
Small Molecule Pytren-4QMn Metal Complex Slows down Huntington’s Disease Progression in Male zQ175 Transgenic Mice

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Assessment of 4QMn Acute and Chronic Tolerability
2.2. 4QMn Can Cross the Blood–Brain Barrier
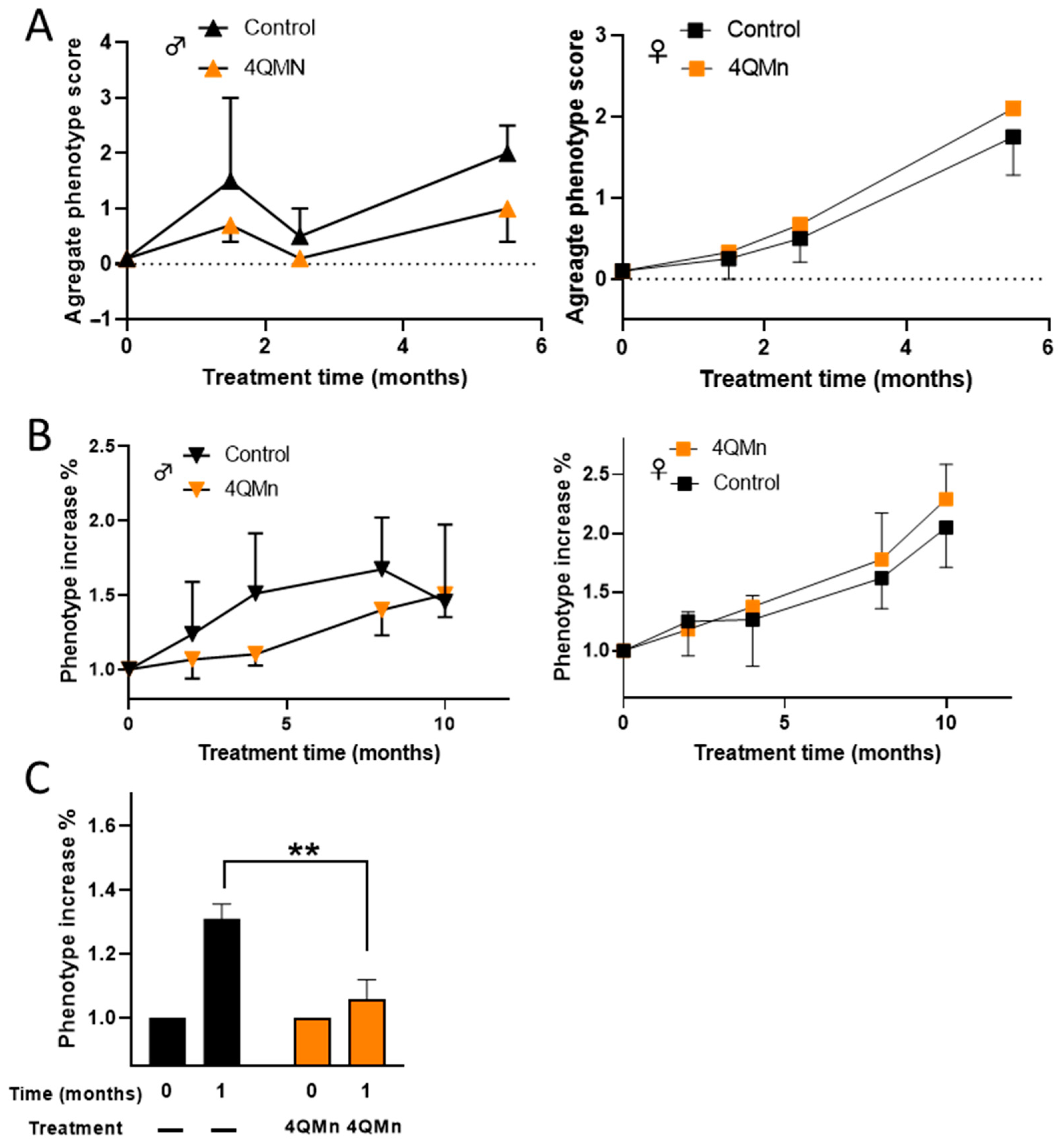
2.3. 4QMn Treatment Ameliorates the Progression of the HD Phenotype in Males
2.4. 4QMn Partially Recovered Darpp32 Expression and Reduced the Number of Nuclei with mHTT Granules in Male Striatum
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Preparation of 4QMn
4.3. Quantification of ALT Activity
4.4. Determination of Pytren-4Q in Tissues
4.5. Immunohistochemistry Evaluation of Huntingtin Aggregates in Striatum
4.6. Quantification of Gene Expression
4.7. Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, F.O. Huntington’s Disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- St-Cyr, S.; Smith, A.R.; Davidson, B.L. Temporal Phenotypic Changes in Huntington’s Disease Models for Preclinical Studies. J. Huntington’s Dis. 2022, 11, 35–57. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.P.; Telenius, H.; Hayden, M.R. The Molecular Genetics of Huntington’s Disease. Curr. Opin. Neurol. 1994, 7, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington Disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, S.; Jing, L.; Huang, L.; Chen, L.; Zhao, X.; Yang, W.; Pan, Y.; Yin, P.; Qin, Z.S.; et al. Truncation of Mutant Huntingtin in Knock-in Mice Demonstrates Exon1 Huntingtin Is a Key Pathogenic Form. Nat. Commun. 2020, 11, 2582. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Estevez-Fraga, C.; van Roon-Mom, W.M.C.; Flower, M.D.; Scahill, R.I.; Wild, E.J.; Muñoz-Sanjuan, I.; Sampaio, C.; Rosser, A.E.; Leavitt, B.R. Potential Disease-Modifying Therapies for Huntington’s Disease: Lessons Learned and Future Opportunities. Lancet Neurol. 2022, 21, 645–658. [Google Scholar] [CrossRef]
- Zheng, S.; Clabough, E.B.D.; Sarkar, S.; Futter, M.; Rubinsztein, D.C.; Zeitlin, S.O. Deletion of the Huntingtin Polyglutamine Stretch Enhances Neuronal Autophagy and Longevity in Mice. PLoS Genet. 2010, 6, e1000838. [Google Scholar] [CrossRef]
- Estevez-Fraga, C.; Rodrigues, F.B.; Tabrizi, S.J.; Wild, E.J. Huntington’s Disease Clinical Trials Corner: April 2022. J. Huntington’s Dis. 2022, 11, 105–118. [Google Scholar] [CrossRef]
- Merino, M.; Sequedo, M.D.; Sánchez-Sánchez, A.V.; Clares, M.P.; García-España, E.; Vázquez-Manrique, R.P.; Mullor, J.L. Mn(II) Quinoline Complex (4QMn) Restores Proteostasis and Reduces Toxicity in Experimental Models of Huntington’s Disease. Int. J. Mol. Sci. 2022, 23, 8936. [Google Scholar] [CrossRef]
- Clares, M.P.; Blasco, S.; Inclán, M.; del Castillo Agudo, L.; Verdejo, B.; Soriano, C.; Doménech, A.; Latorre, J.; García-España, E. Manganese(Ii) Complexes of Scorpiand-like Azamacrocycles as MnSOD Mimics. Chem. Commun. 2011, 47, 5988. [Google Scholar] [CrossRef] [PubMed]
- Clares, M.P.; Serena, C.; Blasco, S.; Nebot, A.; del Castillo, L.; Soriano, C.; Domènech, A.; Sánchez-Sánchez, A.V.; Soler-Calero, L.; Mullor, J.L.; et al. Mn(II) Complexes of Scorpiand-like Ligands. A Model for the MnSOD Active Centre with High in Vitro and in Vivo Activity. J. Inorg. Biochem. 2015, 143, 1–8. [Google Scholar] [CrossRef]
- Serena, C.; Calvo, E.; Clares, M.P.; Diaz, M.L.; Chicote, J.U.; Beltrán-Debon, R.; Fontova, R.; Rodriguez, A.; García-España, E.; García-España, A. Significant In Vivo Anti-Inflammatory Activity of Pytren4Q-Mn a Superoxide Dismutase 2 (SOD2) Mimetic Scorpiand-Like Mn (II) Complex. PLoS ONE 2015, 10, e0119102. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Antioxidants in Huntington’s Disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Essa, M.M.; Moghadas, M.; Ba-Omar, T.; Walid Qoronfleh, M.; Guillemin, G.J.; Manivasagam, T.; Justin-Thenmozhi, A.; Ray, B.; Bhat, A.; Chidambaram, S.B.; et al. Protective Effects of Antioxidants in Huntington’s Disease: An Extensive Review. Neurotox. Res. 2019, 35, 739–774. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; Haller, R.G. Energy Deficit in Huntington Disease: Why It Matters. J. Clin. Investig. 2011, 121, 493–499. [Google Scholar] [CrossRef]
- Melkani, G. Huntington’s Disease-Induced Cardiac Disorders Affect Multiple Cellular Pathways. React. Oxyg. Species 2016, 2, 325. [Google Scholar] [CrossRef]
- Trujillo-Del Río, C.; Tortajada-Pérez, J.; Gómez-Escribano, A.P.; Casterá, F.; Peiró, C.; Millán, J.M.; Herrero, M.J.; Vázquez-Manrique, R.P. Metformin to Treat Huntington Disease: A Pleiotropic Drug against a Multi-System Disorder. Mech. Ageing Dev. 2022, 204, 111670. [Google Scholar] [CrossRef]
- Menalled, L.B.; Kudwa, A.E.; Miller, S.; Fitzpatrick, J.; Watson-Johnson, J.; Keating, N.; Ruiz, M.; Mushlin, R.; Alosio, W.; McConnell, K.; et al. Comprehensive Behavioral and Molecular Characterization of a New Knock-In Mouse Model of Huntington’s Disease: ZQ175. PLoS ONE 2012, 7, e49838. [Google Scholar] [CrossRef]
- Kaye, J.; Reisine, T.; Finkbeiner, S. Huntington’s Disease Mouse Models: Unraveling the Pathology Caused by CAG Repeat Expansion. Fac. Rev. 2021, 10, 77. [Google Scholar] [CrossRef]
- Guyenet, S.J.; Furrer, S.A.; Damian, V.M.; Baughan, T.D.; La Spada, A.R.; Garden, G.A. A Simple Composite Phenotype Scoring System for Evaluating Mouse Models of Cerebellar Ataxia. J. Vis. Exp. 2010, 39, e1787. [Google Scholar] [CrossRef]
- Young, D.; Mayer, F.; Vidotto, N.; Schweizer, T.; Berth, R.; Abramowski, D.; Shimshek, D.R.; van der Putten, P.H.; Schmid, P. Mutant Huntingtin Gene-Dose Impacts on Aggregate Deposition, DARPP32 Expression and Neuroinflammation in HdhQ150 Mice. PLoS ONE 2013, 8, e75108. [Google Scholar] [CrossRef]
- Porro, A.; Mohiuddin, M.; Zurfluh, C.; Spegg, V.; Dai, J.; Iehl, F.; Ropars, V.; Collotta, G.; Fishwick, K.M.; Mozaffari, N.L.; et al. FAN1-MLH1 Interaction Affects Repair of DNA Interstrand Cross-Links and Slipped-CAG/CTG Repeats. Sci. Adv. 2021, 7, eabf7906. [Google Scholar] [CrossRef]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. In Polyglutamine Disorders; Nóbrega, C., Pereira de Almeida, L., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; Volume 1049, pp. 59–83. ISBN 978-3-319-71778-4. [Google Scholar]
- McColgan, P.; Tabrizi, S.J. Huntington’s Disease: A Clinical Review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Impaired Redox Signaling in Huntington’s Disease: Therapeutic Implications. Front. Mol. Neurosci. 2019, 12, 68. [Google Scholar] [CrossRef]
- Gkekas, I.; Gioran, A.; Boziki, M.K.; Grigoriadis, N.; Chondrogianni, N.; Petrakis, S. Oxidative Stress and Neurodegeneration: Interconnected Processes in PolyQ Diseases. Antioxidants 2021, 10, 1450. [Google Scholar] [CrossRef]
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J. Huntington’s Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Bono-Yagüe, J.; Gómez-Escribano, A.P.; Millán, J.M.; Vázquez-Manrique, R.P. Reactive Species in Huntington Disease: Are They Really the Radicals You Want to Catch? Antioxidants 2020, 9, 577. [Google Scholar] [CrossRef] [PubMed]
- Romá-Mateo, C.; García-Giménez, J.L. Oxidative Stress and Rare Diseases: From Molecular Crossroads to Therapeutic Avenues. Antioxidants 2021, 10, 617. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Colson, T.-L.L.; Abd-Elrahman, K.S.; Ferguson, S.S.G. Metabotropic Glutamate Receptor 2/3 Activation Improves Motor Performance and Reduces Pathology in Heterozygous ZQ175 Huntington Disease Mice. J. Pharmacol. Exp. Ther. 2021, 379, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin Treatment Reduces Motor and Neuropsychiatric Phenotypes in the ZQ175 Mouse Model of Huntington Disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Maiese, K. Targeting Molecules to Medicine with MTOR, Autophagy and Neurodegenerative Disorders: MTOR and the Nervous System. Br. J. Clin. Pharmacol. 2016, 82, 1245–1266. [Google Scholar] [CrossRef]
- Huang, Z.-N.; Chen, J.-M.; Huang, L.-C.; Fang, Y.-H.; Her, L.-S. Inhibition of P38 Mitogen–Activated Protein Kinase Ameliorates HAP40 Depletion–Induced Toxicity and Proteasomal Defect in Huntington’s Disease Model. Mol. Neurobiol. 2021, 58, 2704–2723. [Google Scholar] [CrossRef]
- Padovan-Neto, F.E.; Jurkowski, L.; Murray, C.; Stutzmann, G.E.; Kwan, M.; Ghavami, A.; Beaumont, V.; Park, L.C.; West, A.R. Age- and Sex-Related Changes in Cortical and Striatal Nitric Oxide Synthase in the Q175 Mouse Model of Huntington’s Disease. Nitric Oxide 2019, 83, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Colson, T.-L.L.; Abd-Elrahman, K.S.; Ferguson, S.S.G. Metabotropic Glutamate Receptor 5 Antagonism Reduces Pathology and Differentially Improves Symptoms in Male and Female Heterozygous ZQ175 Huntington’s Mice. Front. Mol. Neurosci. 2022, 15, 801757. [Google Scholar] [CrossRef] [PubMed]
- Shang, D.; Wang, L.; Klionsky, D.J.; Cheng, H.; Zhou, R. Sex Differences in Autophagy-Mediated Diseases: Toward Precision Medicine. Autophagy 2021, 17, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, D.; Stawinska-Witoszynska, B. Gender Differences in Non-Sex Linked Disorders: Insights From Huntington’s Disease. Front. Neurol. 2020, 11, 571. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.A.; Collins, F.S. Policy: NIH to Balance Sex in Cell and Animal Studies. Nature 2014, 509, 282–283. [Google Scholar] [CrossRef]
- Dorner, J.L.; Miller, B.R.; Barton, S.J.; Brock, T.J.; Rebec, G.V. Sex Differences in Behavior and Striatal Ascorbate Release in the 140 CAG Knock-in Mouse Model of Huntington’s Disease. Behav. Brain Res. 2007, 178, 90–97. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merino, M.; González, S.; Tronch, M.C.; Sánchez-Sánchez, A.V.; Clares, M.P.; García-España, A.; García-España, E.; Mullor, J.L. Small Molecule Pytren-4QMn Metal Complex Slows down Huntington’s Disease Progression in Male zQ175 Transgenic Mice. Int. J. Mol. Sci. 2023, 24, 15153. https://doi.org/10.3390/ijms242015153
Merino M, González S, Tronch MC, Sánchez-Sánchez AV, Clares MP, García-España A, García-España E, Mullor JL. Small Molecule Pytren-4QMn Metal Complex Slows down Huntington’s Disease Progression in Male zQ175 Transgenic Mice. International Journal of Molecular Sciences. 2023; 24(20):15153. https://doi.org/10.3390/ijms242015153
Chicago/Turabian StyleMerino, Marián, Sonia González, Mª Carmen Tronch, Ana Virginia Sánchez-Sánchez, Mª Paz Clares, Antonio García-España, Enrique García-España, and José L. Mullor. 2023. "Small Molecule Pytren-4QMn Metal Complex Slows down Huntington’s Disease Progression in Male zQ175 Transgenic Mice" International Journal of Molecular Sciences 24, no. 20: 15153. https://doi.org/10.3390/ijms242015153
APA StyleMerino, M., González, S., Tronch, M. C., Sánchez-Sánchez, A. V., Clares, M. P., García-España, A., García-España, E., & Mullor, J. L. (2023). Small Molecule Pytren-4QMn Metal Complex Slows down Huntington’s Disease Progression in Male zQ175 Transgenic Mice. International Journal of Molecular Sciences, 24(20), 15153. https://doi.org/10.3390/ijms242015153