Abstract
The reactions of alkenes with phenyl-N-triflylimino-λ3-iodane PhI=NTf (1) have been studied in different conditions. In methylene chloride, in the presence of N-halosuccinimides, the products of mono and bis-triflamidation were obtained. In MeCN, the product of bromotriflamidation (with NBS) with solvent interception or of bis-triflamidation (with NIS) is formed. The reaction with trans-stilbene in acetonitrile with NBS gave rise to cyclization to 2-methyl-4,5-diphenyl-1-triflyl-4,5-dihydro-1H-imidazole. In contrast, with NIS as an oxidant, both in CH2Cl2 and MeCN, the major product was 2,3-diphenyl-1-triflylaziridine formed in good yield. With NBS, aziridine is also formed but as a minor product, the major one being a mixture of diastereomers of the product of bromotriflamidation. The reaction of compound 1 with vinylcyclohexane in methylene chloride affords the mixtures of regioisomers of the products of halotriflamidation, whereas in acetonitrile, the products of solvent interception and cyclization to the imidazoline are formed. A mechanism explaining the formation of all isolated products is proposed.
1. Introduction
N-Trifluoromethylsulfonyl (triflyl) substituted nitrogen compounds occupy a unique place in organic chemistry [1,2,3]. The triflyl group is a very strong electron-withdrawing substituent responsible for the high NH-acidity of triflamides, their specific catalytic activity, the ability to form strong intra- and intermolecular hydrogen bonds, and a number of specific chemical properties. Low nucleophilicity and high NH-acidity allow triflamides to be used in a variety of organic reactions [1,2,3].
N-Triflylaziridines are of considerable interest from the viewpoint of their further transformations, allowing us to obtain structures that already contain the triflyl group. They have wide areas of application, such as nucleophilic ring opening to give N-protected alkyleneamines [4] and catalytic enantioselective alkylation of the methine carbon of carboxylic acids [5], which are used in the synthesis of tripodal tetradentate C3-symmetric amines [6]. N-Triflylaziridines are also widely employed in the synthesis of ligands for metal complex catalysis [7,8,9,10,11,12,13,14]. However, the methods of their synthesis meet significant difficulties precisely because of specific properties of the triflyl group.
Prior to our studies, several examples of the synthesis of N-triflylaziridines were described in the literature. One of the first examples studied was the classical reaction of triflylazide with electron-rich O-TMS vinyl ethers [15] (Scheme 1, route 1):
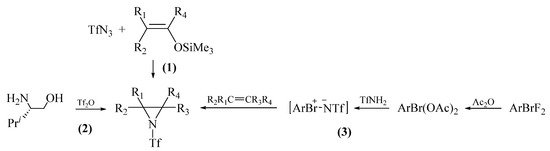
Scheme 1.
The known synthetic routes to N-triflyl aziridines.
The necessity to use an unstable and explosive triflyl azide greatly complicates the reaction. Similar transformations of perfluorosulfonylazides and O-TMS ethers via the formation of N-perfluoromethylsulfonylaziridines give a set of α-[N-(peruoroalkanesulfonyl)]aminoketones and α-amino acids in good yields [16,17]. UV activation of the reaction of butene-2 with the azide H(CF2)2O(CF2)2SO2N3 affords the corresponding aziridine in 61% yield [18].
N-Triflylaziridine was also obtained by treating (2S)-2-amino-3-methylbutan-1-ol with triflic anhydride on strong cooling (Scheme 1, route 2) [19].
One of the most efficient methods for the synthesis of N-triflylaziridines is the reaction of alkenes with N-triflylimino-λ3-bromane 4-CF3C6H4Br=NTf [20,21] prepared in situ or in advance through several steps [21,22] (Scheme 1, route 3). The reagent is formed by the reaction of triflamide with 4-CF3C6H4Br(OAc)2 synthesized, in turn, by treating difluoride 4-CF3C6H4BrF2 with acetic anhydride [21]. Difluoride is obtained by the reaction of 4-CF3C6H4SiMe3 with BrF3 [22]. N-Triflylimino-λ3-bromane is relatively stable and can be stored under argon at 4 °C for several months [21,22]. The reagent can also be obtained directly, bypassing the diacetate formation step, from 4-CF3C6H4BrF2 [23]. Aziridination occurs with high chemoselectivity and in good to excellent yields (75–98%) (Scheme 1, Pathway 2) [20,21]. The scope of substrates included cycloalkenes C6–C8, norbornene, styrenes, and octenes. At the same time, the overall synthesis of N-triflylaziridines in Scheme 1, Pathway 2 involves four separate stages and the use of BrF3, which significantly limits the synthetic applicability of the method.
The addition of N,N-dichloroalkanesulfonamide I(CF2)2O(CF2)2SO2NCl2 to styrene in the presence of EtONa/EtOH affords the corresponding aziridine in 49% yield [24]. A shortcoming of highly reactive N,N-dichloroalkanesulfonamides as reagents is their instability in air moisture and the need for special storage conditions [25].
More stable than triflylimino-λ3-bromane are triflylimino-λ3-iodanes. The N-triflyl-substituted analogs of the latter, N-triflylimino-λ3-iodanes ArI=NTf, were synthesized by the reaction of iodosobenzenes ArI=O with triflamide as late as in 2022 and used for amidation or imidation of phosphines and 1,3-diketones [26]. However, in our recent work [27], we found no aziridines in the reaction of styrene with triflamide and iodosobenzene in the presence of iodine and CuCl.
2. Results and Discussion
Therefore, the synthesis of N-triflylaziridines remains a challenging task. Based on this, we have studied the reactions of alkenes with phenyl-N-triflylimino-λ3-iodane PhI=NTf (1) in different conditions. In methylene chloride, in the absence of oxidants, no reactions occurred with styrene, vinylcyclohexane, dimethyl(divinyl)silane, or tetramethyl(divinyl)siloxane; only the starting reagent 1 was recovered. Given that alkenes readily undergo triflamidation with solvent interception affording N-triflylamidines in the reaction with triflamide in the presence of N-halosuccinimides [27,28,29], we have studied the reactions of compound 1 with styrene in the presence of N-bromo (NBS) and N-iodosuccinimide (NIS). Depending on the solvent, the products of mono and bis-triflamidation of the double bond were obtained. Note that in the products of halotriflamidation 3, 4 the triflamide residue adds to the α-carbon of the styrene molecule (Scheme 2).
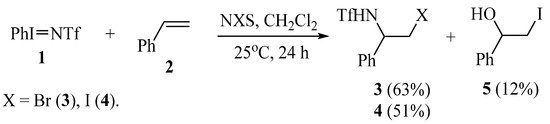
Scheme 2.
Oxidative triflamidation/iodination of styrene with compound 1 in the presence of N-halosuccinimides in methylene chloride.
The structure of the products and, hence, the regioselectivity of addition is proved by the presence of the NH signal at 5.76 (3) and 5.89 ppm (4), multiplets of the methine proton at 4.97 (3) and 4.78 ppm (4), and doublets of doublets at 3.74 (3) and 3.56 (4) of the methylene protons.
Varying the solvent from CH2Cl2 to MeCN changes the course of the reaction; with NBS, N-(2-bromo-1-phenylethyl)-N′-(triflyl)acetimidamide 6 is obtained in moderate yield (Scheme 3).
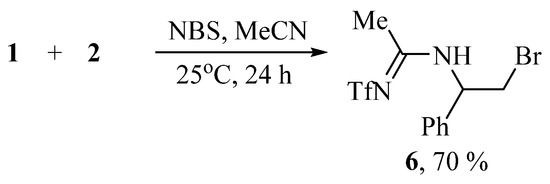
Scheme 3.
Bromotriflamidation of styrene with solvent interception in the presence of NBS.
The structure of amidine 6 was confirmed by the presence of the NH doublet at 7.67 ppm, a doublet of triplets of CHN at 5.34 ppm, two doublets of diastereotopic CH2Br at 3.73 and 3.64 ppm, and the Me group singlet at 2.48 ppm. The 13C NMR spectrum contains the signals at 169 (C=N), 58 (CHN), 34 (CH2Br), and 22.3 (Me). The NMR spectra for compounds 3, 4, 6 is offered in Supplementary Materials (Figures S1–S9).
In the presence of NIS, styrene reacts with N-triflylimino-λ3-iodane 1 in acetonitrile to give the product of bis-triflamidation TfNHCH(Ph)–CH2NHTf 7 in 54% yield, identical to that obtained earlier by the reaction with triflamide in the system t-BuOCl/NaI [30].
In continuation, we examined the reaction of compound 1 with some alkenes in the system I2/NaI/MeCN, which was successfully employed for the synthesis of N-tosylaziridines [31] (vide supra). In contrast, no aziridine was formed with compound 1, but only the aforementioned adduct 7 in a low yield of 25%. Under the same conditions, trans-stilbene and vinylcyclohexane did not react with compound 1. UV activation did not change the course of the reaction and only increased the yield of adduct 7 to 40%.
When the oxidant I2/NaI was replaced by NBS or NIS, the reaction of N-triflylimino-λ3-iodane 1 and trans-stilbene 8 in acetonitrile led to cyclization and formation of 2-methyl-4,5-diphenyl-1-triflyl-4,5-dihydro-1H-imidazole 9 (Scheme 4).
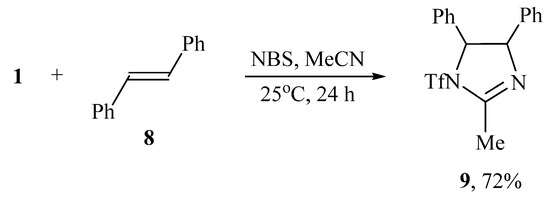
Scheme 4.
Bromotriflamidation with solvent interception and dehydrobrominative cyclization.
The structure and composition of imidazole 9 was confirmed by IR, NMR spectroscopy, and high-resolution mass spectrometry (HRMS), which showed the molecular ion [M + H]+ with m/z 369.08807 corresponding to C17H16F3N2O2S+. When using NIS as the oxidant, imidazole 9 was also formed but as a minor product, the major one being 2,3-diphenyl-1-triflylaziridine 10 isolated in 78% yield (Scheme 5). The formation of aziridine 10 is the first case of the synthesis of N-sulfonylaziridines using N-sulfonylimino-λ3-iodanes as a source of the nitrogen group.
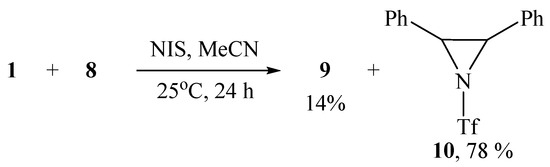
Scheme 5.
Bromotriflamidation with solvent interception and dehydrobrominative cyclization.
The structure of aziridine 10 was proved by IR and NMR spectroscopy and confirmed by HRMS data. In particular, the values of the 1JCH constants in the aziridine moiety are known to be much larger than in saturated aliphatic compounds and lie in the range of 170–180 Hz [32,33,34]. The measured value of 1JCH in product 10 is 177 Hz, which unambiguously proves its structure. The HRMS spectrum shows a molecular ion m/z at 328.06242, corresponding to the molecular formula C15H13F3NO2S+.
Earlier, it was shown that the reaction of styrenes with tosylamide in acetonitrile in the presence of t-BuOI leads to the substituted aziridines [35]. However, no aziridine is formed in the reaction with triflamide under the same conditions, but only the product of bis-triflamidation and disubstituted piperazine, which is, formally, the product of dimerization of the target aziridine [30]. It can be assumed that the formation of piperazine is the result of relative instability of the intermediate 2-phenyl-1-triflylaziridine, because its analog, 2-phenyl-1-tosylaziridine is stable [30]. Presumably, the stability of aziridine 10 is due to the presence of the second phenyl group stabilizing the molecule. In methylene chloride, the NIS-induced reaction also gives aziridine 10 in about the same yield, but with NBS it is the minor product, whereas the major product was identified to be N-(2-bromo-1,2-diphenylethyl)triflamide 11 (Scheme 6).

Scheme 6.
Aziridination and bromotriflamidation of stilbene with N-triflylimino-λ3-iodane 1.
According to the NMR spectroscopy data, compound 11, possessing two chiral carbon atoms, is formed as a mixture of two pairs of diastereomers. Two sets of the NH, CHBr, and CHN signals and two pairs of the CHBr and CHN 13C signals appear in the ratio of 5:1. The NMR and HRMS spectra for compounds 9–11 is offered in Supplementary Materials (Figures S10–S20 and S40–S42).
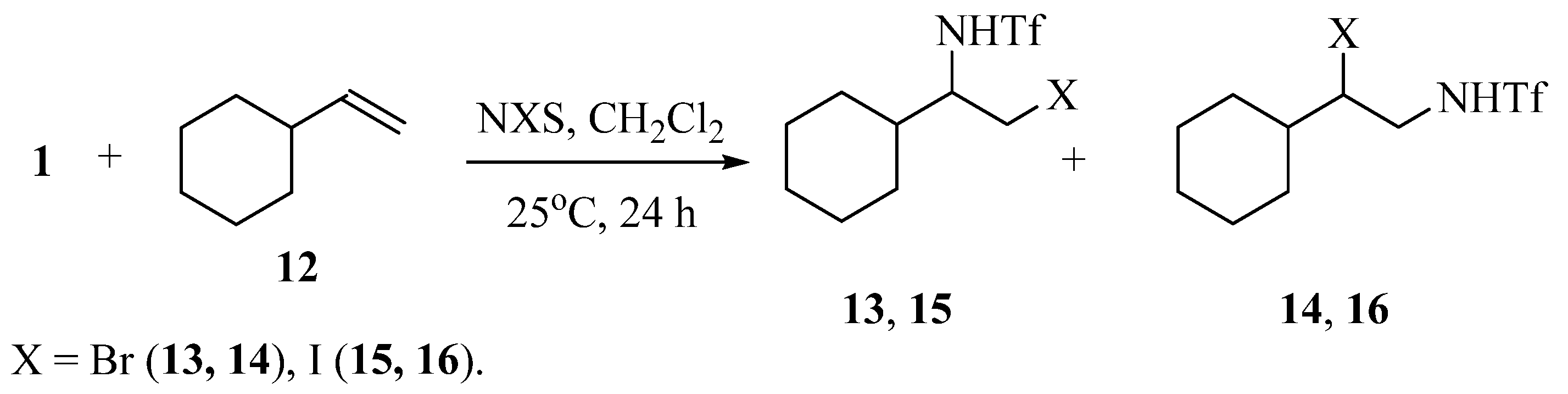
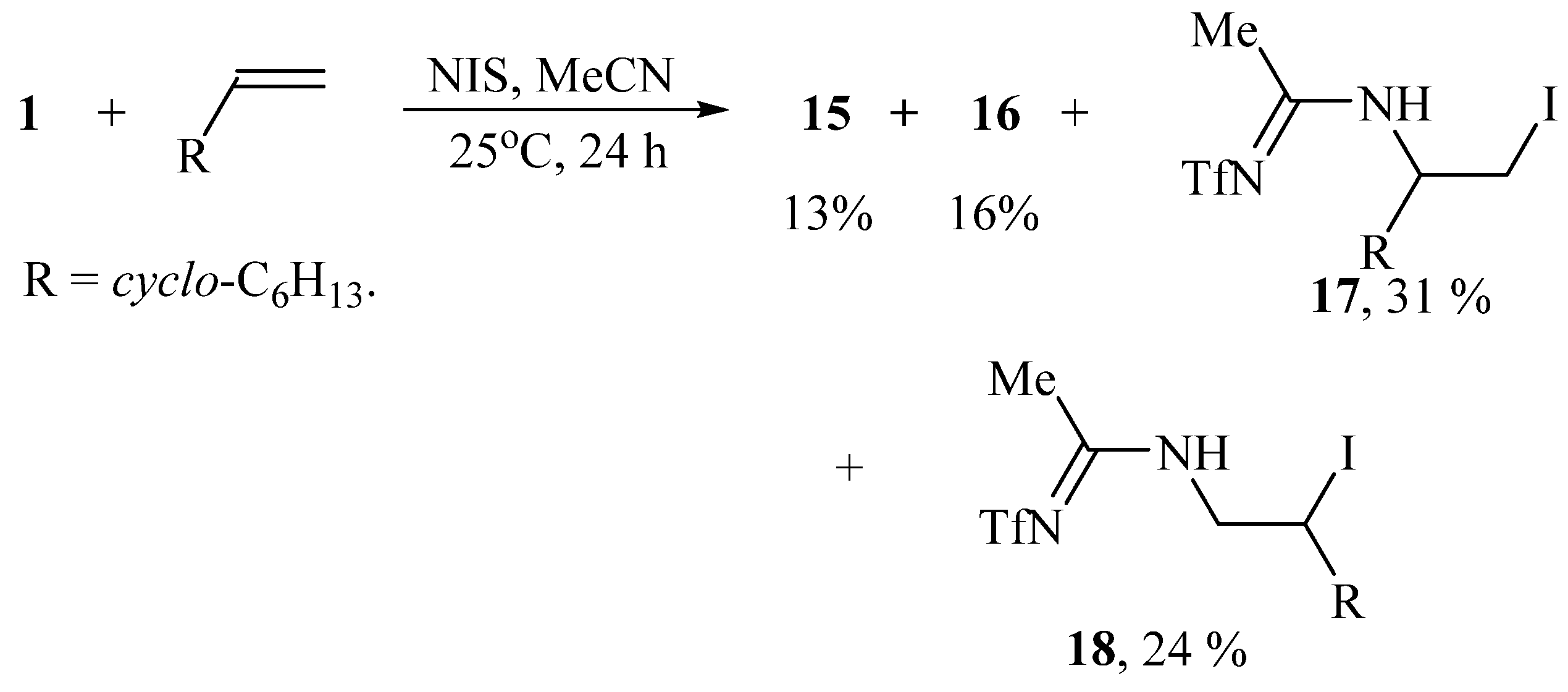
The reaction of vinylcyclohexane 12 with N-triflylimino-λ3-iodane 1 in methylene chloride in the presence of NBS or NIS results in a mixture of the regioisomers of the products of bromo- or iodotriflamidation in the ratio of 1:2 (13:14 = 21:41%) or 1:3 (15:16 = 14:43%) and total yield of 62 or 57% (Scheme 7):
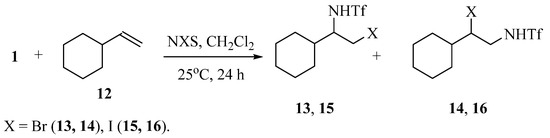
Scheme 7.
Regioselectivity of halotriflamidation of styrene with N-triflylimino-λ3-iodane 1.
The structure of the regioisomers was assigned based on the chemical shifts and multiplicity of the signals in the 1H and 13C NMR spectra. The NH signals in compounds 13 and 15 appear as clearly resolved doublets, whereas in their isomers 14 and 16, they look as an unresolved broad singlet (X = Br) or as a triplet (X = I). In regioisomers 13 and 15 the methine protons of the CH–CH2 spin system resonate in a higher field than the methylene protons, whereas in regioisomers 14 and 16 their relative position is reversed. In the 13C NMR spectra, the most upfield signals belong to the CH2Br (37 ppm) or CH2I carbons (13 ppm).
Replacing the solvent in the reaction in Scheme 7 from methylene chloride to acetonitrile gives rise to the formation of additional products. In the presence of NIS, apart from regioisomers 15 and 16, the isomeric amidines 17 and 18 were isolated as the products of solvent interception (Scheme 8):
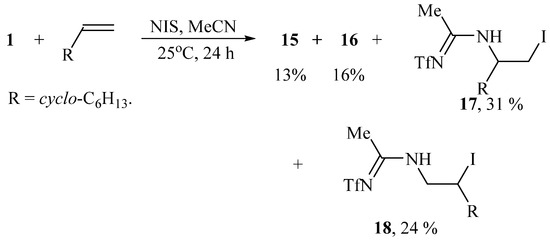
Scheme 8.
Iodotriflamidation without (15, 16) and with solvent interception (17, 18).
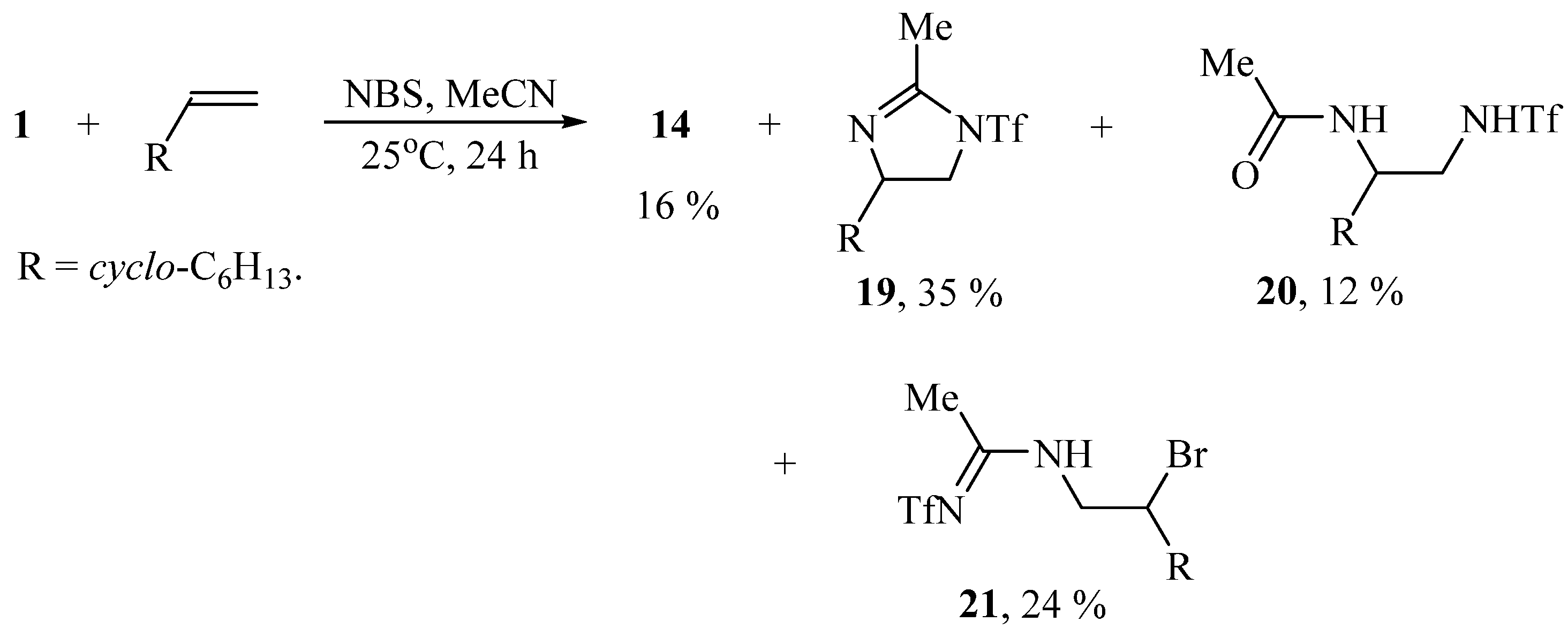
In a similar way the reaction proceeds in the presence of NBS, except for the formation of imidazoline 19 isomeric to the earlier synthesized 5-cyclohexyl-2-methyl-1-triflyl-4,5-dihydro-1H-imidazole [27] and the product of solvent interception/hydrolysis 20 in the total yield of 88% (Scheme 9):
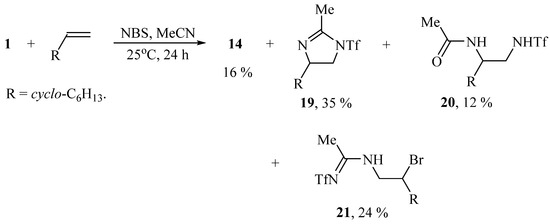
Scheme 9.
The products of the reaction of vinylcyclohexane with iodane 1 and NBS in MeCN.
The NMR and HRMS spectra for compounds 13–19 is offered in Supplementary Materials (Figures S21–S39, S43 and S44).
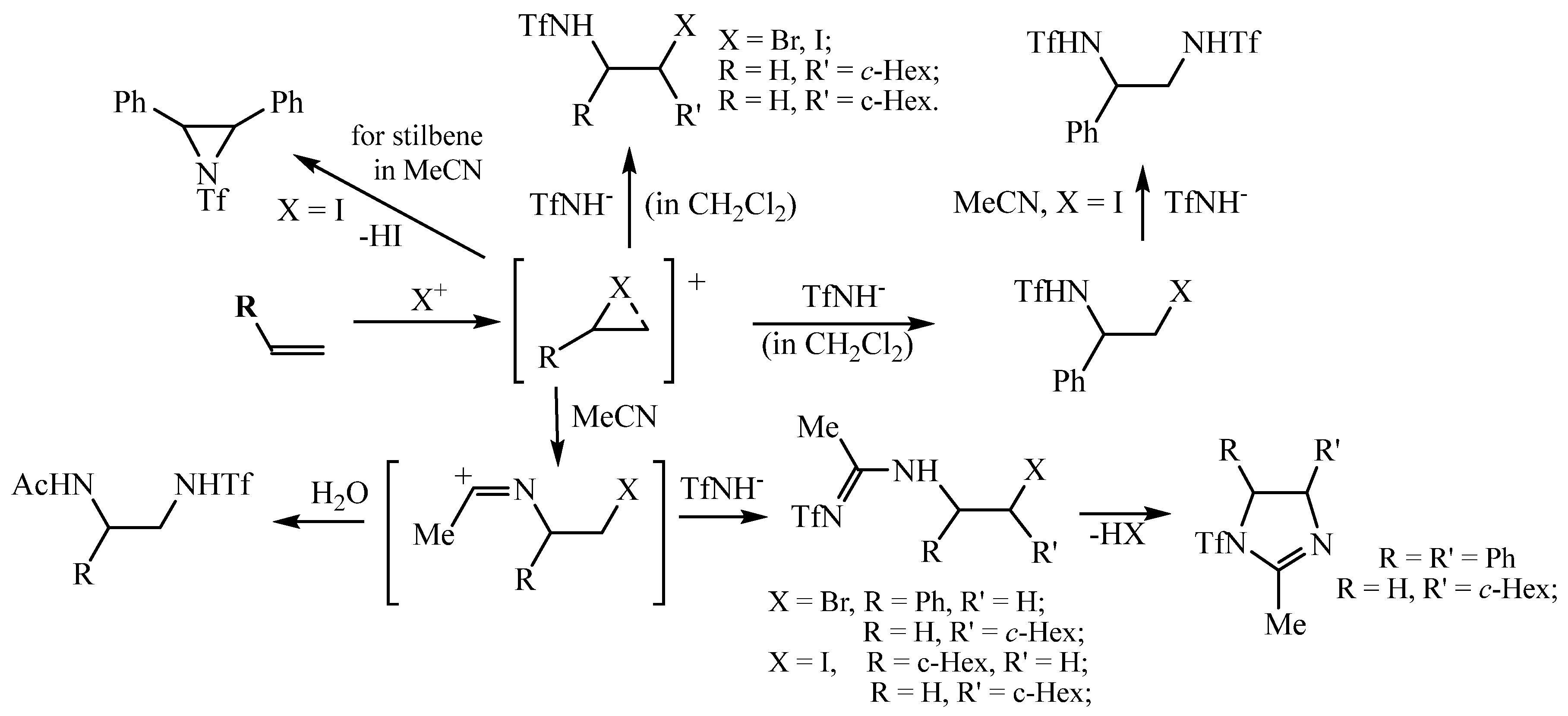
A mechanistic scheme allowing to explain the formation of all products in Scheme 2, Scheme 3, Scheme 4, Scheme 5, Scheme 6, Scheme 7, Scheme 8 and Scheme 9 is presented in Scheme 10. The specific reactivity of triflamide leads to the formation (under the conditions of the reactions) of both N-sulfonylamidines, products containing acetamide and triflamide groups, and products formed without the participation of a solvent as a reagent in the reaction. Since the basicity of acetonitrile (780 kJ/mol) [36] is higher than that of triflamide (740 kJ/mol) [1], acetonitrile is a stronger nucleophile than triflamide, which leads to the formation of amidines (or the corresponding acetamides upon hydrolysis of the corresponding intermediate), as shown in Scheme 10. The use of CH2Cl2 eliminates the involvement of the solvent in the reaction.
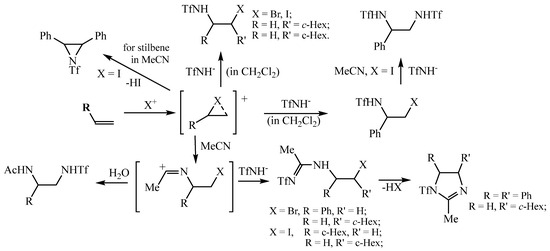
Scheme 10.
A tentative mechanism for the formation of diverse products in the oxidative sulfonamidation of alkenes.
3. Materials and Methods
3.1. General Details
The 1H (400.1 MHz), 13C (100.6 MHz), and 19F (376.0 MHz) NMR spectra were registered on a Bruker DPX-400 spectrometer for 5–10% solutions in CDCl3. 1H and 13C chemical shifts (δ) are reported in parts per million (ppm) relative to the residual solvent peak of CDCl3 (δ = 7.27 (1H) and 77.10 (13C) ppm, respectively). 19F NMR chemical shifts (δ) are given relative to CCl3F. All coupling constants (J) are reported in hertz (Hz). Abbreviations are s, singlet; d, doublet; t, triplet; q, quartet; and brs, broad singlet. The IR spectra (cm−1) were taken on a Bruker Vertex 70 spectrometer. Mass spectra were registered in ESI-TOF-HRMS mode on an Agilent 6210 instrument (Agilent Technologies, Santa Clara, CA, USA). Elemental composition was determined on a Thermo Scientific Flash 2000 CHNS analyzer. Melting points were measured on a Boetius apparatus. TLC was performed on silica 60 plates (0.25 mm, F254, Merck, Rahway, NJ, USA) and visualized by UV lamp. Commercial reagents and solvents were used without further purification unless otherwise mentioned.
3.2. Synthesis of Compounds
3.2.1. Synthesis of Phenyl-N-triflylimino-λ3-iodane 1
Triflamide (0.50 g, 3.4 mmol) was added to a solution of iodosolbenzene (0.75 g, 3.4 mmol) in methylene chloride (10 mL) and stirred for 5 min. Then, the solvent was distilled off to dryness in a vacuum, and the solid white residue was washed with hexane to remove triflamide. The residue was dried in a vacuum to obtain 1.10 g (94%) iminoiodane 1.
3.2.2. Reaction of PhI=NTf 1 with Styrene in the System NXS–CH2Cl2
To 15 mL of freshly distilled CH2Cl2, styrene (0.208 g, 2 mmol) and compound 1 (0.351 g, 1 mmol) were added. The mixture was stirred for 5 min, then NBS (0.178 g, 1 mmol) or NIS (0.225 g, 1 mmol) was added, stirred for 24 h, and the solvent was removed. The residue was washed from unreacted reagents with hexane–ethyl acetate (4:1), the solvent was removed, and the oily residue was purified by column chromatography (silica, 0.06–0.20 mm, Acros Organics) eluting with ether–hexane 1:2 mixture to get N-(2-halo-1-phenylethyl)triflamides 3, 4, and 1:1 mixture to obtain 2-iodo-1-phenylethan-1-ol 5.
N-(2-Bromo-1-phenylethyl)triflamide (3)
Yellow oil, 0.209 g (63%). IR νmax (thin, cm−1): 3303 (NH), 3092, 3067, 3035, 2919, 2852, 2373, 1721, 1602, 1548, 1496, 1455, 1429, 1377, 1231, 1196, 1145, 1067, 1017, 961, 845, 816, 788, 759, 701, 615, 565, 511. 1H NMR (CDCl3): δ 7.40 (m, 3H, Ph), 7.30 (d, J = 7.4 Hz, 2H, Ph), 5.76 (br.s, 1H, NH), 4.97 (q, J = 5.9 Hz, 1H, CHN), 3.78 (dd, J = 10.7, 5.3 Hz, 1H, CH2Br), 3.70 (dd, J = 10.9, 5.8 Hz, 1H, CH2Br). 13C NMR (CDCl3): δ 137.04 (Cu), 129.22 (Cm), 129.13 (Cp), 126.26 (Co), 121.25 (q, J = 321.2 Hz, CF3), 58.99 (CHN), 36.49 (CH2Br). 19F NMR (CDCl3): δF = −77.02. Anal., found: C, 32.60; H, 2.75; N, 4.26; F, 17.20; S, 9.70. Calcd for C9H9BrF3NO2S: C, 32.55; H, 2.73; N, 4.22; F, 17.16; S, 9.65.
N-(2-Iodo-1-phenylethyl)triflamide (4)
Yellow-orange oil, 0.195 g (51%). IR νmax (thin, cm−1): 3297 (NH), 3091, 3066, 3033, 2958, 2920, 1722, 1708, 1693, 1661, 1646, 1628, 1602, 1588, 1547, 1526, 1496, 1455, 1424, 1376, 1270, 1231, 1198, 1143, 1064, 1030, 1003, 945, 915, 878, 841, 761, 699, 652, 609, 588, 508. 1H NMR (CDCl3): δ 7.42 (m, 3H, Ph), 7.29 (d, J = 7.7 Hz, 2H, Ph), 5.89 (d, J = 8.3 Hz, 1H, NH), 4.78 (dt, J = 8.0, 6.5 Hz, 1H, CHN), 3.60 (dd, J = 10.7, 6.1 Hz, 1H, CH2I), 3.53 (dd, J = 10.2, 5.9 Hz, 1H, CH2I). 13C NMR (CDCl3): δ 137.82 (Cu), 129.19 (Cm), 129.05 (Cp), 126.04 (Co), 121.20 (q, J = 321.1 Hz, CF3), 59.33 (CHN), 10.40 (CH2I). 19F NMR (CDCl3): δF −76.95. Anal., found: C, 28.63; H, 2.50; N, 3.73; F, 15.10; S, 8.53. Calcd for C9H9IF3NO2S: C, 28.51; H, 2.39; N, 3.69; F, 15.03; S, 8.46.
2-Iodo-1-phenylethan-1-ol (5)
Violet liquid, 0.06 g (12%). Identical to that in [30].
3.2.3. Reaction of PhI=NTf 1 with Styrene in the System NXS–MeCN
The reaction was performed and treated as above. To the residue, ether was added, kept for 1 h in a fridge, and the precipitated succinimide was filtered off. The ether was removed, and the oily residue was purified by column chromatography eluting with a 1:2 or 2:1 mixture to obtain products 6 and 7.
N-(2-Bromo-1-phenylethyl)-N′-(triflyl)acetimidamide (6)
Yellow oil, 0.263 g (70%). IR νmax (thin, cm−1): 3216 (NH), 3065, 3034, 2954, 2924, 2854, 2649, 2595, 1958, 1885, 1804, 1740, 1654, 1590, 1554, 1495, 1486, 1452, 1432, 1377, 1325, 1229, 1197, 1148, 1093, 1055, 1006, 987, 920, 863, 832, 759, 700, 667, 607, 582, 538, 507. 1H NMR (CDCl3): δ 7.67 (d, J = 7.0 Hz, 1H, NH), 7.35 (m, 3H, Ph), 7.28 (dd, J = 7.4, 2.2 Hz, 2H, Ph), 5.34 (dt, J = 7.7, 5.4 Hz, 1H, CHN), 3.73 (dd, J = 10.7, 8.3 Hz, 1H, CH2Br), 3.64 (dd, J = 11.0, 5.1 Hz, 1H, CH2Br), 2.48 (s, 3H, CH3). 13C NMR (CDCl3): δ 168.71 (C=N), 137.05 (Cu), 129.41 (Cm), 128.94 (Cp), 126.82 (Co), 121.17 (q, J = 320.9 Hz, CF3), 57.67 (CHN), 33.56 (CH2Br), 21.88 (CH3). 19F NMR (CDCl3): δF −79.14. Anal., found: C, 35.46; H, 3.30; N, 7.60; F, 15.30; S, 8.70. Calcd for C10H15BrF3NO3S: C, 35.40; H, 3.24; N, 7.51; F, 15.27; S, 8.59.
N,N′-(1-Phenylethane-1,2-diyl)bis(triflamide) (7)
White powder, 0.215 g (54%). Identical to that described earlier [30].
3.2.4. Reaction of PhI=NTf 1 with Styrene in the System I2–NaI–MeCN
To 15 mL of freshly distilled MeCN, styrene (0.208 g, 2 mmol) and PhI=NTf (0.351 g, 1 mmol) were added, stirred for 5 min, and added I2 (25 mg, 0.1 mmol) and NaI (8 mg, 0.05 mmol). The mixture was stirred for 24 h and poured into aqueous solution of Na2S2O3. The obtained solution was extracted with ether, dried over anhydrous MgSO4, and ether removed in a vacuum. The oily residue was purified by column chromatography eluting with ether-hexane 1:2 to obtain product 7 (0.126 g, 32%).
3.2.5. Reaction of PhI=NTf 1 with Stilbene in the System NXS-MeCN
To 10 mL of freshly distilled MeCN, stilbene (0.180 g, 1 mmol) and PhI=NTf (0.351 g, 1 mmol) were added, stirred for 5 min, and NBS (0.178 g, 1 mmol) or NIS (0.225 g, 1 mmol) was added. The mixture was stirred for 24 h, the solvent removed, the residue was mixed with ether, kept for 1 h in a fridge, and the precipitated succinimide was filtered off. The ether was evaporated, and the oily residue was purified by column chromatography, eluting with hexane to obtain 2,3-diphenyl-1-(triflyl)aziridine 10 and with the mixture ether-hexane 1:2 to obtain 2-methyl-4,5-diphenyl-1-triflyl-4,5-dihydro-1H-imidazole 9.
2-Methyl-4,5-diphenyl-1-((trifluoromethyl)sulfonyl)-4,5-dihydro-1H-imidazole (9)
Colorless oil, 0.264 g (72%). IR νmax (thin, cm−1): 3373, 3308, 3170, 3089, 3066, 3034, 2919, 2852, 1957, 1664, 1603, 1587, 1549, 1496, 1455, 1434, 1405, 1386, 1364, 1227, 1213, 1198, 1153, 1096, 1079, 1062, 1015, 981, 951, 915, 786, 759, 698, 645, 607, 582, 534. 1H NMR (CDCl3): δ 7.41 (m, 6H, Ph), 7.9 (d, J = 7.0 Hz, Ph), 7.17 (d, J = 7.0 Hz, 2H, Ph), 5.12 (d, J = 2.3 Hz, 1H, CHN), 5.06 (d, J = 2.9 Hz, 1H, CHN), 2.56 (s, 3H, CH3). 13C NMR (CDCl3): δ 154.73 (C=N), 140.36 (C′i), 139.98 (Ci), 129.36 (C′m), 129.21 (C′o), 129.03 (C′p) 128.48 (Cp), 126.23 (Cm), 125.91 (Co), 121.40 (q, CF3, J = 323.3 Hz), 77.63 (CHN), 72.75 (CHN), 16.61 (CH3). 19F NMR (CDCl3): δF −79.05. HRMS (ESI): m/z: [M + H]+ calcd for C17H16F3N2O2S+: 369.08845; found: 369.08807.
2,3-Diphenyl-1-(triflyl)aziridine (10)
Colorless oil, 0.254 g (78%). IR νmax (thin, cm−1): 3305, 3087, 3063, 3033, 2983, 2922, 2852, 2362, 1957, 1888, 1810, 1726, 1657, 1600, 1579, 1540, 1495, 1453, 1406, 1378, 1317, 1279, 1226, 1201, 1148, 1071, 1028, 1002, 943, 916, 871, 797, 749, 698, 638, 613, 513. 1H NMR (CDCl3): δ 7.37 (m, 5H, Ph), 3.88 (s, 1H, CH). 13C NMR (CDCl3): δ 137.22 (Ci), 128.65 (Cm), 128.40 (Cp), 125.60 (Co), 121.36 (q, CF3, J = 307.4 Hz), 62.91 (d, CHN, J = 177.7 Hz). 19F NMR (CDCl3): δF = −75.99. Anal., found: C, 35.46; H, 3.30; N, 7.60; F, 15.30; S, 8.70. Calcd for C10H15BrF3NO3S, %: C, 35.40; H, 3.24; N, 7.51; F, 15.27; S, 8.59. HRMS (ESI): m/z: [M + H]+ calcd for C15H13F3NO2S+: 328.06191; found: 328.06242.
3.2.6. Reaction of PhI=NTf 1 with Stilbene in the System NBS(NIS)-CH2Cl2
The reaction was performed and treated as above. After the removal of the solvent, the solid residue was washed with the mixture hexane–ethyl acetate (4:1) to separate the unreacted compounds. The liquid residue was kept in a vacuum, the oily residue was purified by column chromatography eluting with hexane to obtain 2,3-diphenyl-1-triflylaziridine 10, and with a mixture of ether-hexane (1:2) to isolate N-(2-bromo-1,2-diphenylethyl)triflamide 11.
N-(2-Bromo-1,2-diphenylethyl)triflamide (11)
Colorless oil, 0.220 g (45%). IR νmax (thin, cm−1): 3282 (NH), 3108, 3090, 3066, 3011, 2982, 2922, 2854, 1961, 1885, 1807, 1716, 1601, 1588, 1496, 1455, 1367, 1277, 1228, 1197, 1076, 1030, 1002, 972, 940, 916, 866, 818, 762, 698, 613. 1H NMR (CDCl3): δ 7.31 (m, 5H, Ph), 7.09 (m, 2H, Ph), 7.01 (m, 2H, Ph), 5.66 (d, J = 9.5 Hz, 1H, NH) 5.35 (d, J = 5.0 Hz, 1H, CHBr), 5.03 (dd, J = 9.5, 5.0 Hz, 1H, CHN). 13C NMR (CDCl3): δ 135.78 (Ci), 135.77 (Ci′), 129.29 (Cp), 129.00 (Cp′), 128.84 (Cm), 128.63 (Co), 128.41 (Cm′), 127.59 (Co′), 119.48 (q, CF3, J = 321.4 Hz), 64.30 (CHBr), 58.08 (CHN). 19F NMR (CDCl3): δF = −77.10. HRMS (ESI): m/z: [M–Br]+ calcd for C15H13F3NO2S+: 328.061908; found: 328.062180.
3.2.7. Reaction of PhI=NTf 1 with Vinylcyclohexane in the System NBS(NIS)-CH2Cl2
The reaction was performed and treated as above. The solid residue was washed with hexane–ethyl acetate (4:1) to separate unreacted reagents. The solvent was evaporated, and the oily residue was purified by column chromatography eluting with ether-hexane (1:2) to obtain the mixture of regioisomers 13 and 14, or 15 and 16.
N-(2-Bromo-1-cyclohexylethyl)triflamide (13)
Colorless oil, 0.070 g (21%). IR νmax (thin, cm−1): 3321, 3062, 2926, 2855, 1656, 1516, 1444, 1430, 1379, 1225, 1196, 1148, 1080, 1051, 1024, 993, 956, 914, 893, 854,791, 761, 644, 604, 564. 1H NMR (CDCl3): δ 5.15 (d, J = 8.5 Hz, 1H, NH), 3.65 (d, J = 2.8 Hz, 1H, CH2Br), 3.61 (d, J = 3.7 Hz, 1H, CH2Br), 3.43 (m, 1H, CHN), 1.74–1.57 (m, 5H), 1.29–1.11 (m, 6H). 13C NMR (CDCl3): δ 119.69 (q, CF3, J = 320.5 Hz), 60.12 (CHN), 39.54 (CH), 36.07 (CH2Br), 29.23, 28.90, 25.87, 25.74, 25.71 (CH2). 19F NMR (CDCl3): δF −77.23.
N-(2-Bromo-2-cyclohexylethyl)triflamide (14)
Colorless oil, 0.140 g (41%). IR νmax (thin, cm−1): 3326, 3069, 2926, 2855, 1706, 1656, 1516, 1448, 1431, 1374, 1229, 1195, 1149, 1080, 1050, 1025, 993,958, 917, 892, 851,791, 761, 644, 605, 562. 1H NMR (CDCl3): δ 5.46 (br.s, 1H, NH), 3.99 (dt, J = 8.9, 4.0 Hz, 1H, CHBr), 3.73 (dd, J = 14.4, 3.1 Hz, 1H, CH2N), 3.54 (dd, J = 14.4, 9.2 Hz, 1H, CH2N), 1.85–1.72 (m, 5H), 1.29–1.14 (m, 6H). 13C NMR (CDCl3): δ 121.62 (q, CF3, J = 318.8 Hz), 62.34 (CHBr), 48.72 (CH2N), 42.05 (CH), 30.67, 29.85, 26.01, 25.90, 25.80 (CH2). 19F NMR (CDCl3): δF −77.11. Anal., found: C, 31.98; H, 4.50; N, 4.17; F, 16.90; S 9.51. Calcd for C9H15BrF3NO2S: C, 31.96; H, 4.47; N, 4.14; F, 16.85; S, 9.48.
N-(1-Cyclohexyl-2-iodoethyl)triflamide (15)
Light-brown oil, 0.055 g (14%). IR νmax (thin, cm−1): 3303, 2928, 2855, 2798, 2341, 1719, 1429, 1378, 1233, 1192, 1144, 1098, 1071, 1025, 991, 952, 920, 890, 849, 783, 632, 607, 577. 1H NMR (CDCl3): δ 4.91 (d, J = 8.9 Hz, 1H, NH), 3.45 (d, J = 3.4 Hz, 2H, CH2I), 2.92 (dq, J = 8.3, 3.4 Hz, 1H, CHN), 1.72–1.63 (m, 5H), 1.33–1.14 (m, 6H). 13C NMR (CDCl3): δ 119.63 (q, CF3, J = 320.9 Hz), 59.34 (CHN), 41.40 (CH), 29.10, 28.55, 25.85, 25.68 (CH2), 12.07 (CH2I). 19F NMR (CDCl3): δF −77.18.
N-(2-Cyclohexyl-2-iodoethyl)triflamide (16)
Light-brown oil, 0.165 g (43%). IR νmax (thin, cm−1): 3308, 2931, 2857, 2364, 1733, 1448, 1372, 1233, 1193, 1144, 1096, 1080, 1041, 993, 954, 920, 890, 851, 783, 632, 607, 577. 1H NMR (CDCl3): δ 5.30 (t, J = 6.3 Hz, 1H, NH), 4.10 (dt, J = 8.3, 4.8 Hz, 1H, CHI), 3.63 (t, J = 4.9 Hz, 2H, CH2N), 1.83–1.74 (m, 5H), 1.34–1.08 (m, 6H). 13C NMR (CDCl3): δ 119.79 (q, CF3, J = 321.1 Hz), 50.01 (CHI), 44.94 (CH2N), 42.01 (CH), 32.42, 31.67, 26.05, 25.76 (CH2). 19F NMR (CDCl3): δF −77.02. Anal., found: C, 28.10; H, 3.95; N, 3.67; F, 14.82; S 8.35. Calcd for C9H15IF3NO2S: C, 28.06; H, 3.93; N, 3.64; F, 14.80; S, 8.32.
3.2.8. Reaction of PhI=NTf 1 with Vinylcyclohexane in the System NIS-MeCN
The reaction was performed and treated as above. The residue after separation of succinimide and evaporation of ether was purified by column eluting with hexane to obtain a mixture of regioisomers 15 and 16 (50 mg), then with ether-hexane (4:1), to obtain the regioisomers of amidines 17 and 18.
N-(1-Cyclohexyl-2-iodoethyl)-N′-(triflyl)acetimidamide (17)
Brown liquid, 0.133 g (31%). IR νmax (thin, cm−1): 3299, 3065, 2932, 2857, 2672, 1655, 1550, 1440, 1420, 1380, 1301, 1280, 1232, 1198, 1145, 1072, 1050, 1022, 991, 964, 957, 891, 852, 787, 645, 608. 1H NMR (CDCl3): δ 6.64 (d, J = 8.8 Hz, 1H, NH), 4.09 (dt, J = 7.6, 3.4 Hz, 1H, CHN), 3.44 (dd, J = 10.8, 3.6 Hz, 1H, CH2I), 3.30 (dd, J = 10.9, 5.4 Hz, 1H, CH2I), 2.51 (s, 3H, CH3), 1.89–1.61 (m, 6H), 1.35–1.07 (m, 5H). 13C NMR (CDCl3): δ 169.02 (C=N), 119.77 (q, CF3, J = 319.2 Hz), 56.76 (CHN), 41.56 (CH), 32.21, 31.70, 25.68, 25.65 (CH2), 22.15 (CH3), 8.85 (CH2I). 19F NMR (CDCl3): δF −78.86. Anal., found: C, 31.04; H, 4.29; N, 6.59; F, 13.39; S, 7.53. Calcd. for C11H18F3IN2O2S: C, 31.00; H, 4.26; N, 6.57; F, 13.37; S, 7.52.
N-(2-Cyclohexyl-2-iodoethyl)-N′-((trifluoromethyl)sulfonyl)acetimidamide (18)
Brown liquid, 0.10 g (24%). IR νmax (thin, cm−1): 3306, 3069, 2932, 2857, 2672, 1732, 1655, 1518, 1449, 1430, 1377, 1304, 1279, 1232, 1196, 1147, 1079, 1050, 1030, 994, 962, 919, 893, 853, 789, 645, 608. 1H NMR (CDCl3): δ 6.47 (br.s, 1H, NH), 4.20 (dt, J = 9.7, 4.0 Hz, 1H, CHI), 3.94 (ddd, J = 14.5, 6.4, 3.6 Hz, 1H, CH2N), 3.55 (ddd, J = 14.5, 9.7, 4.9 Hz, 1H, CH2N), 2.52 (s, 3H, CH3), 1.85–1.62 (m, 6H), 1.35–1.09 (m, 5H). 13C NMR (CDCl3): δ 168.59 (C=N), 119.88 (q, CF3, J = 320.6 Hz), 48.65 (CH2N), 43.82 (CH), 43.17 (CHI), 32.05, 32.03, 26.09, 25.91 (CH2), 22.47 (CH3). 19F NMR (CDCl3): δF −79.07. HRMS (ESI): m/z: [M–I]+ calcd for C11H18F3N2O2S +: 299.104109; found: 299.104370.
3.2.9. Reaction of PhI=NTf 1 with Vinylcyclohexane in the System NBS-MeCN
The reaction was performed and treated as above. The residue after solvent removal was poured with ether, kept for 1 h in a fridge, and the precipitated succinimide was filtered off. The ether was evaporated, and the oily residue was purified by column eluting with hexane to afford 14 (0.05 g), then with ether-hexane (1:2) to obtain 20, then with ether-hexane (2:1) to obtain 19 and, finally, with ether-hexane (4:1) to get 21.
4-Cyclohexyl-2-methyl-1-triflyl-4,5-dihydro-1H-imidazole (19)
White powder, 0.105 g (35%). Mp = 163.6 °C. IR νmax (thin, cm−1): 3013, 2933, 2637, 2588, 1675, 1638, 1483, 1449, 1415, 1378, 1271, 1235, 1207, 1156, 1092, 1047, 1030, 979, 896, 864, 843, 772, 685, 619, 580, 531. 1H NMR (CDCl3): δ 4.51 (dd, J = 15.2, 10.7 Hz, 2H, CH2N), 4.08 (dd, J = 15.6, 12.8 Hz, 1H, CHN), 2.89 (s, 3H, CH3), 1.88–1.59 (m, 7H), 1.34–1.11 (m, 4H). 13C NMR (CDCl3): δ 168.95 (C=N), 118.59 (q, J = 323.6 Hz, CF3), 63.14 (CHN), 52.81 (CH2N), 41.03 (CH), 28.36, 27.93, 25.64, 25.76, 25.30 (CH2), 14.73 (CH3). 19F NMR (CDCl3): δF −74.13. HRMS (ESI): m/z: [M + H]+ calcd for C11H18F3N2O2S+: 299.104109; found: 299.10376.
N-(1-cyclohexyl-2-(triflamidoethyl)acetamide (20) (white powder, 0.03 g (12%) and N-(2-bromo-2-cyclohexylethyl)-N′-(triflyl)acetimidamide (21) (yellow liquid, 0.092 g (24%) are identical to the obtained and characterized earlier [27].
4. Conclusions
To summarize, the reactions of alkenes with phenyl-N-triflylimino-λ3-iodane, which has only recently been introduced in organic synthesis, in the presence of N-halosuccinimides as oxidants, have been investigated. Depending on the substrate, solvent, and oxidant, a variety of linear and heterocyclic products of triflamidation were obtained. Thus, in methylene chloride, the products of halotriflamidation were obtained with styrene and vinylcyclohexane, and the regioselectivity of the reaction was determined. In acetonitrile, the solvent competes with weak triflamide anion in addition to the C=C double bond and leads to the formation of amidines. The reaction with stilbene was shown to be the first example of the synthesis of N-triflylaziridines using N-triflylimino-λ3-iodane. Remarkably, in the reaction with NIS, N-triflylaziridine is formed in equally good yield in methylene chloride and in acetonitrile, while with NBS it is formed in much lower yield and only in methylene chloride; in acetonitrile, N-triflylimidazoline was obtained in a good yield as a result of bromotriflamidation followed by dehydrobromination with cyclization. A tentative mechanism starting with the addition of electrophilic halogen to alkene is proposed that accounts for the formation of all observed products.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms242115947/s1.
Author Contributions
Conceptualization, A.S.G., M.Y.M., B.A.S.; methodology, A.S.G., M.Y.M.; investigation, A.S.G.; writing—original draft preparation, A.S.G., M.Y.M.; writing—review and editing, B.A.S.; supervision, M.Y.M.; project administration, M.Y.M.; funding acquisition, B.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Russian Science Foundation (RSF Grant no. 22-13-00036).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
Spectral measurements were performed on the equipment of the Baikal Center for Collective Use, SB RAS. We thank A.V. Kuzmin for HRMS analysis performed on the Research Facilities for Physical and Chemical Ultramicroanalysis, Limnological Institute, SB RAS.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shainyan, B.A.; Tolstikova, L.L. Trifluoromethanesulfonamides and Related Compounds. Chem. Rev. 2013, 113, 699–733. [Google Scholar] [CrossRef] [PubMed]
- Moskalik, M.Y.; Astakhova, V.V. Triflamides and Triflimides: Synthesis and Applications. Molecules 2022, 27, 5201. [Google Scholar] [CrossRef] [PubMed]
- Shainyan, B.A. Recent trends in the chemistry of triflamides. Tetrahedron 2023, 147, 133662. [Google Scholar] [CrossRef]
- Moss, T.A.; Barber, D.M.; Kyle, A.F.; Dixon, D.J. Catalytic Asymmetric Alkylation Reactions for the Construction of Protected Ethylene-Amino and Propylene-Amino Motifs Attached to Quaternary Stereocentres. Chem. Eur. J. 2013, 19, 3071–3081. [Google Scholar] [CrossRef]
- Moss, T.A.; Fenwick, D.R.; Dixon, D.J. Enantio- and Diastereoselective Catalytic Alkylation Reactions with Aziridines. J. Am. Chem. Soc. 2008, 130, 10076–10077. [Google Scholar] [CrossRef]
- Cernerud, M.; Adolfsson, H.; Moberg, C. C3-symmetric tripodal tetra-amines—Preparation from chiral amino alcohols via aziridines. Tetrahedron Asymmetry 1997, 8, 2655–2662. [Google Scholar] [CrossRef]
- Pandey, P.; Bera, J.K. Hydrosilylative reduction of primary amides to primary amines catalyzed by a terminal [Ni–OH] complex. Chem. Commun. 2021, 57, 9204–9207. [Google Scholar] [CrossRef]
- Sarbajna, A.; Pandey, P.; Rahaman, S.M.W.; Singh, K.; Tyagi, A.; Dixneuf, P.H.; Bera, J.K. A Triflamide-Tethered N-Heterocyclic Carbene–Rhodium(I) Catalyst for Hydroalkoxylation Reactions: Ligand-Promoted Nucleophilic Activation of Alcohols. ChemCatChem 2017, 9, 1397–1401. [Google Scholar] [CrossRef]
- Liu, Z. (S)-N-Trifluoromethylsulfonyl-2-isopropylaziridine. In Encyclopedia of Reagents for Organic Synthesis (EROS); Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Duncan, A.P.; Leighton, J.L. Enantioselective Cu-Catalyzed Conjugate Addition of Diethylzinc to Acyclic Aliphatic Enones. Org. Lett. 2004, 6, 4117–4119. [Google Scholar] [CrossRef]
- Nelson, S.G.; Zhu, C.; Shen, X. Catalytic Asymmetric Acyl Halide−Aldehyde Cyclocondensation Reactions of Substituted Ketenes. J. Am. Chem. Soc. 2004, 126, 14–15. [Google Scholar] [CrossRef]
- Krauss, I.J.; Leighton, J.L. Highly Practical and Enantioselective Cu-Catalyzed Conjugate Addition of Alkylzinc Reagents to Cyclic Enones at Ambient Temperature. Org. Lett. 2003, 5, 3201–3203. [Google Scholar] [CrossRef] [PubMed]
- Lake, F.; Moberg, C. Sulfonamide Ligands from Chiral Aziridines—Application to the Titanium-Mediated Addition of Diethylzinc to Benzaldehyde. Eur. J. Org. Chem. 2002, 2002, 3179–3188. [Google Scholar] [CrossRef]
- Cernerud, M.; Skrinning, A.; Bérgère, I.; Moberg, C. Bis(ethylsulfonamide)amines via nucleophilic ring-opening of chiral aziridines. Application to Ti-mediated addition of diethylzinc to benzaldehyde. Tetrahedron Asymmetry 1997, 8, 3437–3441. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, S. A new route to fluorine-containing aziridines and α-amino esters. Tetrahedron 2001, 57, 669–674. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, G.; Zhu, S.; Zhu, G.; Jia, Y.; Huang, Q. Reactions of fluoroalkanesulfonyl azides with trimethylsilyl enol ethers. J. Fluor. Chem. 1999, 96, 79–85. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, S. A Novel Method for the Synthesis of N-Per(poly)fluoroalkanesulfonyl Amino Acids. Synthesis 2001, 2001, 0690–0692. [Google Scholar] [CrossRef]
- Zhu, S. Synthesis of fluoroalkanesulfonyl azides and their reactions as fluoroalkanesulfonyl nitrene precursors. Tetrahedron Lett. 1992, 33, 6503–6504. [Google Scholar]
- Nelson, S.G.; Mills, P.M. Catalytic Asymmetric Acyl Halide-Aldehyde Cyclocondensation Reaction. Org. Synth. 2005, 82, 170–178. [Google Scholar]
- Ochiai, M.; Miyamoto, K.; Hayashi, S.; Nakanishi, W. Hypervalent N-sulfonylimino-λ3-bromane: Active nitrenoid species at ambient temperature under metal-free conditions. Chem. Commun. 2010, 46, 511–521. [Google Scholar] [CrossRef]
- Hoque, M.M.; Miyamoto, K.; Tada, N.; Shiro, M.; Ochiai, M. Synthesis and Structure of Hypervalent Diacetoxybromobenzene and Aziridination of Olefins with Imino-λ3-bromane Generated in Situ under Metal-Free Conditions. Org. Lett. 2011, 13, 5428–5431. [Google Scholar] [CrossRef]
- Ochiai, M.; Nishi, Y.; Goto, S.; Shiro, M.; Frohn, H.J. Synthesis, Structure, and Reaction of 1-Alkynyl(aryl)-λ3-bromanes. J. Am. Chem. Soc. 2003, 125, 15304–15305. [Google Scholar] [CrossRef]
- Ochiai, M.; Kaneaki, T.; Tada, N.; Miyamoto, K.; Chuman, H.; Shiro, M.; Hayashi, S.; Nakanishi, W. A New Type of Imido Group Donor: Synthesis and Characterization of Sulfonylimino-λ3-bromane that Acts as a Nitrenoid in the Aziridination of Olefins at Room Temperature under Metal-Free Conditions. J. Am. Chem. Soc. 2007, 129, 12938–12939. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.-Z. Synthesis and reactions of fluoroalkanesulfonyl azides and N,N-dichlorofluoroalkanesulfonamides. J. Chem. Soc. Perkin Transs 1 1994, 15, 2077–2081. [Google Scholar] [CrossRef]
- Zhu, S.-Z.; Zhou, C.-M.; Li, A.-W.; Xu, B. Synthesis and reactions of N,N-dichloroperfluoroalkanesulfonylamides. J. Fluor. Chem. 1994, 67, 7–8. [Google Scholar] [CrossRef]
- Sunagawa, S.; Morisaki, F.; Baba, T.; Tsubouchi, A.; Yoshimura, A.; Miyamoto, K.; Uchiyama, M.; Saito, A. In Situ Generation of N-Triflylimino-λ3-iodanes: Application to Imidation of Phosphines and Catalytic α-Amidation of 1,3-Dicarbonyl Compounds. Org. Lett. 2022, 24, 5230–5234. [Google Scholar] [CrossRef] [PubMed]
- Moskalik, M.Y.; Garagan, I.A.; Astakhova, V.V.; Sterkhova, I.V.; Shainyan, B.A. Solvent-dependent oxidative triflamidation of alkenes and N(O)-Heterocyclization of the products. Tetrahedron 2021, 88, 132145. [Google Scholar] [CrossRef]
- Ganin, A.S.; Moskalik, M.Y.; Astakhova, V.V.; Sterkhova, I.V.; Shainyan, B.A. Heterocyclization and solvent interception upon oxidative triflamidation of allyl ethers, amines and silanes. Tetrahedron 2020, 76, 131374. [Google Scholar] [CrossRef]
- Ganin, A.S.; Moskalik, M.Y.; Garagan, I.A.; Astakhova, V.V.; Shainyan, B.A. Triflamidation of Allyl-Containing Substances:Unusual Dehydrobromination vs. Intramolecular Heterocyclization. Molecules 2022, 27, 6910. [Google Scholar] [CrossRef]
- Shainyan, B.A.; Moskalik, M.Y.; Starke, I.; Schilde, U. Formation of unexpected products in the attempted aziridination of styrene with trifluoromethanesulfonyl nitrene. Tetrahedron 2010, 66, 8383–8386. [Google Scholar] [CrossRef]
- Kiyokawa, K.; Kosaka, T.; Minakata, S. Metal-Free Aziridination of Styrene Derivatives with Iminoiodinane Catalyzed by a Combination of Iodine and Ammonium Iodide. Org. Lett. 2013, 15, 4858–4861. [Google Scholar] [CrossRef]
- Knipe, A.C.; Khandelwal, Y.; McAuley, I.E.; Brown, N.M.D. Substituent effects on 13C chemical shifts of 1-arylsulphonyl-2-arylaziridines. Magn. Res. Chem. 1985, 23, 177–180. [Google Scholar] [CrossRef]
- Forni, A.; Moretti, I.; Mucci, A.; Prati, F.; Schenetti, L. Invertomers at nitrogen in aziridine carboxylates by mltltinuclear (1H, 13C, 17O, and 15N) NMR study. Chem. Het. Comp. 1995, 31, 1071–1078. [Google Scholar] [CrossRef]
- Joly, G.J.; Peeters, K.; Mao, H.; Brossette, T.; Hoornaert, G.J.; Compernolle, F. Use of the N-tosyl-activated aziridine 1,2-dideoxy-1,2-iminomannitol as a synthon for 1-deoxymannojirimycin analogues. Tetrahedron Lett. 2000, 41, 2223–2226. [Google Scholar] [CrossRef]
- Minakata, S.; Morino, Y.; Oderaotoshi, Y.; Komatsu, M. Novel aziridination of olefins: Direct synthesis from sulfonamides using t-BuOI. Chem. Commun. 2006, 31, 3337–3339. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.I.; Denault, J.W.; Cooks, R.G. Proton affinity of deuterated acetonitrile estimated by the kinetic method with full entropy analysis. Dedicated to Professor N.M.M. Nibbering on the occasion of his retirement and in recognition of his many contributions to gas-phase ion chemistry, his leadership in mass spectrometry, and his friendship. Int. J. Mass Spectr. 2001, 210–211, 133–146. [Google Scholar]
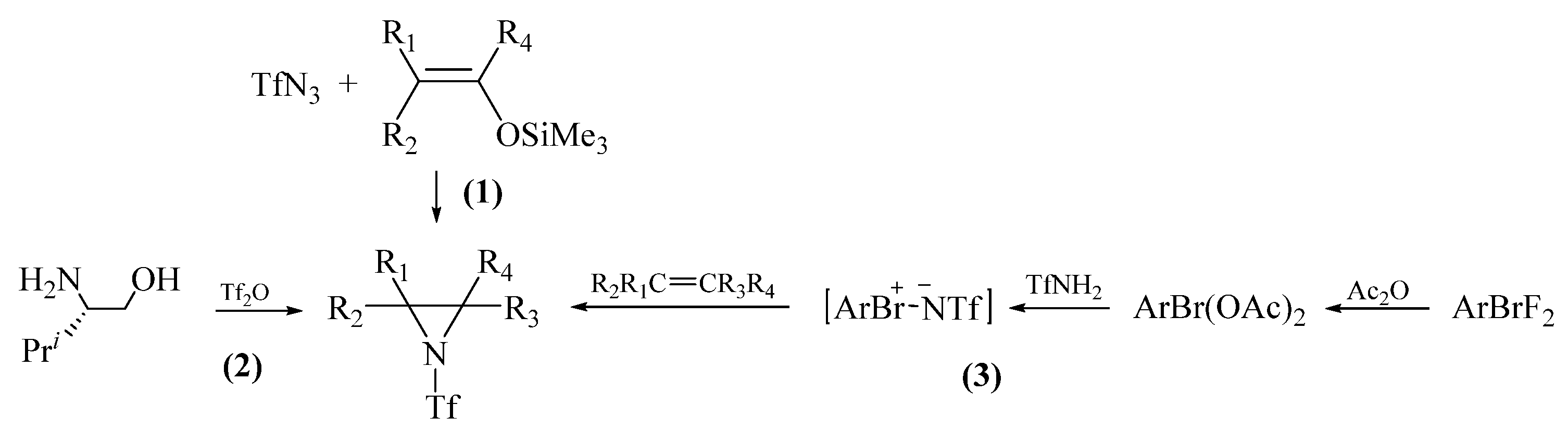
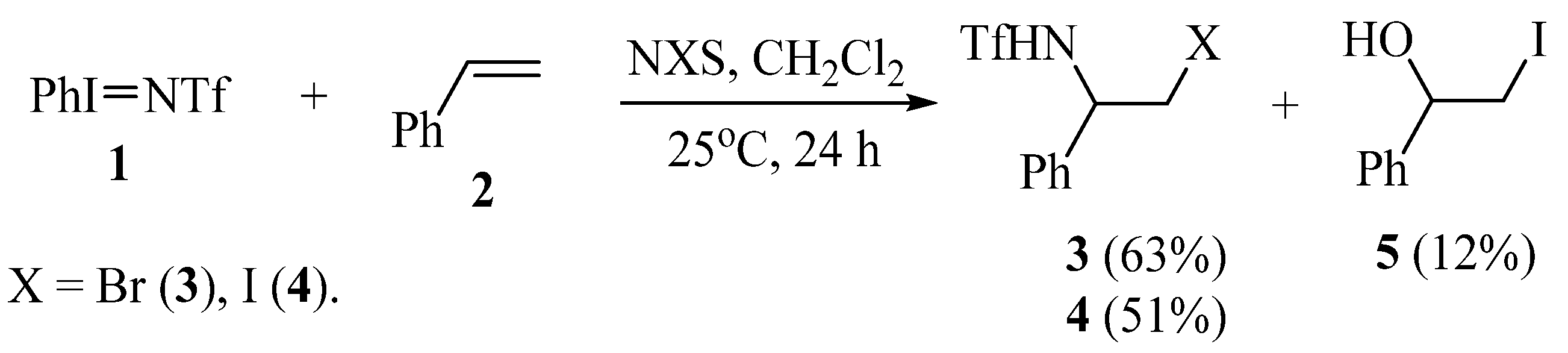
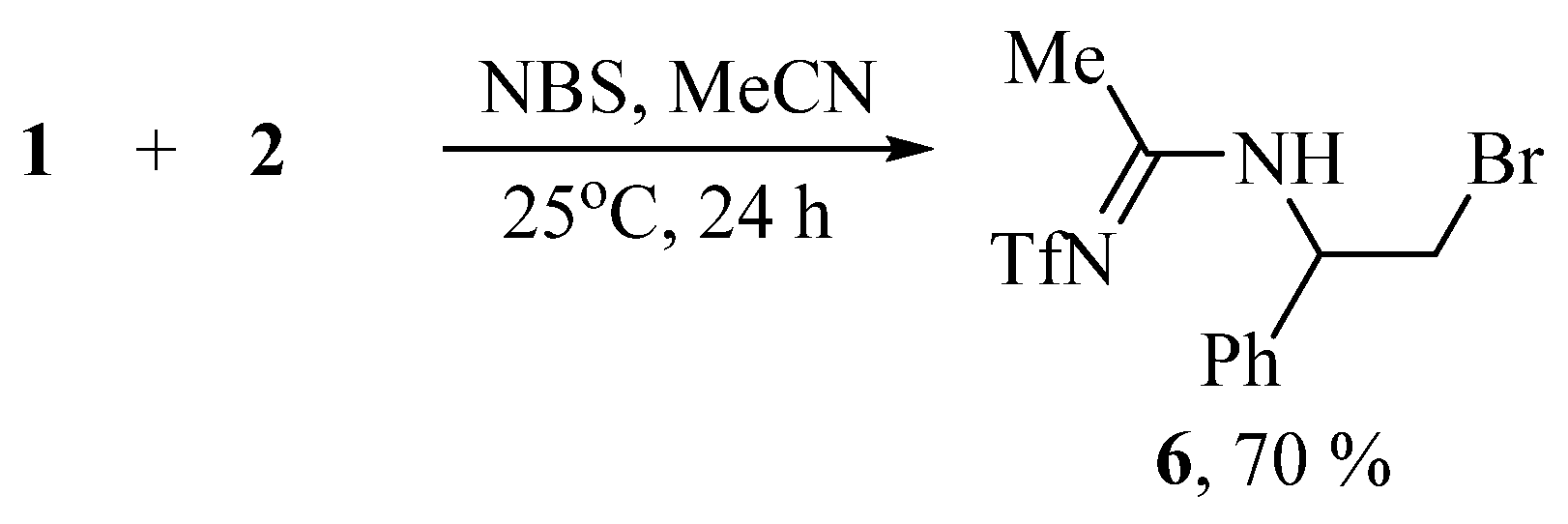
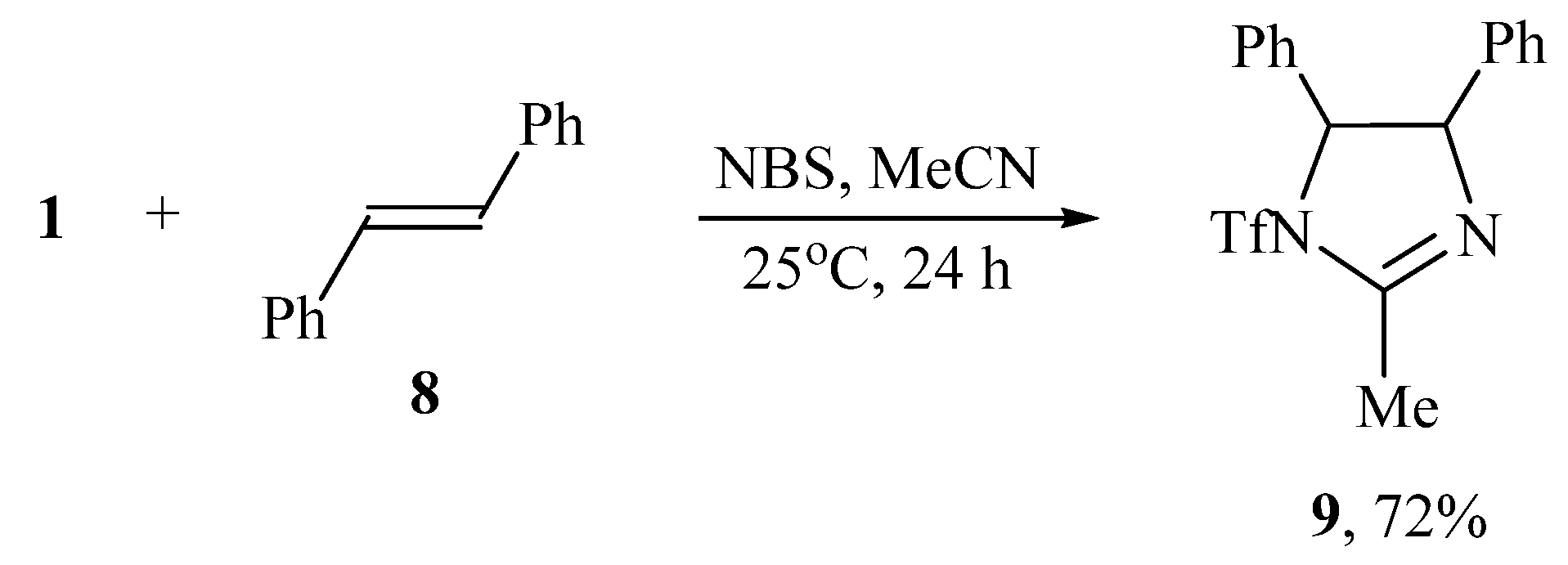
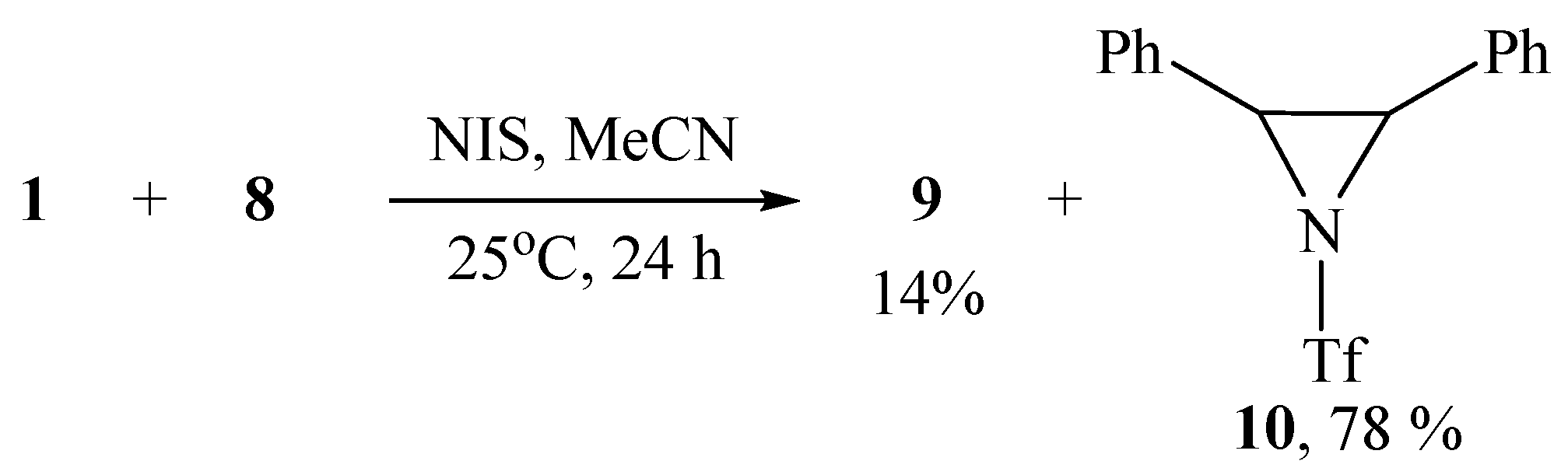
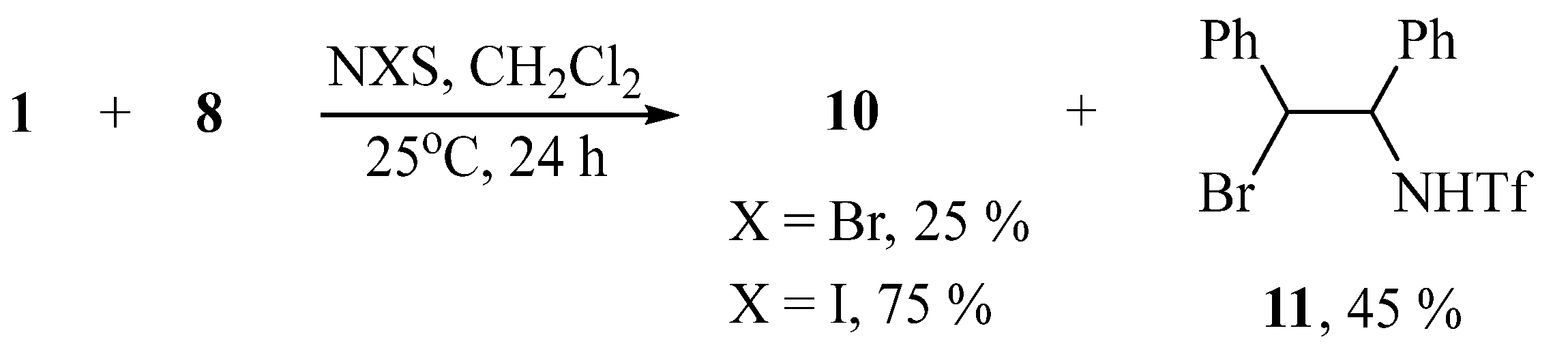
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).