
The Integration of Genome-Wide DNA Methylation and Transcriptomics Identifies the Potential Genes That Regulate the Development of Skeletal Muscles in Ducks



Abstract
:1. Introduction
2. Results
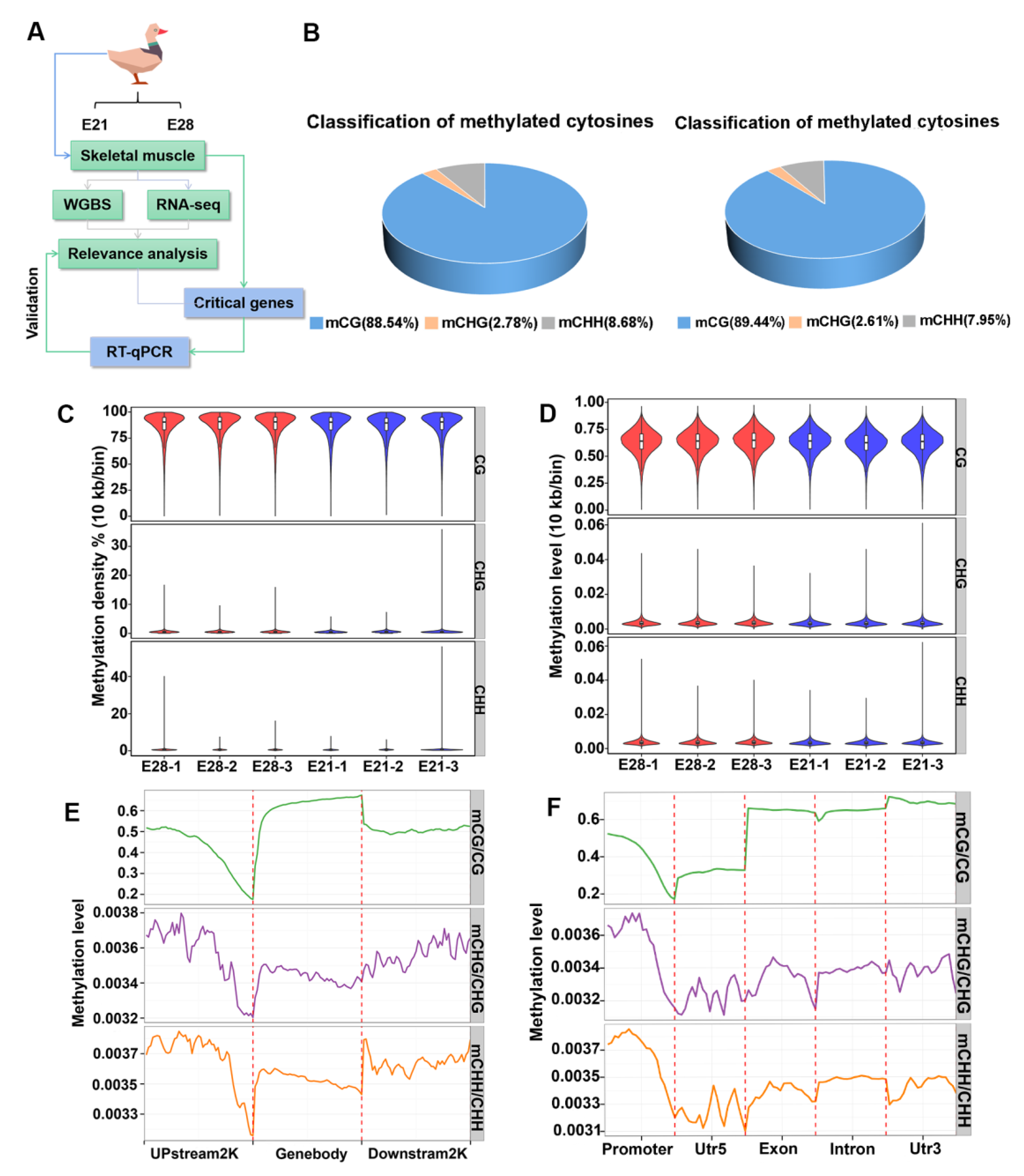
2.1. DNA Methylation Pattern in Duck Skeletal Muscle
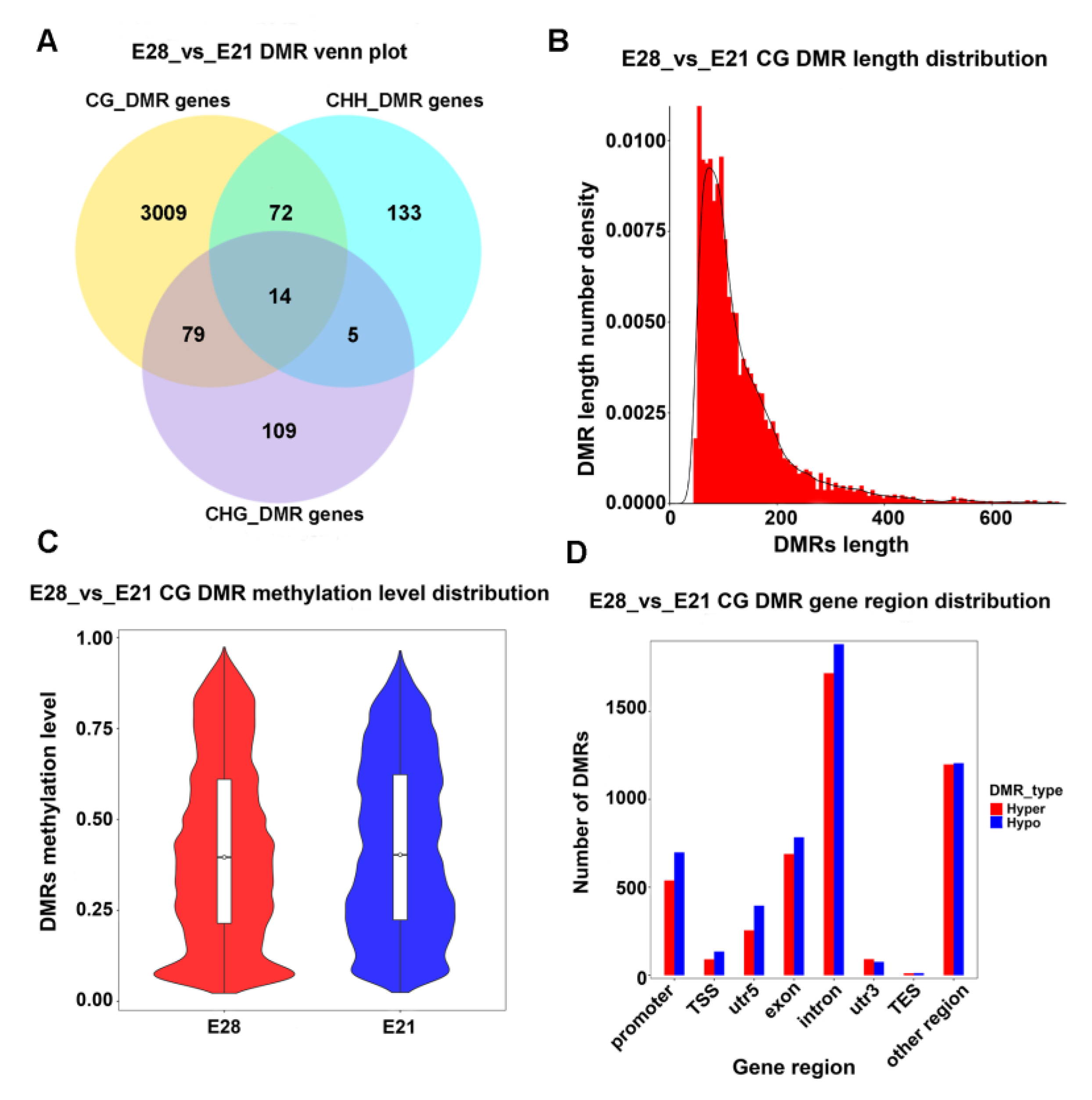
2.2. Identification of Differentially Methylated Regions (DMRs)
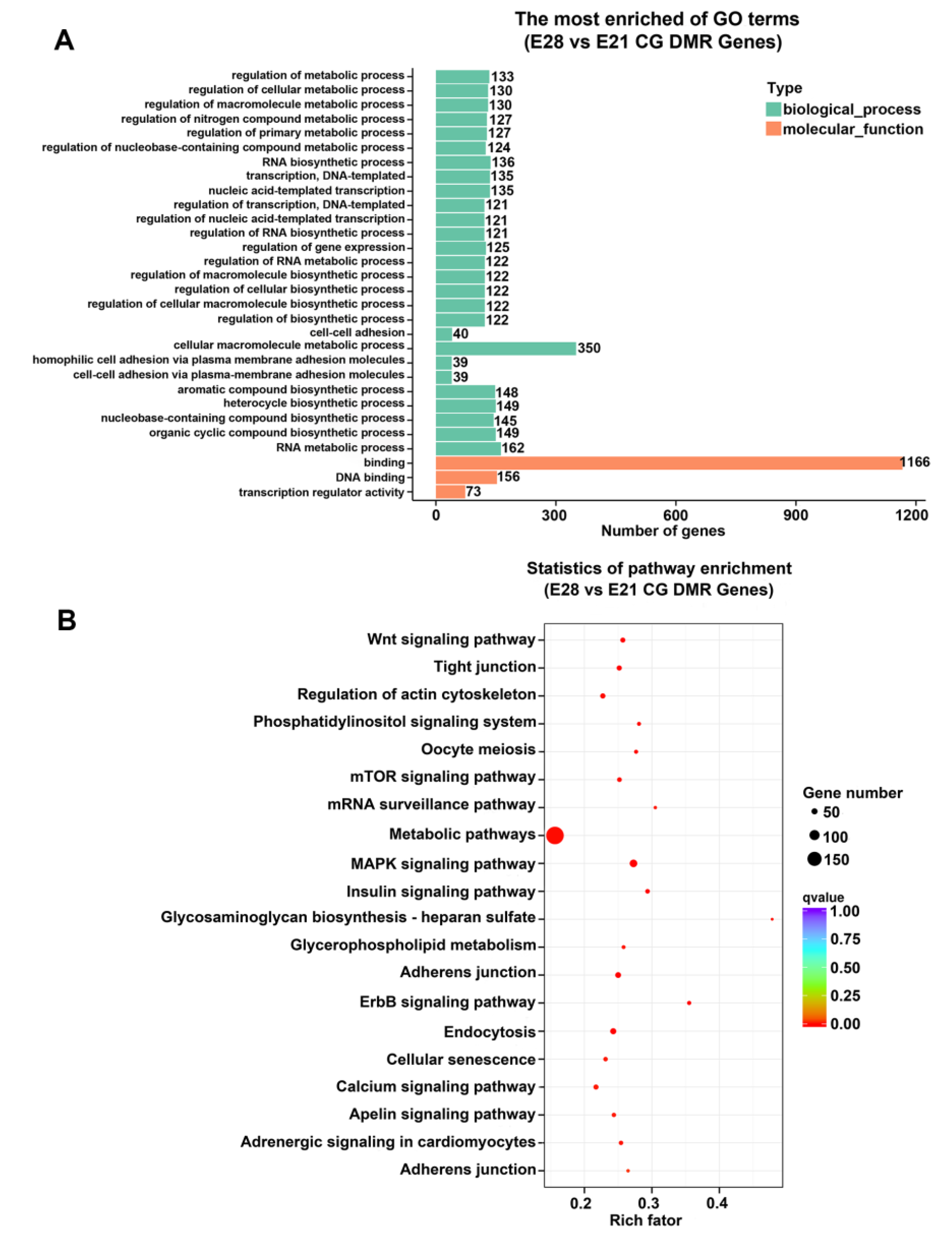
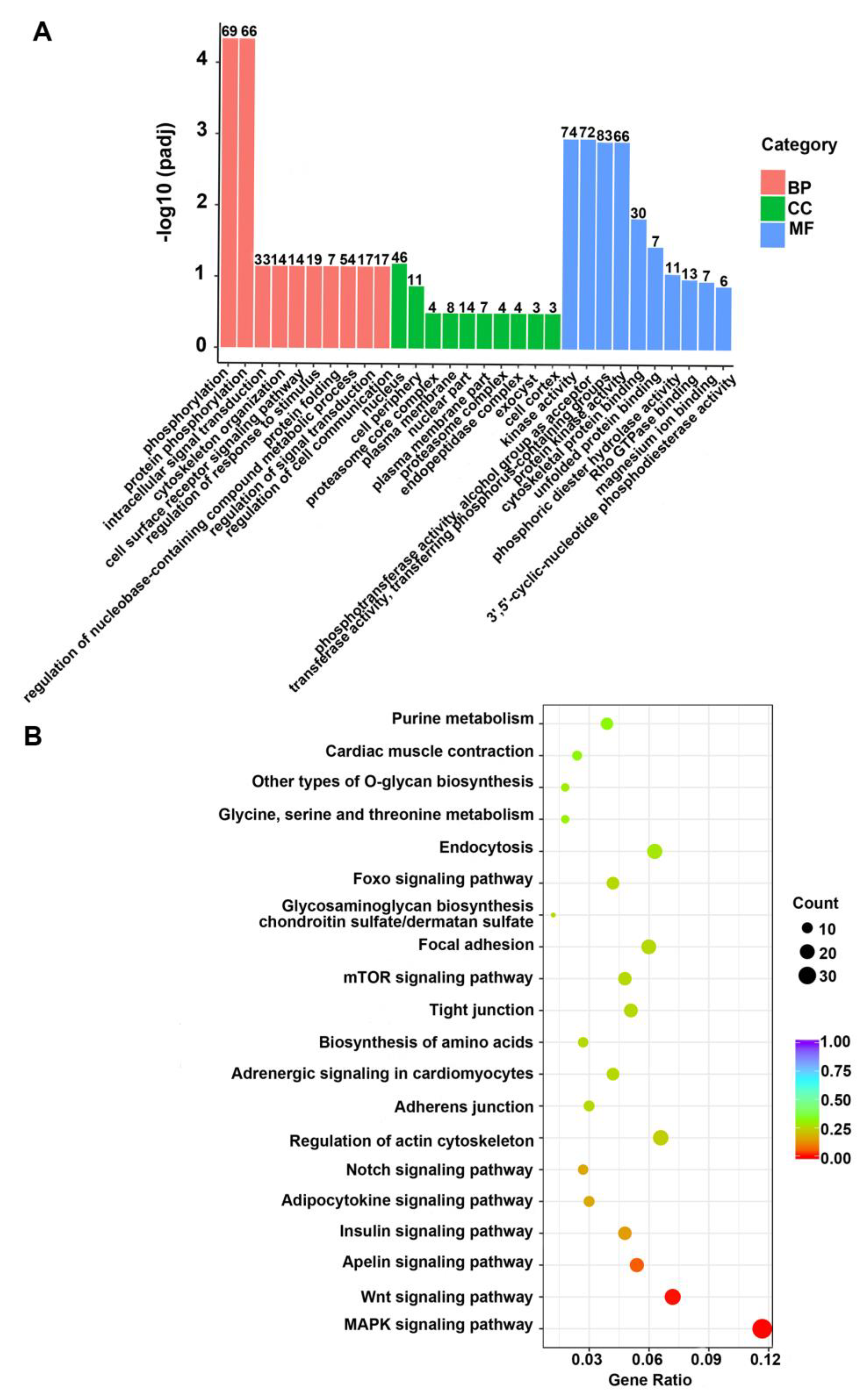
2.3. Functional Enrichment of Genes with Methylation
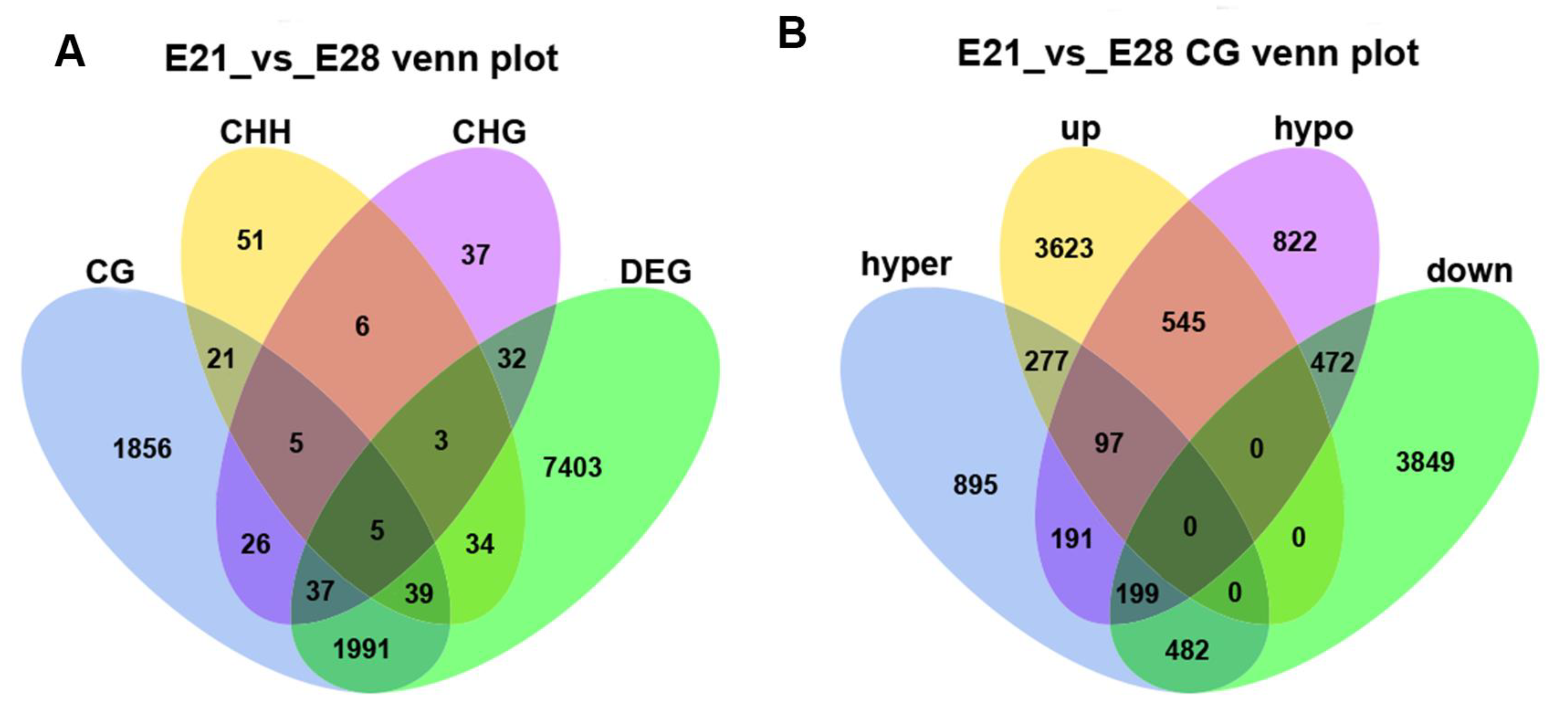
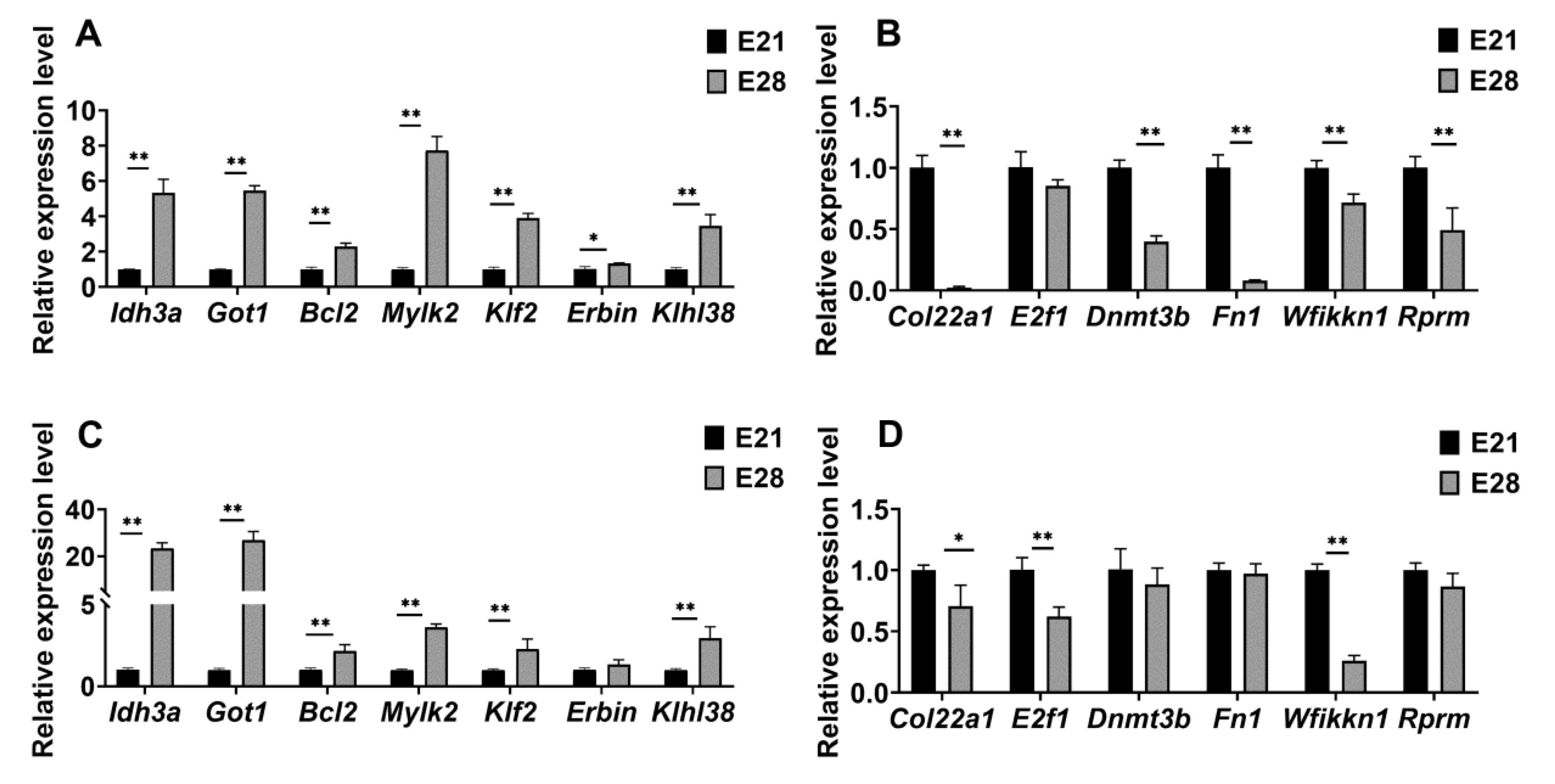
2.4. Association Analysis of DNA Methylation and Gene Expression
2.5. The Key Pathways Enriched the DMGs Negatively Associated with DEGs
2.6. Identification of Genes Implicated in the Development of Skeletal Muscles
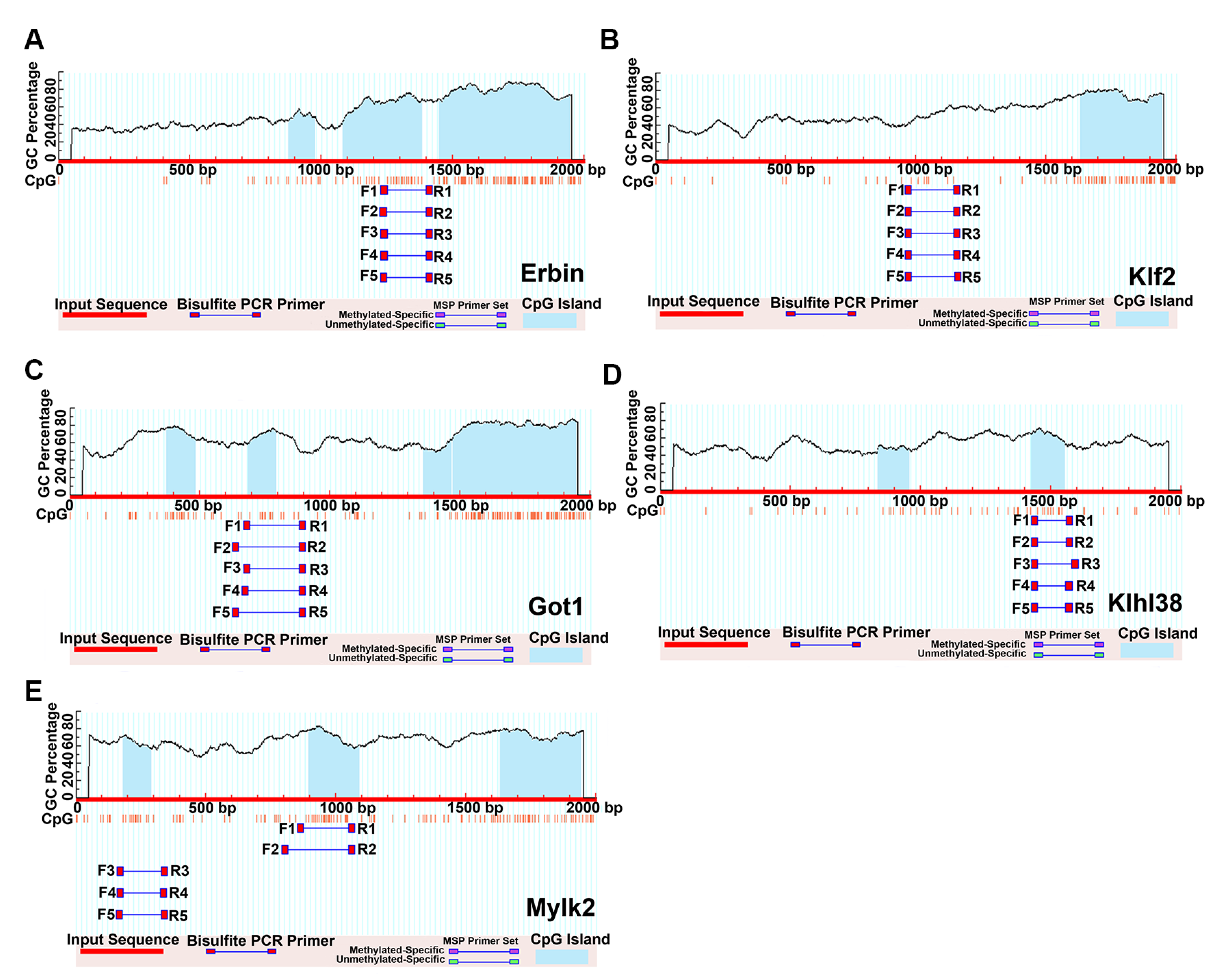
2.7. Prediction of the CpG Islands on the Promoters
3. Discussion
4. Materials and Methods
4.1. Tissue Sampling
4.2. DNA and RNA Extraction
4.3. RNA-Seq
4.4. Genome-Wide DNA Methylation Profiling
4.5. Gene Functional Enrichment Analysis
4.6. Verification of Critical Gene Expression Level
4.7. CpG Island Prediction of Key Genes
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ran, J.S.; Li, J.J.; Yin, L.Q.; Zhang, D.H.; Yu, C.L.; Du, H.R.; Jiang, X.S.; Yang, C.W.; Liu, Y.P. Comparative analysis of skeletal muscle DNA methylation and transcriptome of the chicken embryo at different developmental stages. Front. Physiol. 2021, 12, 697121. [Google Scholar] [CrossRef]
- Ji, G.G.; Shu, J.T.; Zhang, M.; Ju, X.J.; Shan, Y.J.; Liu, Y.F.; Tu, Y.J. Transcriptional regulatory region and DNA methylation analysis of TNNI1 gene promoters in Gaoyou duck skeletal muscle (Anas platyrhynchos domestica). Br. Poult. Sci. 2019, 60, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Han, S.S.; Shen, X.X.; Wang, Y.; Cui, C.; He, H.R.; Chen, Y.Q.; Zhao, J.; Li, D.; Zhu, Q.; et al. The landscape of DNA methylation associated with the transcriptomic network in layers and broilers generates insight into embryonic muscle development in chicken. Int. J. Biol. Sci. 2019, 15, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.H.; Song, W.T.; Tao, Z.Y.; Liu, H.X.; Xu, W.J.; Zhang, S.J.; Li, H.F. Deep RNA sequencing of pectoralis muscle transcriptomes during late-term embryonic to neonatal development in indigenous Chinese duck breeds. PLoS ONE 2017, 12, e0180403. [Google Scholar] [CrossRef]
- Carrio, E.; Diez-Villanueva, A.; Lois, S.; Mallona, I.; Cases, I.; Forn, M.; Peinado, M.A.; Suelves, M. Deconstruction of DNA methylation patterns during myogenesis reveals specific epigenetic events in the establishment of the skeletal muscle lineage. Stem Cells 2015, 33, 2025–2036. [Google Scholar] [CrossRef]
- Pan, Y.X.; Chen, L.; Cheng, J.; Zhu, X.; Wu, P.; Bao, L.S.; Chu, W.Y.; He, S.; Liang, X.F.; Zhang, J.S. Genome-wide DNA methylation profiles provide insight into epigenetic regulation of red and white muscle development in Chinese perch Siniperca chuatsi. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2021, 256, 110647. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wu, P.X.; Wang, S.J.; Ji, X.; Chen, D.; Jiang, A.A.; Xiao, W.H.; Jiang, Y.Z.; Li, Z.; Zeng, Y.S.; et al. Epigenome-wide DNA methylation analysis reveals differentially methylation patterns in skeletal muscle between Chinese Chenghua and Qingyu pigs. J. Integr. Agric. 2022, 21, 1731–1739. [Google Scholar] [CrossRef]
- Li, X.J.; Liu, L.Q.; Dong, H.; Yang, J.J.; Wang, W.W.; Zhang, Q.; Wang, C.L.; Zhou, J.; Chen, H.Q. Comparative genome-wide methylation analysis of longissimus dorsi muscles in Yorkshire and Wannanhua pigs. Anim. Genet. 2021, 52, 78–89. [Google Scholar] [CrossRef]
- Zhang, X.M.; Li, Y.; Zhu, C.Y.; Li, F.D.; Liu, Z.Y.; Li, X.J.; Shen, X.; Wu, Z.P.; Fu, M.S.; Xu, D.N.; et al. DNA demethylation of myogenic genes may contribute to embryonic leg muscle development differences between Wuzong and Shitou Geese. Int. J. Mol. Sci. 2023, 24, 7188. [Google Scholar] [CrossRef]
- Sun, L.M.; Bai, M.; Xiang, L.J.; Zhang, G.S.; Ma, W.; Jiang, H.Z. Comparative transcriptome profiling of longissimus muscle tissues from Qianhua Mutton Merino and Small Tail Han sheep. Sci. Rep. 2016, 6, 33586. [Google Scholar] [CrossRef]
- Li, H.; Zhu, C.; Tao, Z.; Xu, W.; Song, W.; Hu, Y.; Zhu, W.; Song, C. MyoD and Myf6 gene expression patterns in skeletal muscle during embryonic and posthatch development in the domestic duck (Anas platyrhynchos domestica). J. Anim. Breed. Genet. 2014, 131, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.X.; Liang, Y.X.; Deng, K.P.; Zhang, Z.; Zhang, G.M.; Zhang, Y.L.; Wang, F. Analysis of DNA methylation profiles during sheep skeletal muscle development using whole-genome bisulfite sequencing. BMC Genom. 2020, 21, 327. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.J.; Wang, J.K.; Shen, Y.F.; Zhang, J.L.; Liu, J.; Xiao, A.P.; Liu, Y.S.; Eer, H.H.; Zhang, Q.E. Whole-genome DNA methylation profiling reveals epigenetic signatures in developing muscle in Tan and Hu sheep and their offspring. Front. Vet. Sci. 2023, 10, 1186040. [Google Scholar] [CrossRef]
- Zhang, D.; Wu, S.M.; Zhang, X.X.; Ren, S.Q.; Tang, Z.L.; Gao, F. Coordinated transcriptional and post-transcriptional epigenetic regulation during skeletal muscle development and growth in pigs. J. Anim. Sci. Biotechnol. 2022, 13, 146. [Google Scholar] [CrossRef]
- Yang, Y.L.; Fan, X.H.; Yan, J.Y.; Chen, M.Y.; Zhu, M.; Tang, Y.J.; Liu, S.Y.; Tang, Z.L. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021, 49, 1313–1329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, W.J.; He, L.J.; Wang, L.Q.; Qiu, K.; Yin, J.D. Global DNA methylation pattern involved in the modulation of differentiation potential of adipogenic and myogenic precursors in skeletal muscle of pigs. Stem Cell Res. Ther. 2020, 11, 536. [Google Scholar] [CrossRef]
- Li, D.F.; Ge, J.Y.; Wang, H.B.; He, Z.L.; Jin, T.; Yu, M.L. Differences in skeletal muscle development among Pekin duck, Liancheng white duck and their hybrid F_5 resource groups. J. Nanjing Agric. Univ. 2022, 45, 1235–1245. [Google Scholar] [CrossRef]
- Hu, Z.G.; Cao, J.T.; Zhang, J.Q.; Ge, L.Y.; Zhang, H.L.; Liu, X.L. Skeletal muscle transcriptome analysis of Hanzhong Ma Duck at different growth stages using RNA-Seq. Biomolecules 2021, 11, 315. [Google Scholar] [CrossRef]
- Hu, Z.; Liu, X. Integration of transcriptomics and non-targeted metabolomics reveals the underlying mechanism of skeletal muscle development in duck during embryonic stage. Int. J. Mol. Sci. 2023, 24, 5214. [Google Scholar] [CrossRef]
- Davegårdh, C.; Hall Wedin, E.; Broholm, C.; Henriksen, T.I.; Pedersen, M.; Pedersen, B.K.; Scheele, C.; Ling, C. Sex influences DNA methylation and gene expression in human skeletal muscle myoblasts and myotubes. Stem Cell Res. Ther. 2019, 10, 26. [Google Scholar] [CrossRef]
- Kanzleiter, T.; Jähnert, M.; Schulze, G.; Selbig, J.; Hallahan, N.; Schwenk, R.W.; Schürmann, A. Exercise training alters DNA methylation patterns in genes related to muscle growth and differentiation in mice. Am. J. Physiol.-Endocrinol. Metab. 2015, 308, E912–E920. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.M. The role of AMPK in the regulation of skeletal muscle size, hypertrophy, and regeneration. Int. J. Mol. Sci. 2018, 19, 3125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yan, F.B.; Li, F.; Jiang, K.R.; Li, D.H.; Han, R.L.; Li, Z.J.; Jiang, R.R.; Liu, X.J.; Kang, X.T.; et al. Genome-wide DNA methylation profiles reveal novel candidate genes associated with meat quality at different age stages in hens. Sci. Rep. 2017, 7, 45564. [Google Scholar] [CrossRef]
- Zhang, M.; Li, D.H.; Zhai, Y.H.; Wang, Z.Z.; Ma, X.F.; Zhang, D.Y.; Li, G.X.; Han, R.L.; Jiang, R.R.; Li, Z.J.; et al. The landscape of DNA methylation associated with the transcriptomic network of intramuscular adipocytes generates insight into intramuscular fat deposition in chicken. Front. Cell Dev. Biol. 2020, 8, 206. [Google Scholar] [CrossRef]
- Yang, Y.L.; Liang, G.M.; Niu, G.L.; Zhang, Y.Y.; Zhou, R.; Wang, Y.F.; Mu, Y.L.; Tang, Z.L.; Li, K. Comparative analysis of DNA methylome and transcriptome of skeletal muscle in lean-, obese-, and mini-type pigs. Sci. Rep. 2017, 7, 39883. [Google Scholar] [CrossRef]
- Son, J.S.; Chae, S.A.; Zhao, L.; Wang, H.Y.; de Avila, J.M.; Zhu, M.J.; Jiang, Z.H.; Du, M. Maternal exercise intergenerationally drives muscle-based thermogenesis via activation of apelin-AMPK signaling. Ebiomedicine 2022, 76, 103842. [Google Scholar] [CrossRef]
- Zhang, W.Y.; Zhang, S.X.; Xu, Y.Y.; Ma, Y.L.; Zhang, D.X.; Li, X.Y.; Zhao, S.H. The DNA methylation status of Wnt and Tgfβ signals is a key factor on functional regulation of skeletal muscle satellite cell development. Front. Genet. 2019, 10, 220. [Google Scholar] [CrossRef]
- Zhang, D.H.; Yin, H.D.; Li, J.J.; Wang, Y.; Yang, C.W.; Jiang, X.S.; Du, H.R.; Liu, Y.P. KLF5 regulates chicken skeletal muscle atrophy via the canonical Wnt/β-catenin signaling pathway. Exp. Anim. 2020, 69, 430–440. [Google Scholar] [CrossRef]
- Li, Z.H.; Cai, B.L.; Abdalla, B.A.; Zhu, X.N.; Zheng, M.; Han, P.G.; Nie, Q.H.; Zhang, X.Q. LncIRS1 controls muscle atrophy via sponging miR-15 family to activate IGF1-PI3K/AKT pathway. J. Cachexia Sarcopenia Muscle 2019, 10, 391–410. [Google Scholar] [CrossRef]
- Mudry, J.M.; Lassiter, D.G.; Nylén, C.; García-Calzón, S.; Näslund, E.; Krook, A.; Zierath, J.R. Insulin and glucose alter death-associated protein kinase 3 (DAPK3) DNA methylation in human skeletal muscle. Diabetes 2016, 66, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Tohtong, R.; Yamashita, H.; Graham, M.; Haeberle, J.; Simcox, A.; Maughan, D. Impairment of muscle function caused by mutations of phosphorylation sites in myosin regulatory light chain. Nature 1995, 374, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Kravic, B.; Huraskin, D.; Frick, A.D.; Jung, J.; Redai, V.; Palmisano, R.; Marchetto, S.; Borg, J.-P.; Mei, L.; Hashemolhosseini, S. LAP proteins are localized at the post-synaptic membrane of neuromuscular junctions and appear to modulate synaptic morphology and transmission. J. Neurochem. 2016, 139, 381–395. [Google Scholar] [CrossRef]
- Ehrlich, K.C.; Baribault, C.; Ehrlich, M. Epigenetics of muscle- and brain-specific expression of KLHL family genes. Int. J. Mol. Sci. 2020, 21, 8394. [Google Scholar] [CrossRef]
- Sunadome, K.; Yamamoto, T.; Ebisuya, M.; Kondoh, K.; Sehara-Fujisawa, A.; Nishida, E. ERK5 regulates muscle cell fusion through Klf transcription factors. Dev. Cell 2011, 20, 192–205. [Google Scholar] [CrossRef]
- Manoharan, P.; Song, T.; Radzyukevich, T.L.; Sadayappan, S.; Lingrel, J.B.; Heiny, J.A. KLF2 in myeloid lineage cells regulates the innate immune response during skeletal muscle injury and regeneration. Iscience 2019, 17, 334–346. [Google Scholar] [CrossRef]
- Valdés-Hernández, J.; Ramayo-Caldas, Y.; Passols, M.; Sebastià, C.; Criado-Mesas, L.; Crespo-Piazuelo, D.; Esteve-Codina, A.; Castelló, A.; Sánchez, A.; Folch, J.M. Global analysis of the association between pig muscle fatty acid composition and gene expression using RNA-Seq. Sci. Rep. 2023, 13, 535. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Log2 Fold Change | Diff.Methy | C Context | Region |
|---|---|---|---|---|
| Klhl38 | 6.341923585 | −0.161196584 | CG | promoter |
| Mylk2 | 3.680553968 | −0.353681294 | CG | promoter |
| Got1 | 2.956657678 | −0.209114374 | CG | promoter |
| Klf2 | 1.683077721 | −0.291024055 | CG | promoter |
| Erbin | 1.483720868 | −0.310759514 | CG | promoter |
| Idh3a | 3.115334047 | −0.126427439 | CG | exon |
| Bcl2 | 1.555105211 | −0.228206336 | CG | exon |
| E2f1 | −2.0475711 | 0.125491948 | CG | exon |
| Fn1 | −2.834137149 | 0.353384159 | CG | exon |
| Wfikkn1 | −3.111804698 | 0.161546317 | CG | exon |
| Dnmt3b | −3.684262185 | 0.269994984 | CG | exon |
| Col22a1 | −4.123660418 | 0.35941219 | CG | exon |
| Rprm | −5.226699842 | 0.122454333 | CG | exon |
| Gene Name | Primer Sequences | Product Length | Gene Accession Number |
|---|---|---|---|
| Bcl2 | F: 5′-TGTGCGTGGAGAGCGTCAAC-3′ | 79 | XM_027451680.2 |
| R: 5′-CGGTTCAGGTACTCGGTCATCC-3′ | |||
| Klf2 | F: 5′-GCCTGCCCTTACAACCCTCTG-3′ | 172 | XM_027472014.2 |
| R: 5′-TGCGAACTCTTGGTGTAGGTCTTC-3′ | |||
| Fn1 | F: 5′-GGAGGTAGTCACAGTTGGCAATAC-3′ | 71 | XM_038182473.1 |
| R: 5′-TGTCGTAGCAGGTGTCGTCAG-3′ | |||
| Klhl38 | F: 5′-TCCAGGCAAGACAAGGCTACATC-3′ | 122 | XM_038174946.1 |
| R: 5′-GCAAGTGGGTGAAGACTGAACAAG-3′ | |||
| Erbin | F: 5′-AGCAATATTCAGCCGGAAGC-3′ | 153 | XM_038169994.1 |
| R: 5′-AACAAGTGCAGCCATCTGTG-3′ | |||
| Mylk2 | F: 5′-AACGACACCGAGACGCTGAAC-3′ | 173 | XM_038166457.1 |
| R: 5′-AGGTTGTTGAGCCAGGGATGC-3′ | |||
| Dnmt3b | F: 5′-TCTGGGGAAAGATCAAAGGTT-3′ | 191 | XM_038166400.1 |
| R: 5′-AAACGTGGCAGAATTGAAATG-3′ | |||
| Wfikkn1 | F: 5′-GGCTCTGACTGCGACATCTGG-3′ | 128 | XM_027468956.2 |
| R: 5′-TCGGCGTCCATGTAGCACTTG-3′ | |||
| Got1 | F: 5′-TTGCCGAGTGGAAGGACAACG-3′ | 138 | XM_027460734.2 |
| R: 5′-AGCTGAACATGCCGATCTGGTC-3′ | |||
| Idh3a | F: 5′-TGCTGGATTGATTGGGGGTC-3′ | 146 | XM_027465914.2 |
| R: 5′-CAGCACTCAGAAGAAGGGCA-3′ | |||
| Col22a1 | F: 5′-ATTGACAAGTACGGCATACCACAG-3′ | 106 | XM_038175073.1 |
| R: 5′-AAACCACTCTCACGGCATCTTTG-3′ | |||
| Rprm | F: 5′-TGGTGCAGATCGCCGTCATG-3′ | 73 | XM_038182513.1 |
| R: 5′-AGGTTGCAGCCGAGGAAGAAG-3′ | |||
| E2f1 | F: 5′-CTCGCTGAACCTCACCACCAAG-3′ | 113 | XM_038166109.1 |
| R: 5′-AGATTCGCCTCTTCTGCACCTTC-3′ | |||
| β-actin | F: 5′-TGCGTGACATCAAGGAGAAG-3′ | 300 | NM_001310421.1 |
| R: 5′-TGCCCGGGTACATTGTGGTA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Zhou, J.; Li, F.; Cao, H.; Zhang, X.; Yu, D.; He, Z.; Ji, H.; Lv, K.; Wu, G.; et al. The Integration of Genome-Wide DNA Methylation and Transcriptomics Identifies the Potential Genes That Regulate the Development of Skeletal Muscles in Ducks. Int. J. Mol. Sci. 2023, 24, 15476. https://doi.org/10.3390/ijms242015476
Lu Y, Zhou J, Li F, Cao H, Zhang X, Yu D, He Z, Ji H, Lv K, Wu G, et al. The Integration of Genome-Wide DNA Methylation and Transcriptomics Identifies the Potential Genes That Regulate the Development of Skeletal Muscles in Ducks. International Journal of Molecular Sciences. 2023; 24(20):15476. https://doi.org/10.3390/ijms242015476
Chicago/Turabian StyleLu, Yinglin, Jing Zhou, Fan Li, Heng Cao, Xingyu Zhang, Debing Yu, Zongliang He, Hongjie Ji, Kunpeng Lv, Guansuo Wu, and et al. 2023. "The Integration of Genome-Wide DNA Methylation and Transcriptomics Identifies the Potential Genes That Regulate the Development of Skeletal Muscles in Ducks" International Journal of Molecular Sciences 24, no. 20: 15476. https://doi.org/10.3390/ijms242015476
APA StyleLu, Y., Zhou, J., Li, F., Cao, H., Zhang, X., Yu, D., He, Z., Ji, H., Lv, K., Wu, G., & Yu, M. (2023). The Integration of Genome-Wide DNA Methylation and Transcriptomics Identifies the Potential Genes That Regulate the Development of Skeletal Muscles in Ducks. International Journal of Molecular Sciences, 24(20), 15476. https://doi.org/10.3390/ijms242015476