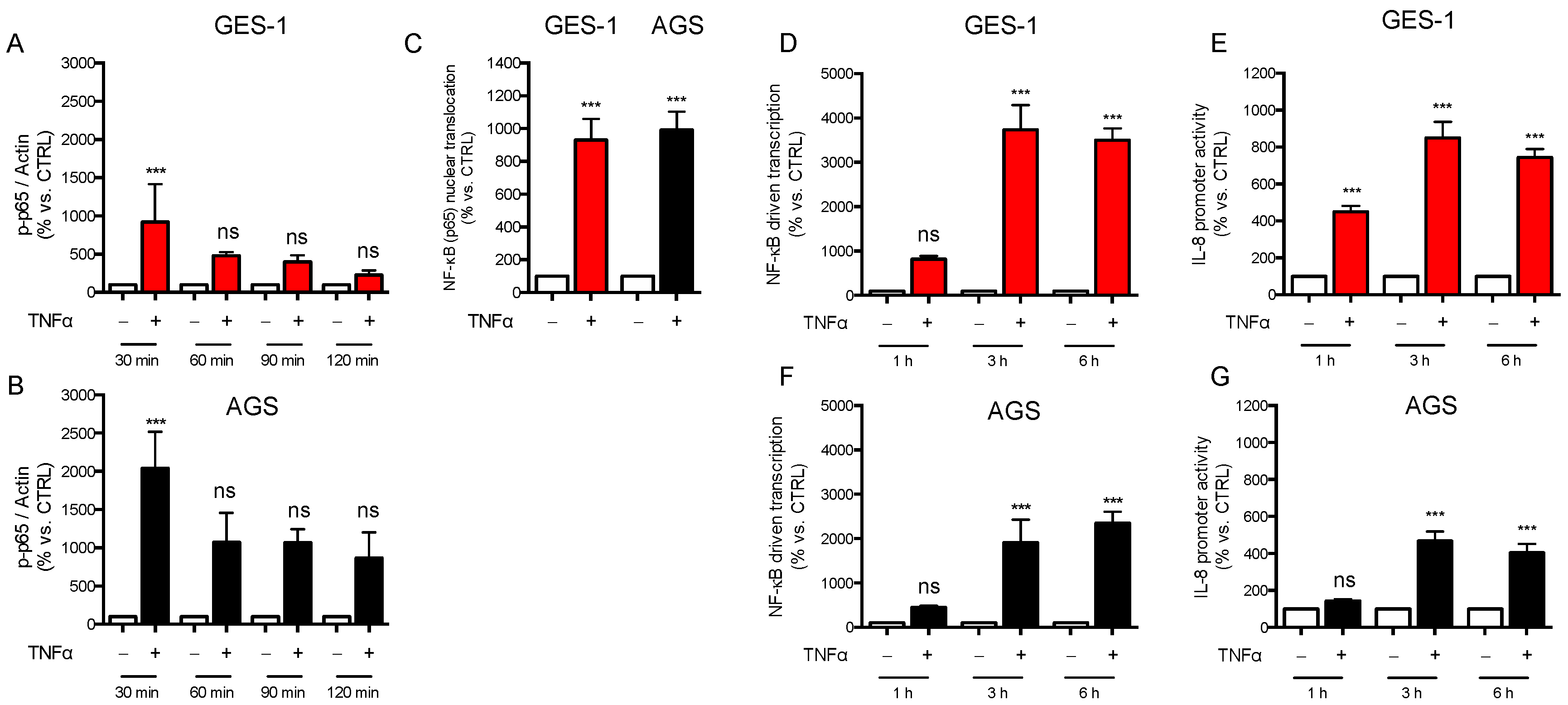
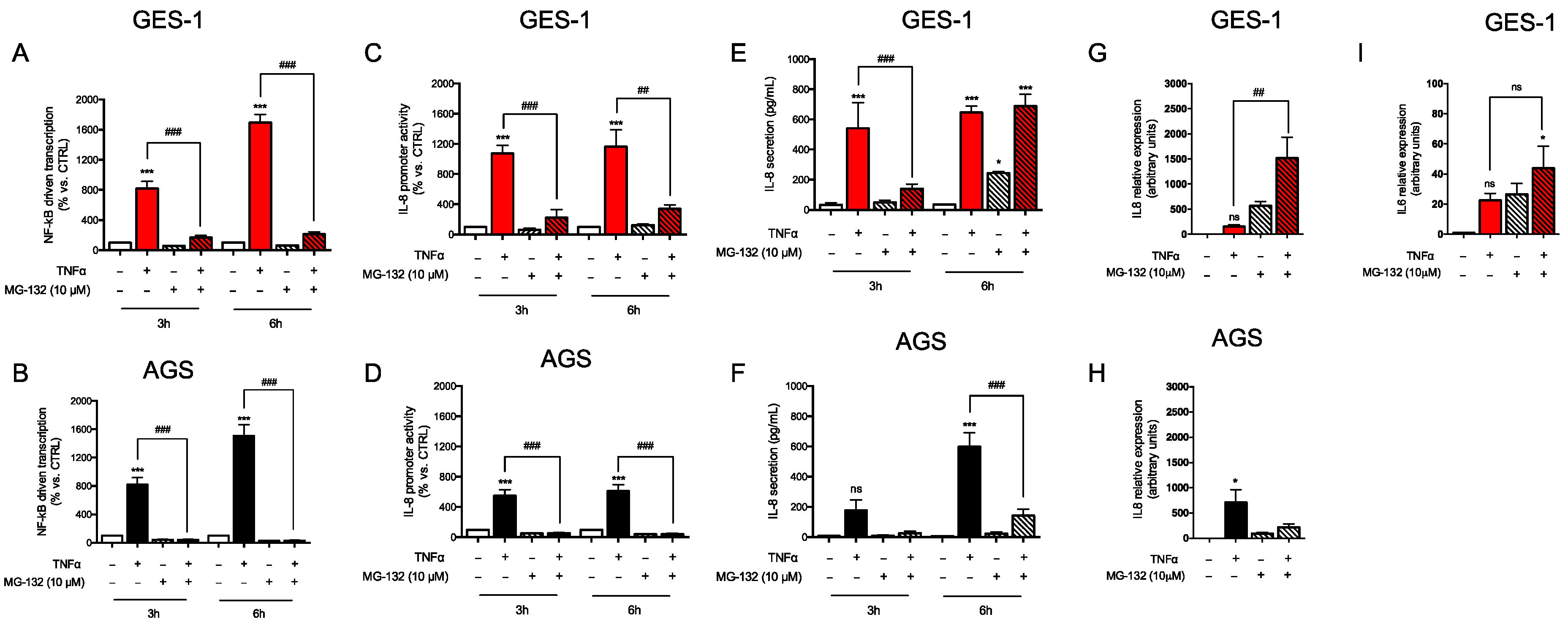
2.1. Response of AGS and GES-1 Cells to Inflammatory Mediators Linked to Gastric Diseases
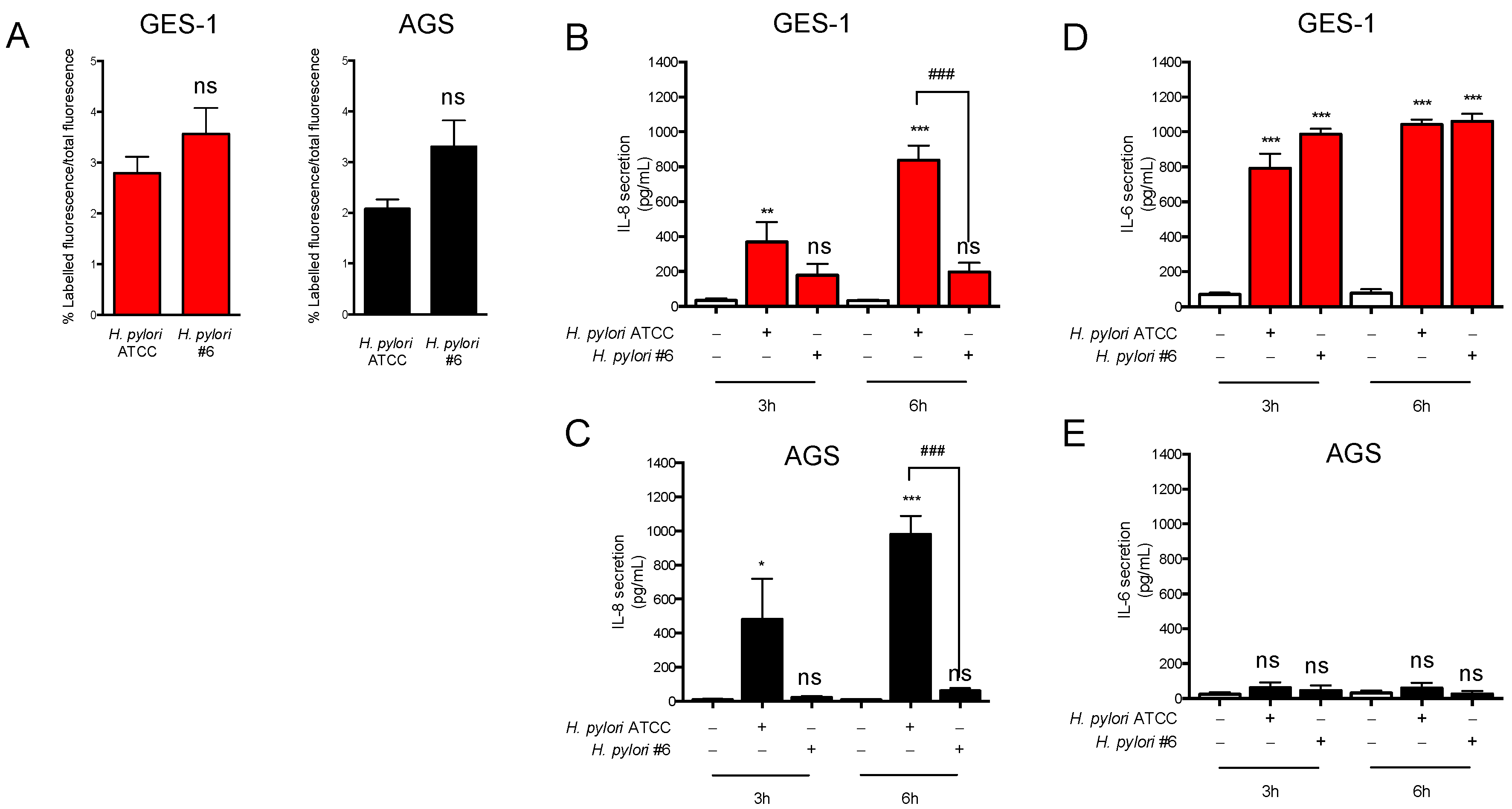
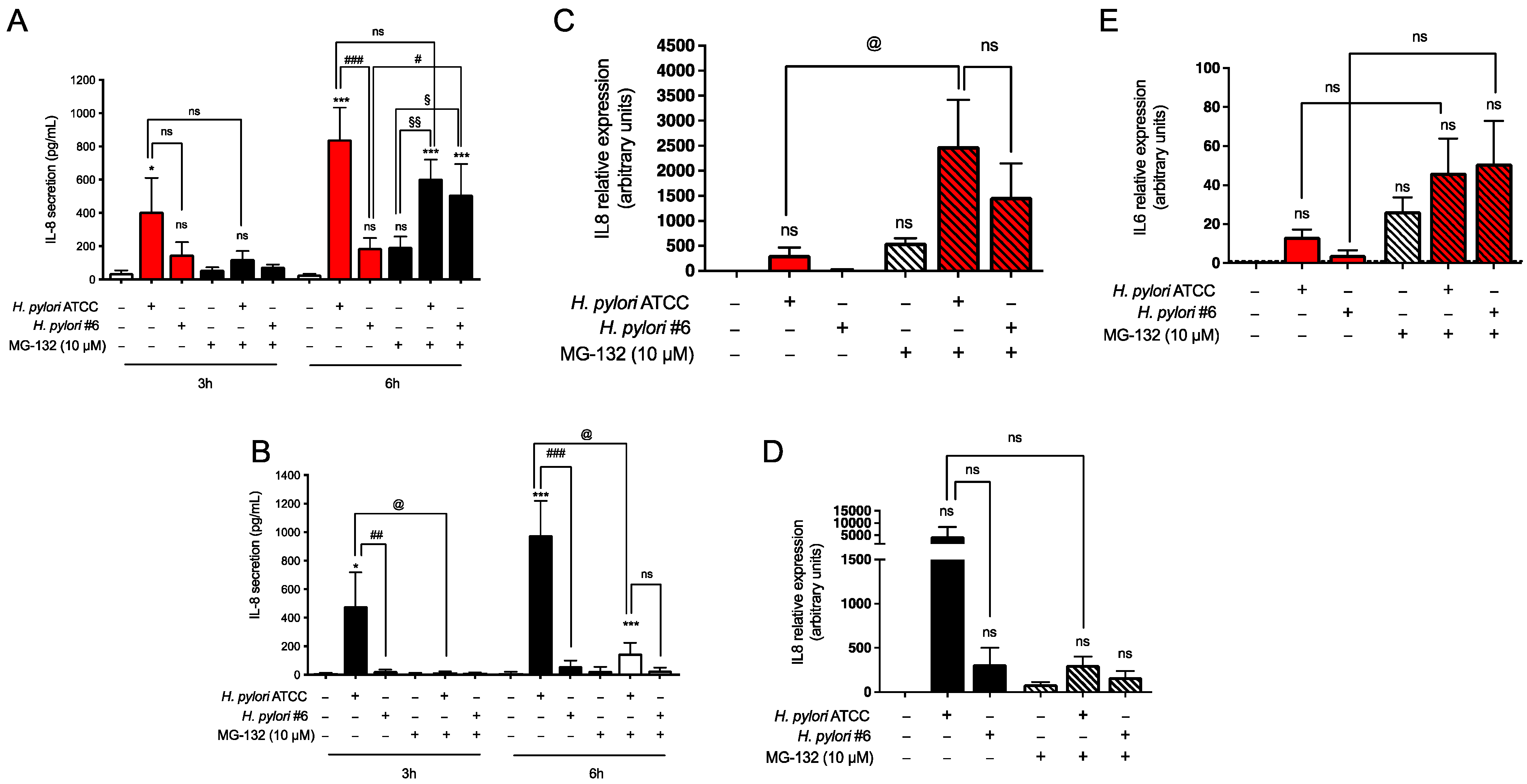
2.2. H. pylori-Induced Inflammatory Response in GES-1 and AGS Cells
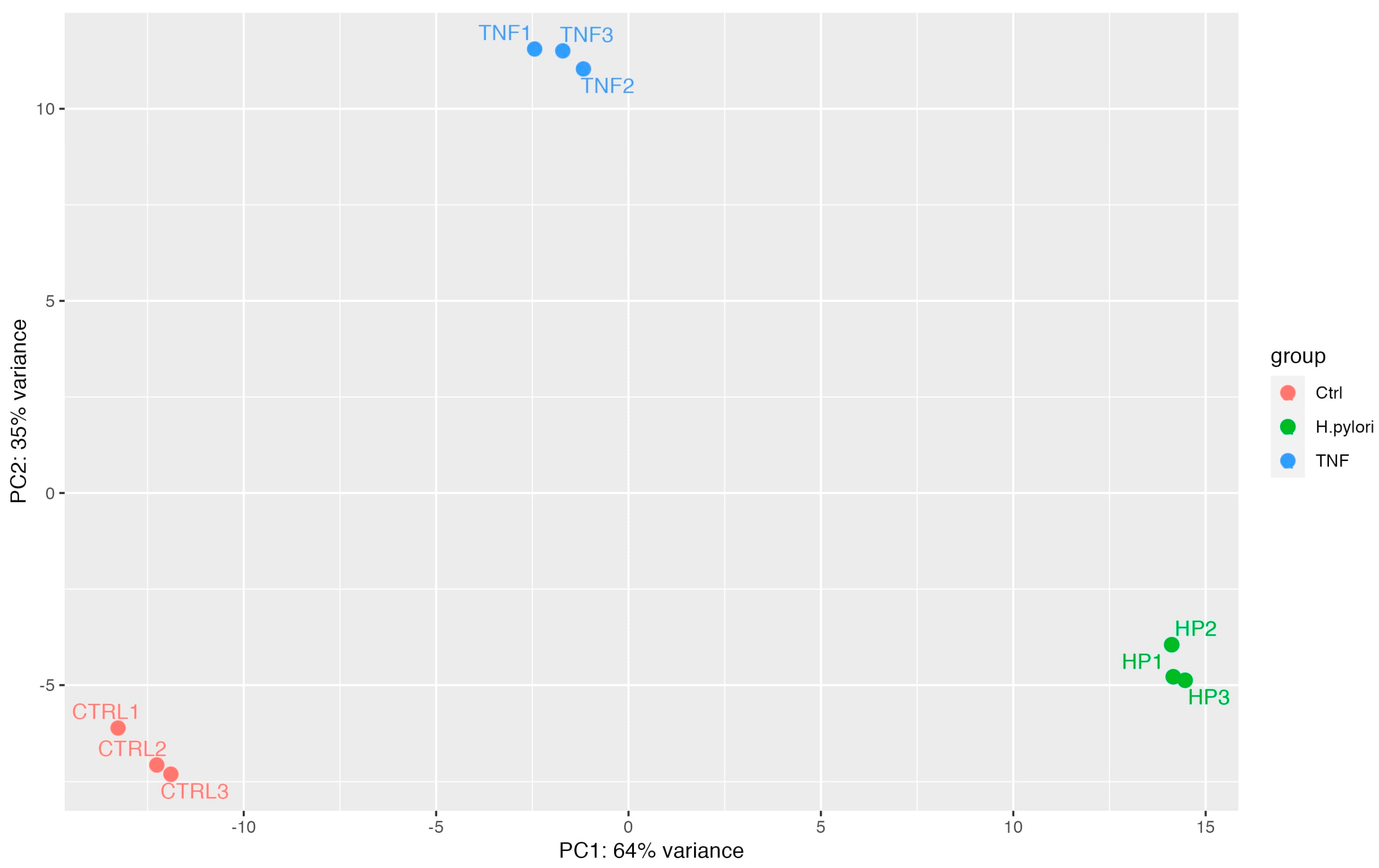
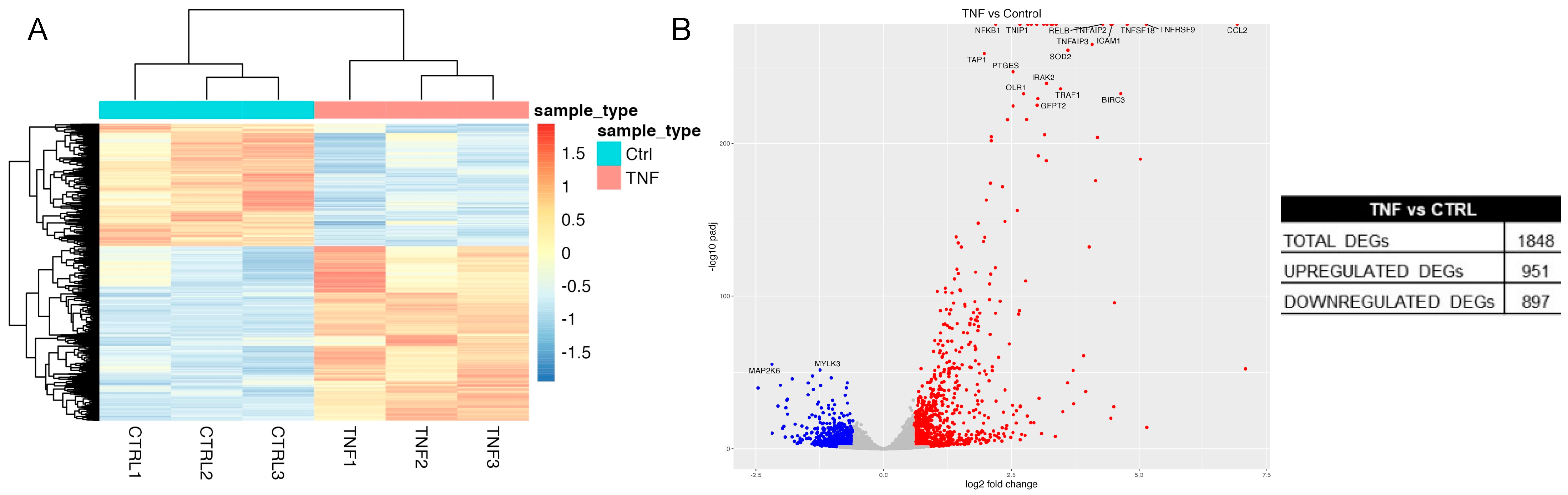
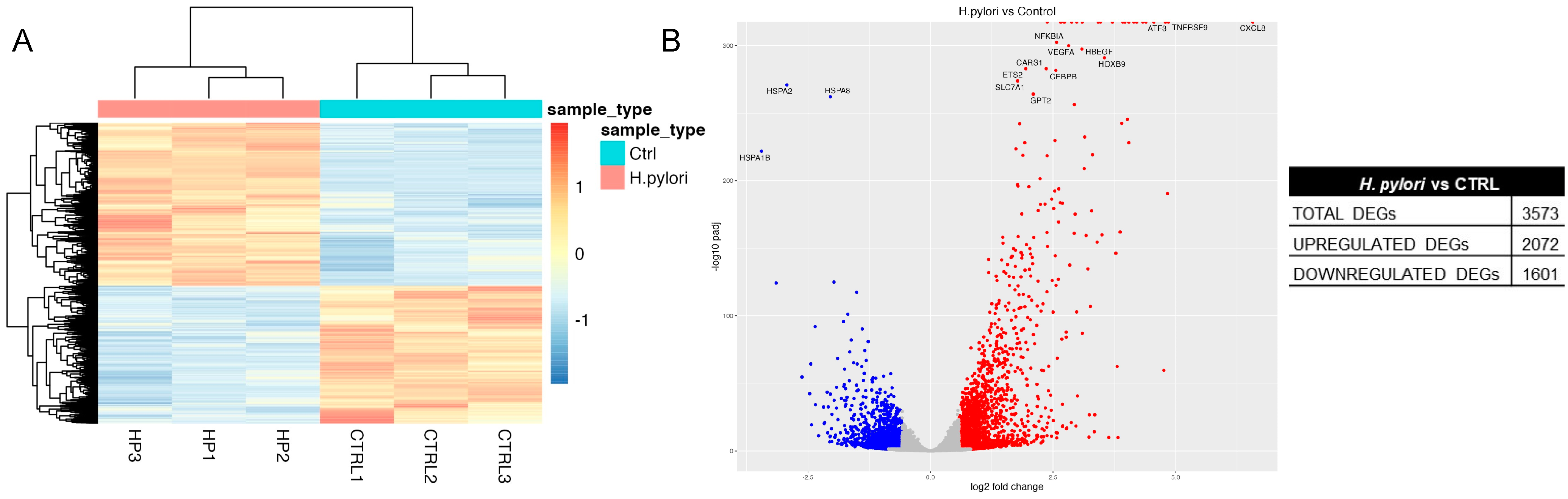
2.3. Transcriptome Analysis of H. pylori Infection in GES-1 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatments
4.3. Cytokine Assays
4.4. H. pylori Strains Infection and Gastric Cell Infection
4.5. Bacterial Adhesion to Gastric Cells
4.6. Gene Expression
4.7. Promoter Activity Assays
4.8. NF-κB Pathway
4.8.1. Western Blot Analysis
4.8.2. Immunofluorescence Analysis
4.9. RNA-Sequencing
4.10. RNA-Sequencing Data Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malfertheiner, P.; Camargo, M.C.; El-Omar, E.; Liou, J.M.; Peek, R.; Schulz, C.; Smith, S.I.; Suerbaum, S. Helicobacter pylori infection. Nat. Rev. Dis. Primers 2023, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program—15th Report on Carcinogens. Available online: https://ntp.niehs.nih.gov/whatwestudy/assessments/cancer/roc (accessed on 1 August 2023).
- Keates, S.; Hitti, Y.S.; Upton, M.; Kelly, C.P. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology 1997, 113, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Isomoto, H.; Mizuta, Y.; Miyazaki, M.; Takeshima, F.; Omagari, K.; Murase, K.; Nishiyama, T.; Inoue, K.; Murata, I.; Kohno, S. Implication of NF-kappaB in Helicobacter pylori-associated gastritis. Am. J. Gastroenterol. 2000, 95, 2768–2776. [Google Scholar] [CrossRef]
- Zimmermann, S.; Pfannkuch, L.; Al-Zeer, M.A.; Bartfeld, S.; Koch, M.; Liu, J.; Rechner, C.; Soerensen, M.; Sokolova, O.; Zamyatina, A.; et al. ALPK1- and TIFA-Dependent Innate Immune Response Triggered by the Helicobacter pylori Type IV Secretion System. Cell Rep. 2017, 20, 2384–2395. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Tegtmeyer, N.; Fischer, W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 2015, 10, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L. Tracking bacterial effector protein delivery into host cells. Mol. Microbiol. 2021, 116, 724–728. [Google Scholar] [CrossRef]
- Backert, S.; Haas, R.; Gerhard, M.; Naumann, M. The Helicobacter pylori Type IV Secretion System Encoded by the cag Pathogenicity Island: Architecture, Function, and Signaling. Curr. Top. Microbiol. Immunol. 2017, 413, 187–220. [Google Scholar] [CrossRef]
- Palrasu, M.; Zaika, E.; El-Rifai, W.; Que, J.; Zaika, A.I. Role of Bacterial and Viral Pathogens in Gastric Carcinogenesis. Cancers 2021, 13, 1878. [Google Scholar] [CrossRef]
- Seidlitz, T.; Koo, B.K.; Stange, D.E. Gastric organoids-an in vitro model system for the study of gastric development and road to personalized medicine. Cell Death Differ. 2021, 28, 68–83. [Google Scholar] [CrossRef]
- Piazza, S.; Martinelli, G.; Fumagalli, M.; Pozzoli, C.; Maranta, N.; Giavarini, F.; Colombo, L.; Nicotra, G.; Vicentini, S.F.; Genova, F.; et al. Ellagitannins from Castanea sativa Mill. Leaf Extracts Impair H. pylori Viability and Infection-Induced Inflammation in Human Gastric Epithelial Cells. Nutrients 2023, 15, 1504. [Google Scholar] [CrossRef]
- Yu, Q.; Shi, H.; Ding, Z.; Wang, Z.; Yao, H.; Lin, R. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation in Helicobacter pylori-associated gastritis by regulating ROS and autophagy. Cell Commun. Signal 2023, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; He, T.; Liu, J.; Jia, S.; Li, B.; Xu, W.; Liao, M.; Guo, L. Pharmacological and molecular analysis of the effects of Huangqi Jianzhong decoction on proliferation and apoptosis in GES-1 cells infected with H. pylori. Front. Pharmacol. 2022, 13, 1009705. [Google Scholar] [CrossRef] [PubMed]
- Nwakiban, A.P.A.; Fumagalli, M.; Piazza, S.; Magnavacca, A.; Martinelli, G.; Beretta, G.; Magni, P.; Tchamgoue, A.D.; Agbor, G.A.; Kuiate, J.R.; et al. Dietary Cameroonian Plants Exhibit Anti-Inflammatory Activity in Human Gastric Epithelial Cells. Nutrients 2020, 12, 3787. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Thakur, M.; Klattenhoff, A.W.; Kidane, D. Aberrant DNA Polymerase Beta Enhances H. pylori Infection Induced Genomic Instability and Gastric Carcinogenesis in Mice. Cancers 2019, 11, 843. [Google Scholar] [CrossRef]
- Wang, J.; Yao, Y.; Zhang, Q.; Li, S.; Tang, L. Inflammatory responses induced by Helicobacter pylori on the carcinogenesis of gastric epithelial GES-1 cells. Int. J. Oncol. 2019, 54, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Milo, R. The distribution of cellular turnover in the human body. Nat. Med. 2021, 27, 45–48. [Google Scholar] [CrossRef]
- Bertaux-Skeirik, N.; Feng, R.; Schumacher, M.A.; Li, J.; Mahe, M.M.; Engevik, A.C.; Javier, J.E.; Peek, R.M., Jr.; Ottemann, K.; Orian-Rousseau, V.; et al. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog. 2015, 11, e1004663. [Google Scholar] [CrossRef]
- Bartfeld, S.; Bayram, T.; van de Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, R.; Peters, P.J.; Clevers, H. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 2015, 148, 126–136.e6. [Google Scholar] [CrossRef]
- Ke, Y.; Ning, T.; Wang, B. Establishment and characterization of a SV40 transformed human fetal gastric epithelial cell line-GES-1. Zhonghua Zhong Liu Za Zhi 1994, 16, 7–10. [Google Scholar]
- Barranco, S.C.; Townsend, C.M., Jr.; Casartelli, C.; Macik, B.G.; Burger, N.L.; Boerwinkle, W.R.; Gourley, W.K. Establishment and characterization of an in vitro model system for human adenocarcinoma of the stomach. Cancer Res. 1983, 43, 1703–1709. [Google Scholar]
- Lamb, A.; Yang, X.D.; Tsang, Y.H.; Li, J.D.; Higashi, H.; Hatakeyama, M.; Peek, R.M.; Blanke, S.R.; Chen, L.F. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009, 10, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- NF-kB Target Genes. Available online: http://www.bu.edu/nf-kb/gene-resources/target-genes/ (accessed on 1 August 2023).
- The NFkB target Gene Sets—Broad Institute. Available online: https://www.yumpu.com/en/document/view/8327926/the-nfkb-target-gene-sets-are-listed-below-broadinstitute (accessed on 1 August 2023).
- Gonzalez-Santamarta, M.; Bouvier, C.; Rodriguez, M.S.; Xolalpa, W. Ubiquitin-chains dynamics and its role regulating crucial cellular processes. Semin. Cell Dev. Biol. 2022, 132, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Barve, S.; Barve, S.S.; Butt, W.; Klein, J.; McClain, C.J. Inhibition of proteasome function leads to NF-kappaB-independent IL-8 expression in human hepatocytes. Hepatology 2003, 38, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Chmiela, M.; Kupcinskas, J. Review: Pathogenesis of Helicobacter pylori infection. Helicobacter 2019, 24 (Suppl. S1), e12638. [Google Scholar] [CrossRef]
- Shimoyama, T.; Crabtree, J.E. Mucosal chemokines in Helicobacter pylori infection. J. Physiol. Pharmacol. 1997, 48, 315–323. [Google Scholar] [PubMed]
- Maeda, S.; Yoshida, H.; Ogura, K.; Mitsuno, Y.; Hirata, Y.; Yamaji, Y.; Akanuma, M.; Shiratori, Y.; Omata, M. H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology 2000, 119, 97–108. [Google Scholar] [CrossRef]
- Feige, M.H.; Vieth, M.; Sokolova, O.; Tager, C.; Naumann, M. Helicobacter pylori induces direct activation of the lymphotoxin beta receptor and non-canonical nuclear factor-kappa B signaling. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 545–550. [Google Scholar] [CrossRef]
- Peek, R.M., Jr.; Fiske, C.; Wilson, K.T. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol. Rev. 2010, 90, 831–858. [Google Scholar] [CrossRef]
- Wedel, A.; Ziegler-Heitbrock, H.W. The C/EBP family of transcription factors. Immunobiology 1995, 193, 171–185. [Google Scholar] [CrossRef]
- Sterken, B.A.; Ackermann, T.; Muller, C.; Zuidhof, H.R.; Kortman, G.; Hernandez-Segura, A.; Broekhuis, M.; Spierings, D.; Guryev, V.; Calkhoven, C.F. C/EBPbeta isoform-specific regulation of migration and invasion in triple-negative breast cancer cells. NPJ Breast Cancer 2022, 8, 11. [Google Scholar] [CrossRef]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschella, G. Zinc-finger proteins in health and disease. Cell Death Discov. 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, X.; Lin, X.; Chen, H. Zinc Finger Proteins in the War on Gastric Cancer: Molecular Mechanism and Clinical Potential. Cells 2023, 12, 1314. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.G.; King, J.C. The Molecular Basis for Zinc Bioavailability. Int. J. Mol. Sci. 2023, 24, 6561. [Google Scholar] [CrossRef]
- Allison, C.C.; Kufer, T.A.; Kremmer, E.; Kaparakis, M.; Ferrero, R.L. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J. Immunol. 2009, 183, 8099–8109. [Google Scholar] [CrossRef]
- Luo, M.; Chen, Y.J.; Xie, Y.; Wang, Q.R.; Xiang, Y.N.; Long, N.Y.; Yang, W.X.; Zhao, Y.; Zhou, J.J. Dickkopf-related protein 1/cytoskeleton-associated protein 4 signaling activation by Helicobacter pylori-induced activator protein-1 promotes gastric tumorigenesis via the PI3K/AKT/mTOR pathway. World J. Gastroenterol. 2022, 28, 6769–6787. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.J.; Gorrell, R.J.; Tafreshi, M.; Hatakeyama, M.; Kwok, T.; Price, J.T. The Helicobacter pylori cytotoxin CagA is essential for suppressing host heat shock protein expression. Cell Stress. Chaperones 2016, 21, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Baird, M.; Woon Ang, P.; Clark, I.; Bishop, D.; Oshima, M.; Cook, M.C.; Hemmings, C.; Takeishi, S.; Worthley, D.; Boussioutas, A.; et al. The unfolded protein response is activated in Helicobacter-induced gastric carcinogenesis in a non-cell autonomous manner. Lab. Investig. 2013, 93, 112–122. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, K.; Jin, Y.; Sheng, X. Endoplasmic reticulum proteostasis control and gastric cancer. Cancer Lett. 2019, 449, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, J.; Yin, X.Y.; Hou, J.X.; Chen, J.; Xie, B.; Chen, G.; Wang, Q.F.; Wang, L.N.; Wang, X.Y.; et al. Vacuolating Cytotoxin A Triggers Mitophagy in Helicobacter pylori-Infected Human Gastric Epithelium Cells. Front. Oncol. 2022, 12, 881829. [Google Scholar] [CrossRef]
- Messing, J.; Thole, C.; Niehues, M.; Shevtsova, A.; Glocker, E.; Boren, T.; Hensel, A. Antiadhesive properties of Abelmoschus esculentus (Okra) immature fruit extract against Helicobacter pylori adhesion. PLoS ONE 2014, 9, e84836. [Google Scholar] [CrossRef]
- Piazza, S.; Pacchetti, B.; Fumagalli, M.; Bonacina, F.; Dell’Agli, M.; Sangiovanni, E. Comparison of Two Ginkgo biloba L. Extracts on Oxidative Stress and Inflammation Markers in Human Endothelial Cells. Mediat. Inflamm. 2019, 2019, 6173893. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC. A Quality Control Tool for High throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 November 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GES-1 | AGS | ||||
|---|---|---|---|---|---|
| Gene Symbol | Nominal p Value | Fold Change | Nominal p Value | Fold Change | NF-κB Target * |
| C5 | 6.69 × 10−3 | 0.525 | >0.05 | NA | No |
| CCL11 | 6.88 × 10−3 | 14.655 | >0.05 | NA | No |
| CCL2 | 9.52 × 10−5 | 189.581 | >0.05 | NA | Yes |
| CCL20 | 2.95 × 10−3 | 785.064 | 4.78 × 10−3 | 294.747 | Yes |
| CCL7 | 3.87 × 10−4 | 21.407 | >0.05 | NA | Yes |
| CSF1 | 1.05 × 10−3 | 10.339 | 3.20 × 10−3 | 3.045 | No |
| CSF2 | 1.90 × 10−2 | 22.523 | >0.05 | NA | No |
| CX3CL1 | 1.20 × 10−2 | 10.079 | >0.05 | NA | No |
| CXCL1 | 4.50 × 10−3 | 61.963 | >0.05 | NA | Yes |
| CXCL10 | 8.55 × 10−3 | 12.729 | >0.05 | NA | Yes |
| CXCL2 (GRO gamma) | 8.22 × 10−4 | 15.207 | 9.61 × 10−5 | 10.778 | Yes |
| CXCL3 | 6.23 × 10−4 | 4.834 | >0.05 | NA | No |
| CXCL5 | 3.70 × 10−2 | 12.098 | 3.43 × 10−3 | 55.715 | Yes |
| CXCL6 | 2.28 × 10−3 | 16.951 | >0.05 | NA | No |
| CXCL8 | 1.01 × 10−3 | 53.569 | 8.12 × 10−4 | 77.708 | Yes |
| IL15 | 3.40 × 10−3 | 2.451 | >0.05 | NA | No |
| IL1B | 2.99 × 10−2 | 8.938 | >0.05 | NA | Yes |
| IL6 | 1.30 × 10−3 | 28.624 | >0.05 | NA | Yes |
| IL7 | 4.56 × 10−2 | 40.039 | >0.05 | NA | No |
| LTB | 4.79 × 10−4 | 581.378 | 2.56 × 10−5 | 3026.288 | Yes |
| MMP9 | 1.47 × 10−3 | 45.932 | >0.05 | NA | Yes |
| NAMPT | 2.52 × 10−3 | 4.103 | >0.05 | NA | No |
| TNF | 7.11 × 10−4 | 69.712 | 4.56 × 10−3 | 63.705 | Yes |
| TNFRSF11B | 3.08 × 10−2 | 1.853 | >0.05 | NA | No |
| VEGFA | 6.56 × 10−3 | 2.676 | >0.05 | NA | No |
| Term | Overlap | p Value | Adjusted p Value |
|---|---|---|---|
| Enriched pathways with upregulated genes | |||
| cytokine-mediated signaling pathway (GO:0019221) | 113 | 3.27 × 10−38 | 1.21 × 10−34 |
| cellular response to cytokine stimulus (GO:0071345) | 82 | 9.47 × 10−26 | 1.75 × 10−22 |
| regulation of inflammatory response (GO:0050727) | 40 | 3.91 × 10−15 | 4.82 x10−12 |
| inflammatory response (GO:0006954) | 42 | 7.62 × 10−15 | 6.94 × 10−12 |
| cellular response to lipopolysaccharide (GO:0071222) | 30 | 9.50 × 10−15 | 6.94 × 10−12 |
| regulation of cell population proliferation (GO:0042127) | 85 | 1.13 × 10−14 | 6.94 × 10−12 |
| cellular response to tumor necrosis factor (GO:0071356) | 38 | 1.43 × 10−14 | 7.53 × 10−12 |
| positive regulation of cytokine production (GO:0001819) | 49 | 3.33 × 10−13 | 1.42 × 10−10 |
| regulation of I-kappaB kinase/NF-kappaB signaling (GO:0043122) | 39 | 3.46 × 10−13 | 1.42 × 10−10 |
| cellular response to interferon-gamma (GO:0071346) | 28 | 5.90 × 10−13 | 2.18 × 10−10 |
| Enriched pathways with downregulated genes | |||
| anterior/posterior pattern specification (GO:0009952) | 12 | 6.22 × 10−6 | 1.68 × 10−2 |
| generation of neurons (GO:0048699) | 22 | 2.05 × 10−5 | 1.92 × 10−2 |
| neuron differentiation (GO:0030182 | 20 | 2.20 × 10−5 | 1.92 × 10−2 |
| ameboidal-type cell migration (GO:0001667) | 10 | 3.37 × 10−5 | 1.92 × 10−2 |
| neural crest cell migration (GO:0001755) | 8 | 8.63 × 10−5 | 3.69 × 10−2 |
| negative regulation of axon extension involved in axon guidance (GO:0048843) | 6 | 9.56 × 10−5 | 3.69 × 10−2 |
| endocardial cushion development (GO:0003197) | 6 | 1.29 × 10−4 | 4.37 × 10−2 |
| regulation of branching involved in ureteric bud morphogenesis (GO:0090189) | 5 | 2.65 × 10−4 | 6.31 × 10−2 |
| axonogenesis (GO:0007409) | 22 | 2.89 × 10−4 | 6.31 × 10−2 |
| regulation of axon extension involved in axon guidance (GO:0048841) | 6 | 2.89 × 10−4 | 6.31 × 10−2 |
| Term | Overlap | p Value | Adjusted p Value |
|---|---|---|---|
| Enriched pathways with upregulated genes | |||
| regulation of transcription by RNA polymerase II (GO:0006357) | 337 | 1.35 × 10−20 | 6.17 × 10−17 |
| regulation of transcription, DNA-templated (GO:0006355) | 327 | 8.36 × 10−17 | 1.91 × 10−13 |
| positive regulation of transcription by RNA polymerase II (GO:0045944) | 162 | 7.86 × 10−16 | 1.20 × 10−12 |
| positive regulation of transcription, DNA-templated (GO:0045893) | 195 | 3.28 × 10−15 | 3.75 × 10−12 |
| negative regulation of transcription by RNA polymerase II (GO:0000122) | 127 | 6.45 × 10−14 | 5.89 × 10−11 |
| negative regulation of transcription, DNA-templated (GO:0045892) | 151 | 7.70 × 10−11 | 5.87 × 10−8 |
| cytokine-mediated signaling pathway (GO:0019221) | 109 | 1.41 × 10−10 | 9.19 × 10−8 |
| cellular response to cytokine stimulus (GO:0071345) | 89 | 5.32 × 10−10 | 3.04 × 10−7 |
| regulation of gene expression (GO:0010468) | 39 | 1.41 × 10−9 | 7.19 × 10−7 |
| positive regulation of pri-miRNA transcription by RNA polymerase II (GO:1902895) | 28 | 1.78 × 10−9 | 8.13 × 10−7 |
| Enriched pathways with downregulated genes | |||
| chaperone cofactor-dependent protein refolding (GO:0051085) | 11 | 3.82 × 10−7 | 1.37 × 10−3 |
| ‘de novo’ posttranslational protein folding (GO:0051084) | 11 | 3.06 × 10−6 | 5.48 × 10−3 |
| response to unfolded protein (GO:0006986) | 13 | 1.51 × 10−5 | 1.81 × 10−2 |
| cellular response to unfolded protein (GO:0034620) | 8 | 2.94 × 10−4 | 1.36 × 10−1 |
| cellular response to steroid hormone stimulus (GO:0071383) | 7 | 3.15 × 10−4 | 1.36 × 10−1 |
| aminoglycan biosynthetic process (GO:0006023) | 11 | 3.42 × 10−4 | 1.36 × 10−1 |
| embryonic organ morphogenesis (GO:0048562) | 10 | 5.68 × 10−4 | 2.00 × 10−1 |
| negative regulation of viral process (GO:0048525) | 13 | 7.28 × 10−4 | 2.18 × 10−1 |
| regulation of cyclin-dependent protein kinase activity (GO:1904029) | 11 | 8.23 × 10−4 | 2.19 × 10−1 |
| odontogenesis (GO:0042476) | 9 | 9.46 × 10−4 | 2.19 × 10−1 |
| Term | Overlap | p Value | Adjusted p Value |
|---|---|---|---|
| Enriched pathways with upregulated genes | |||
| regulation of transcription, DNA-templated (GO:0006355) | 316 | 6.04 × 10−16 | 2.55 × 10−12 |
| regulation of transcription by RNA polymerase II (GO:0006357) | 307 | 8.66 × 10−15 | 1.82 × 10−11 |
| negative regulation of transcription by RNA polymerase II (GO:0000122) | 118 | 1.13 × 10−11 | 1.58 × 10−8 |
| positive regulation of transcription, DNA-templated (GO:0045893) | 169 | 2.87 × 10−9 | 3.02 × 10−6 |
| negative regulation of transcription, DNA-templated (GO:0045892) | 141 | 4.93 × 10−9 | 4.16 × 10−6 |
| positive regulation of transcription by RNA polymerase II (GO:0045944) | 135 | 1.09 × 10−8 | 7.65 × 10−6 |
| regulation of gene expression (GO:0010468) | 152 | 4.75 × 10−8 | 2.86 × 10−5 |
| intrinsic apoptotic signaling pathway in response to endoplasmic reticulum stress (GO:0070059) | 12 | 4.10 × 10−6 | 2.16 × 10−3 |
| intrinsic apoptotic signaling pathway (GO:0097193) | 23 | 4.12 × 10−5 | 1.93 × 10−2 |
| regulation of cellular macromolecule biosynthetic process (GO:2000112) | 68 | 1.01 × 10−4 | 3.65 × 10−2 |
| Enriched pathways with downregulated genes | |||
| cytokine-mediated signaling pathway (GO:0019221) | 80 | 5.59 × 10−8 | 2.25 × 10−4 |
| chaperone cofactor-dependent protein refolding (GO:0051085) | 11 | 5.05 × 10−7 | 9.87 × 10−3 |
| positive regulation of I-kappaB kinase/NF-kappaB signaling (GO:0043123) | 31 | 7.34 × 10−7 | 9.87 × 10−3 |
| ‘de novo’ posttranslational protein folding (GO:0051084) | 11 | 4.00 × 10−6 | 2.70 × 10−3 |
| negative regulation of viral process (GO:0048525) | 17 | 4.22 × 10−6 | 2.70 × 10−3 |
| regulation of I-kappaB kinase/NF-kappaB signaling (GO:0043122) | 35 | 5.22 × 10−6 | 2.70 × 10−3 |
| cellular response to type I interferon (GO:0071357) | 16 | 6.68 × 10−6 | 2.70 × 10−3 |
| type I interferon signaling pathway (GO:0060337) | 16 | 6.68 × 10−6 | 2.70 × 10−3 |
| mitochondrial respiratory chain complex I assembly (GO:0032981) | 15 | 6.71 × 10−6 | 2.70 × 10−3 |
| NADH dehydrogenase complex assembly (GO:0010257) | 15 | 6.71 × 10−6 | 2.70 × 10−3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinelli, G.; Fumagalli, M.; Piazza, S.; Maranta, N.; Genova, F.; Sperandeo, P.; Sangiovanni, E.; Polissi, A.; Dell’Agli, M.; De Fabiani, E. Investigating the Molecular Mechanisms Underlying Early Response to Inflammation and Helicobacter pylori Infection in Human Gastric Epithelial Cells. Int. J. Mol. Sci. 2023, 24, 15147. https://doi.org/10.3390/ijms242015147
Martinelli G, Fumagalli M, Piazza S, Maranta N, Genova F, Sperandeo P, Sangiovanni E, Polissi A, Dell’Agli M, De Fabiani E. Investigating the Molecular Mechanisms Underlying Early Response to Inflammation and Helicobacter pylori Infection in Human Gastric Epithelial Cells. International Journal of Molecular Sciences. 2023; 24(20):15147. https://doi.org/10.3390/ijms242015147
Chicago/Turabian StyleMartinelli, Giulia, Marco Fumagalli, Stefano Piazza, Nicole Maranta, Francesca Genova, Paola Sperandeo, Enrico Sangiovanni, Alessandra Polissi, Mario Dell’Agli, and Emma De Fabiani. 2023. "Investigating the Molecular Mechanisms Underlying Early Response to Inflammation and Helicobacter pylori Infection in Human Gastric Epithelial Cells" International Journal of Molecular Sciences 24, no. 20: 15147. https://doi.org/10.3390/ijms242015147
APA StyleMartinelli, G., Fumagalli, M., Piazza, S., Maranta, N., Genova, F., Sperandeo, P., Sangiovanni, E., Polissi, A., Dell’Agli, M., & De Fabiani, E. (2023). Investigating the Molecular Mechanisms Underlying Early Response to Inflammation and Helicobacter pylori Infection in Human Gastric Epithelial Cells. International Journal of Molecular Sciences, 24(20), 15147. https://doi.org/10.3390/ijms242015147