
The Impact of Data on Structure-Based Binding Affinity Predictions Using Deep Neural Networks

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
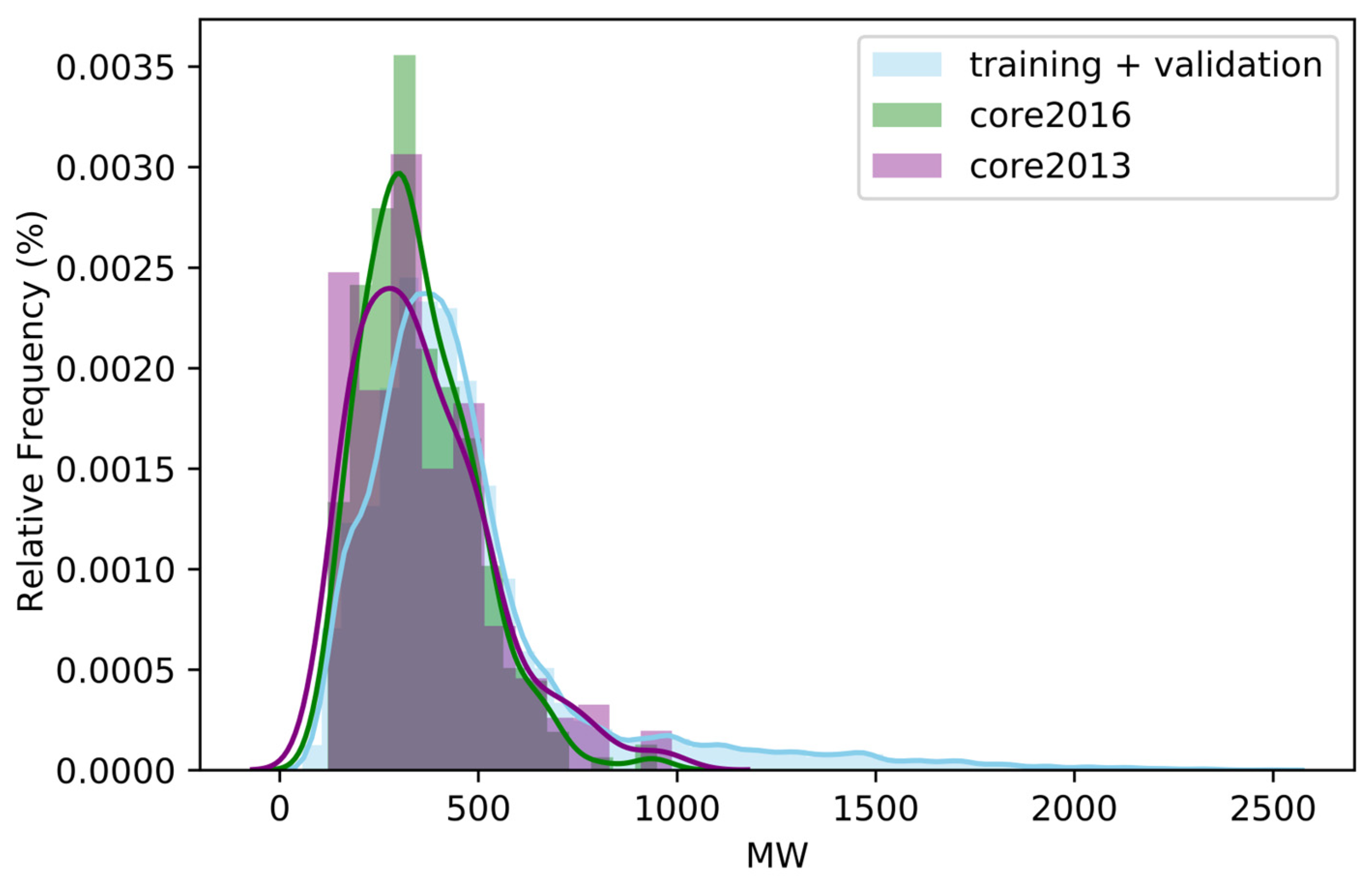
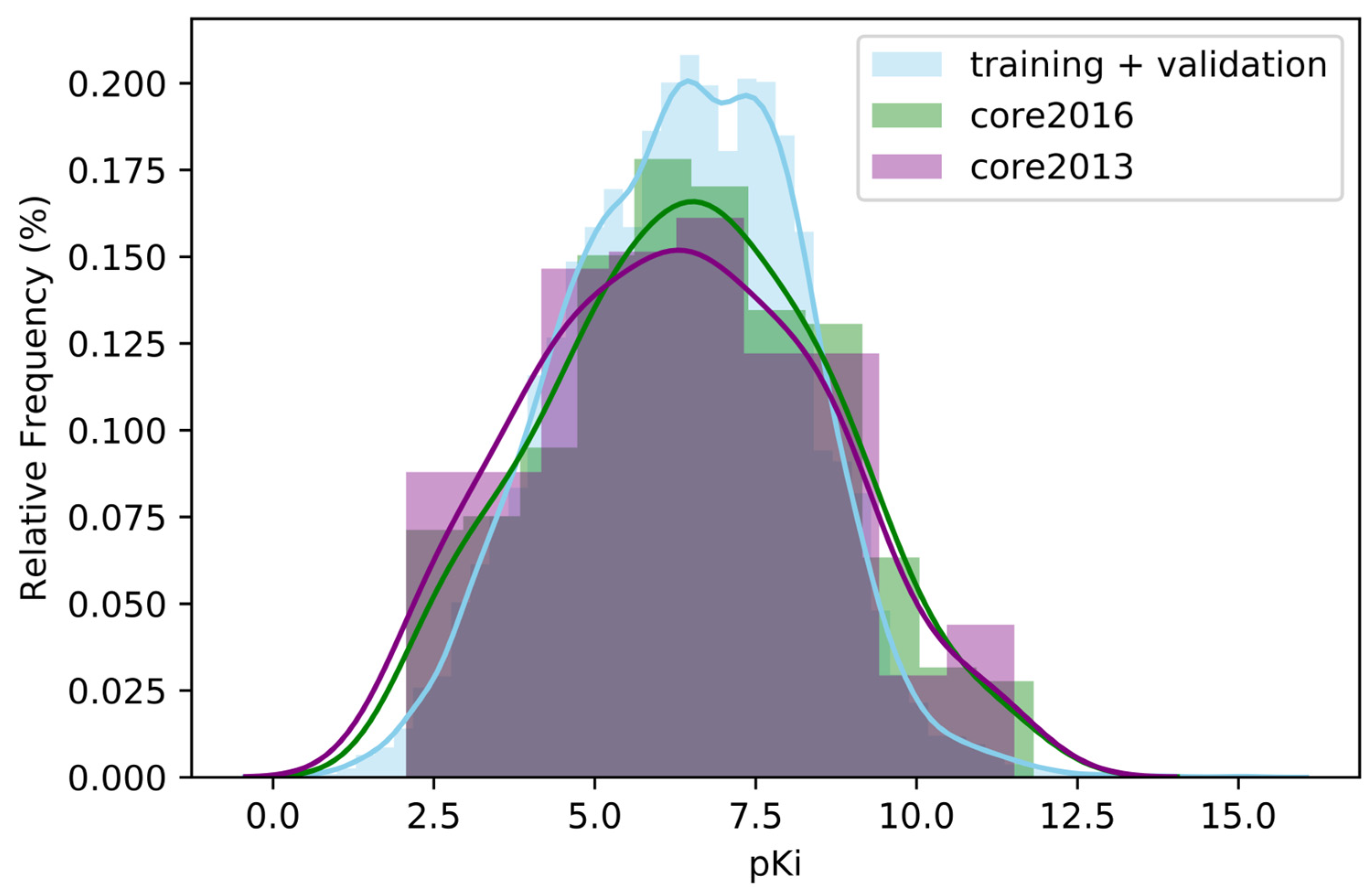
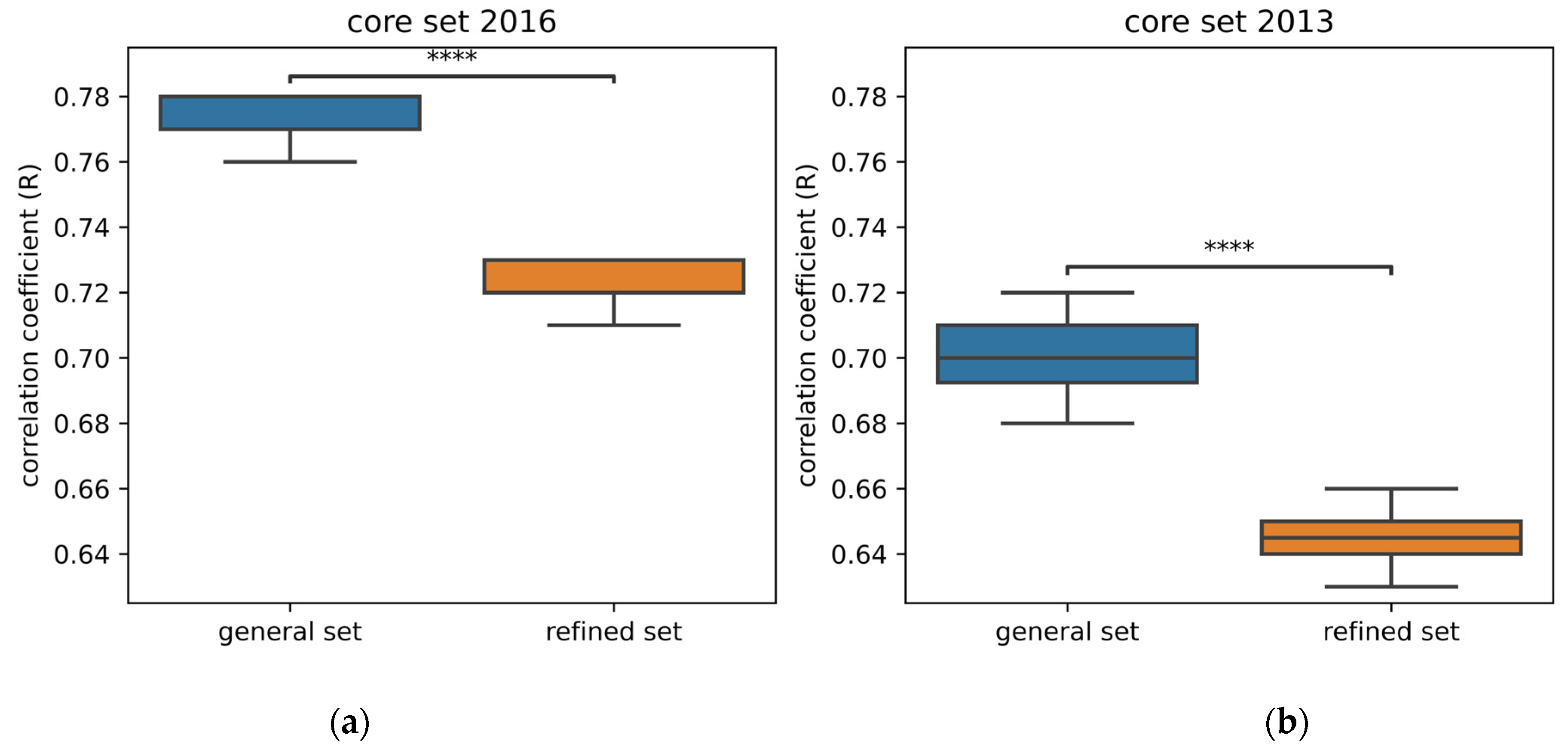
2.1. Impact of the Amount of Data on Performance
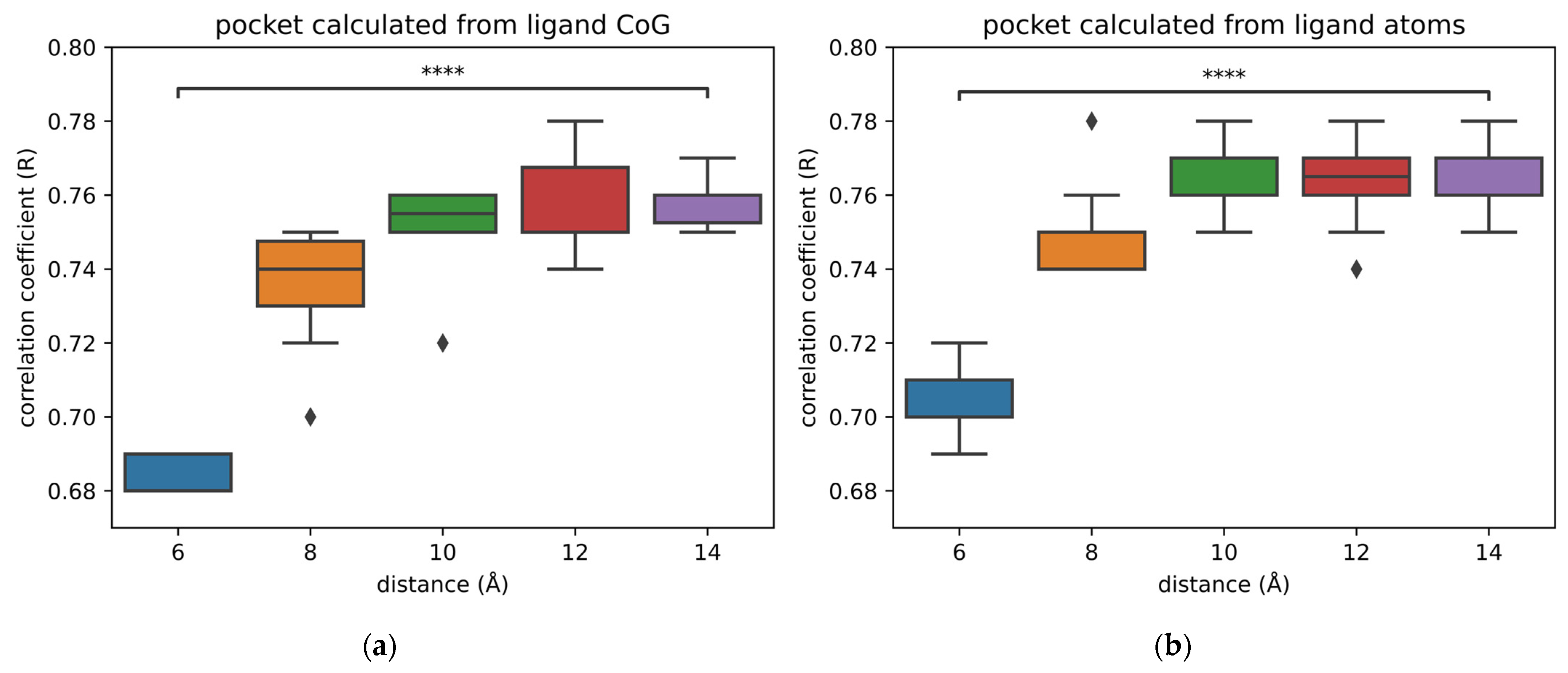
2.2. Size of Pockets
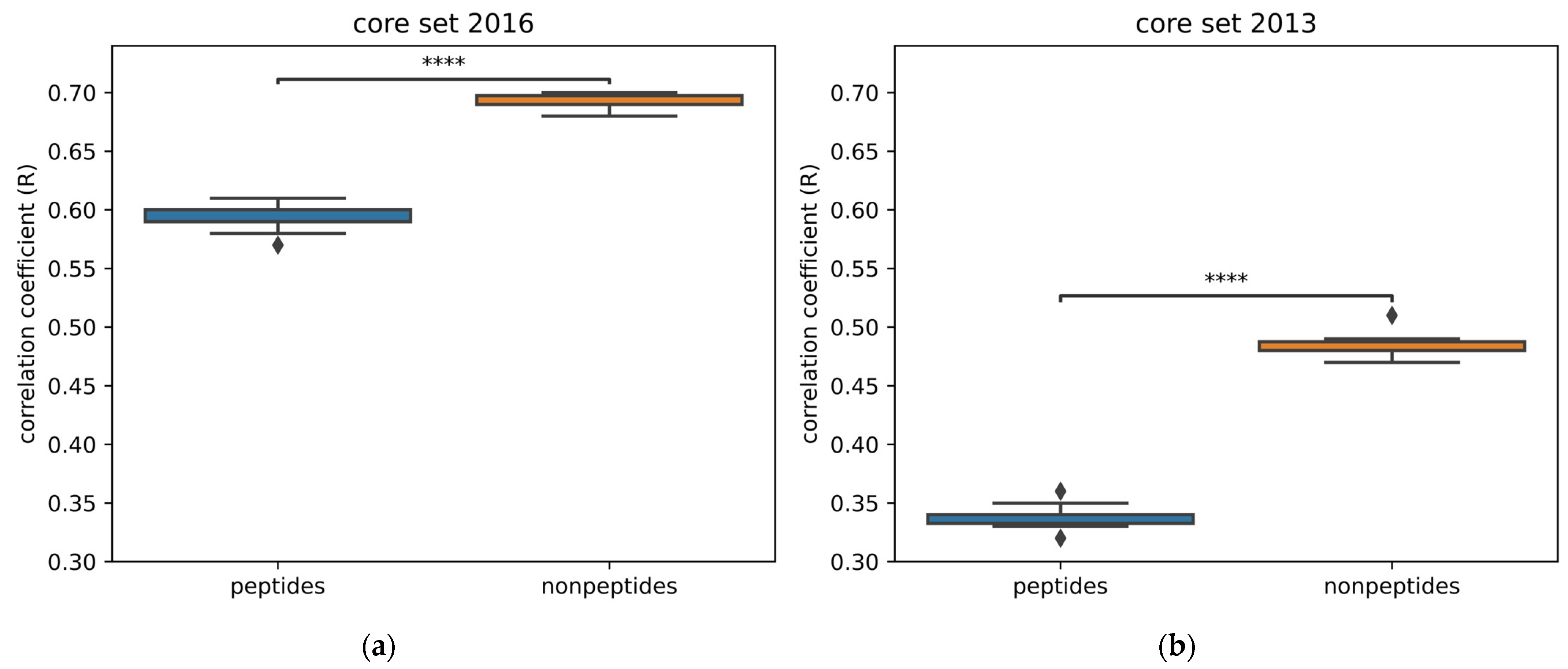
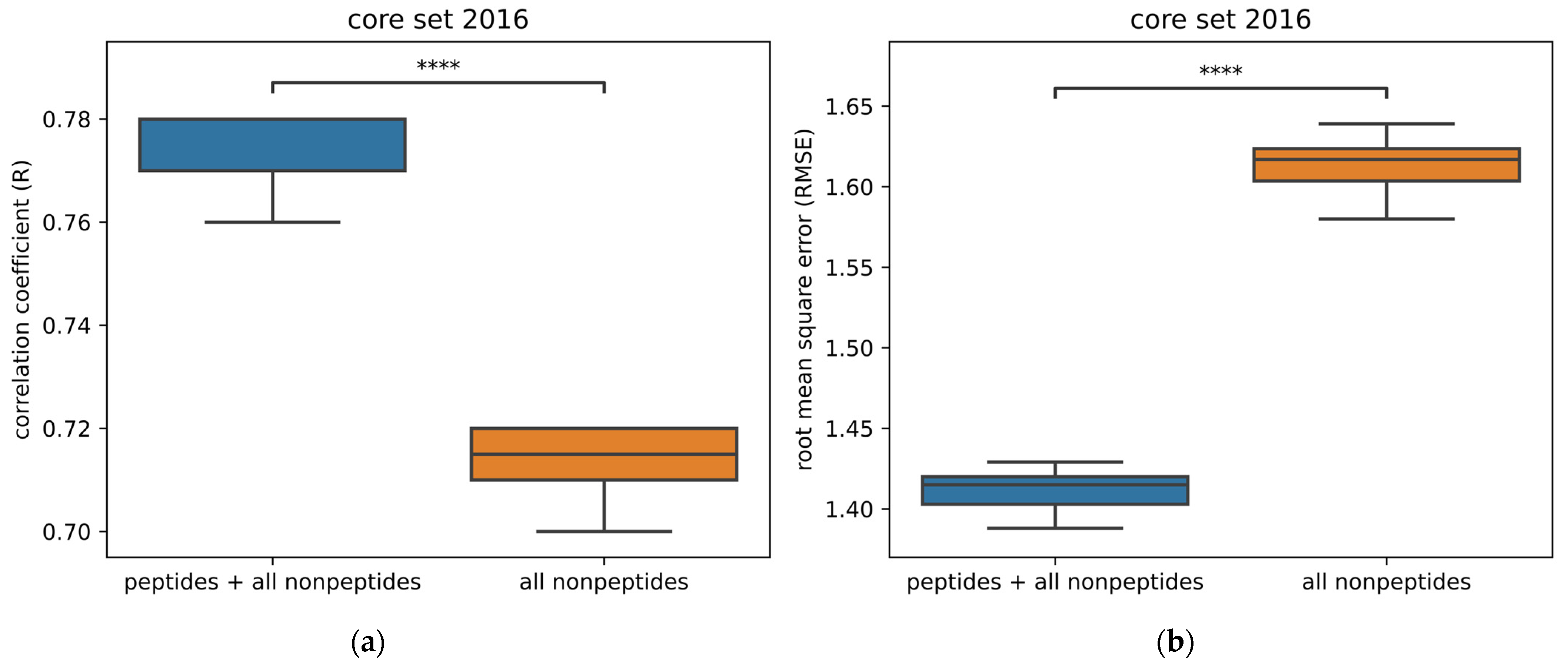
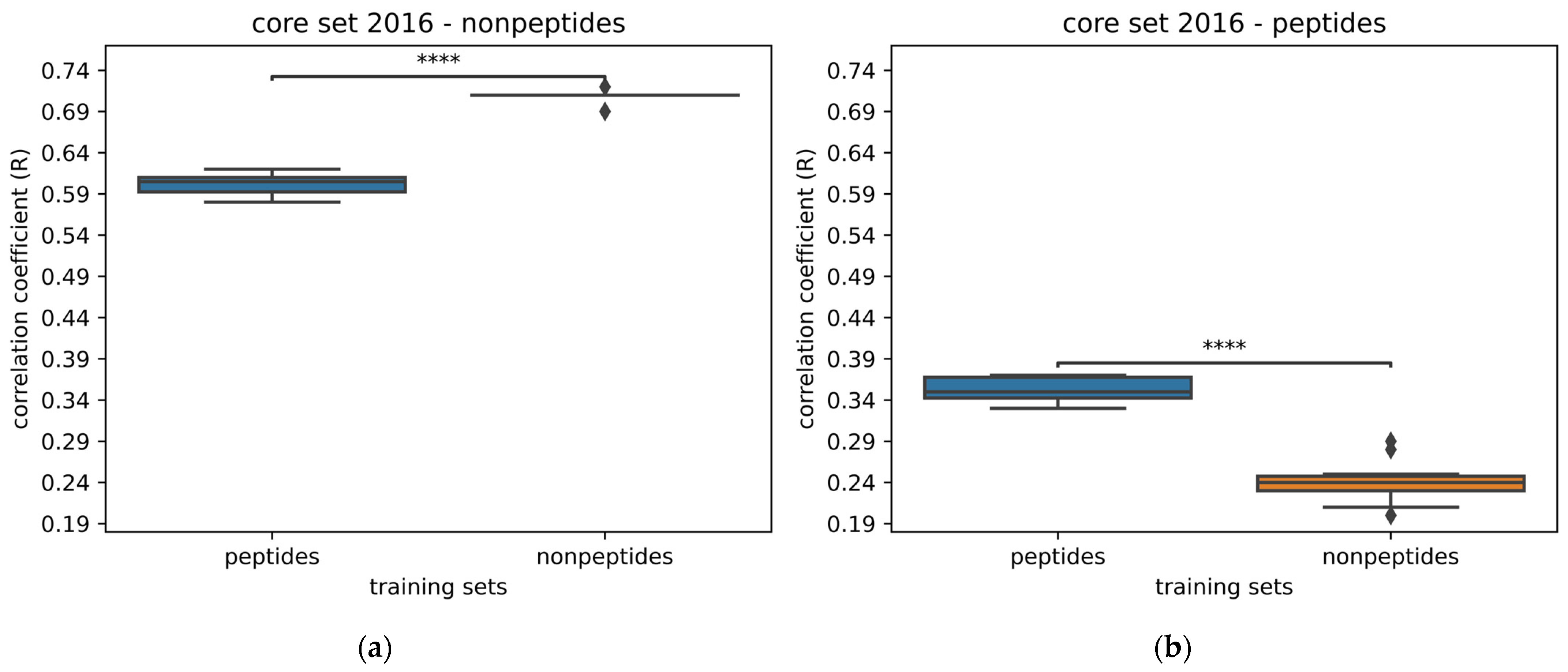
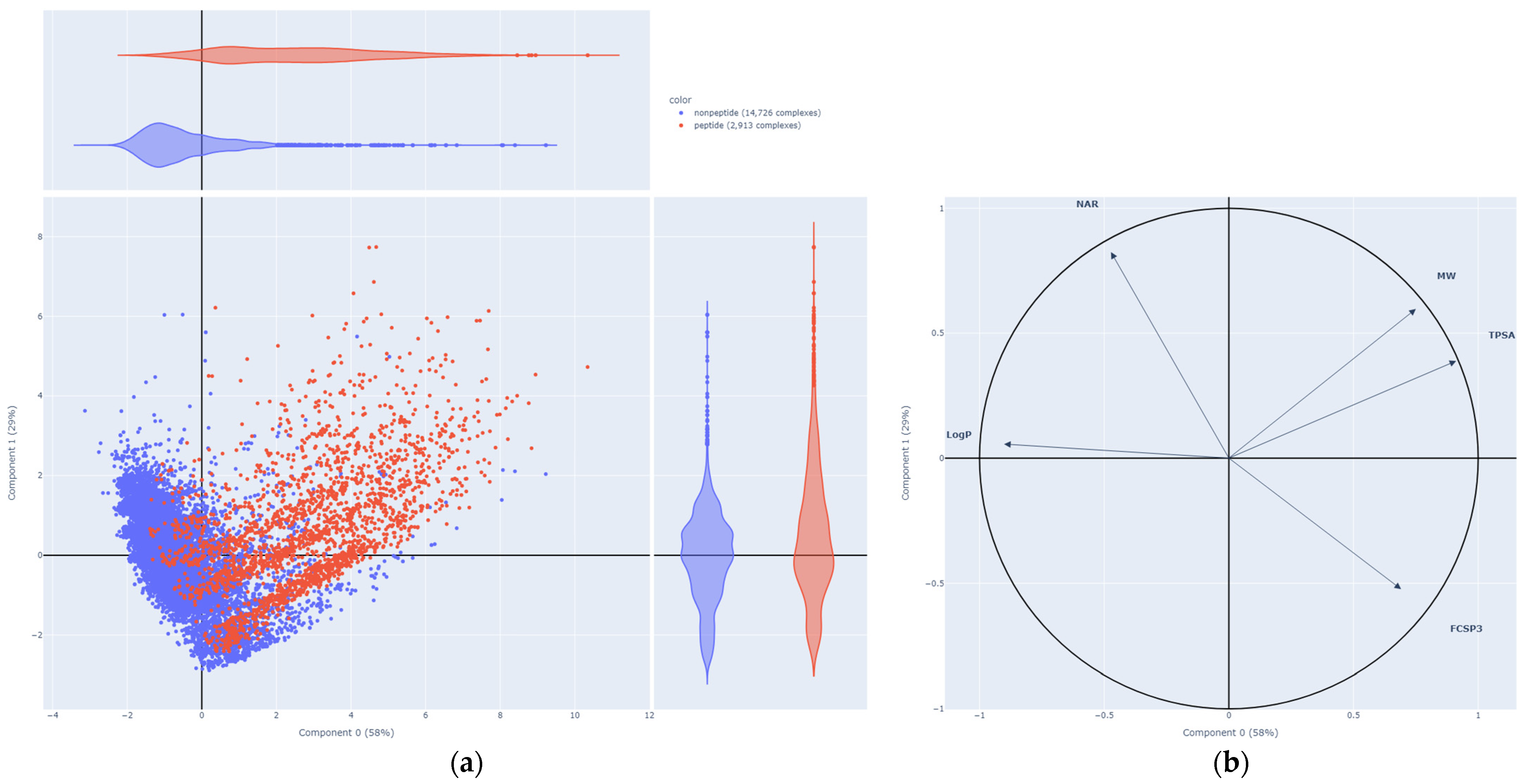
2.3. Peptide vs. Nonpeptide
- Crystallographic complexes with a resolution lower than 2.5 Å
- Known binding affinity (KD/Ki)
- Ligands that do not have protein chain and are not DNA/RNA
2.4. Replication of Results
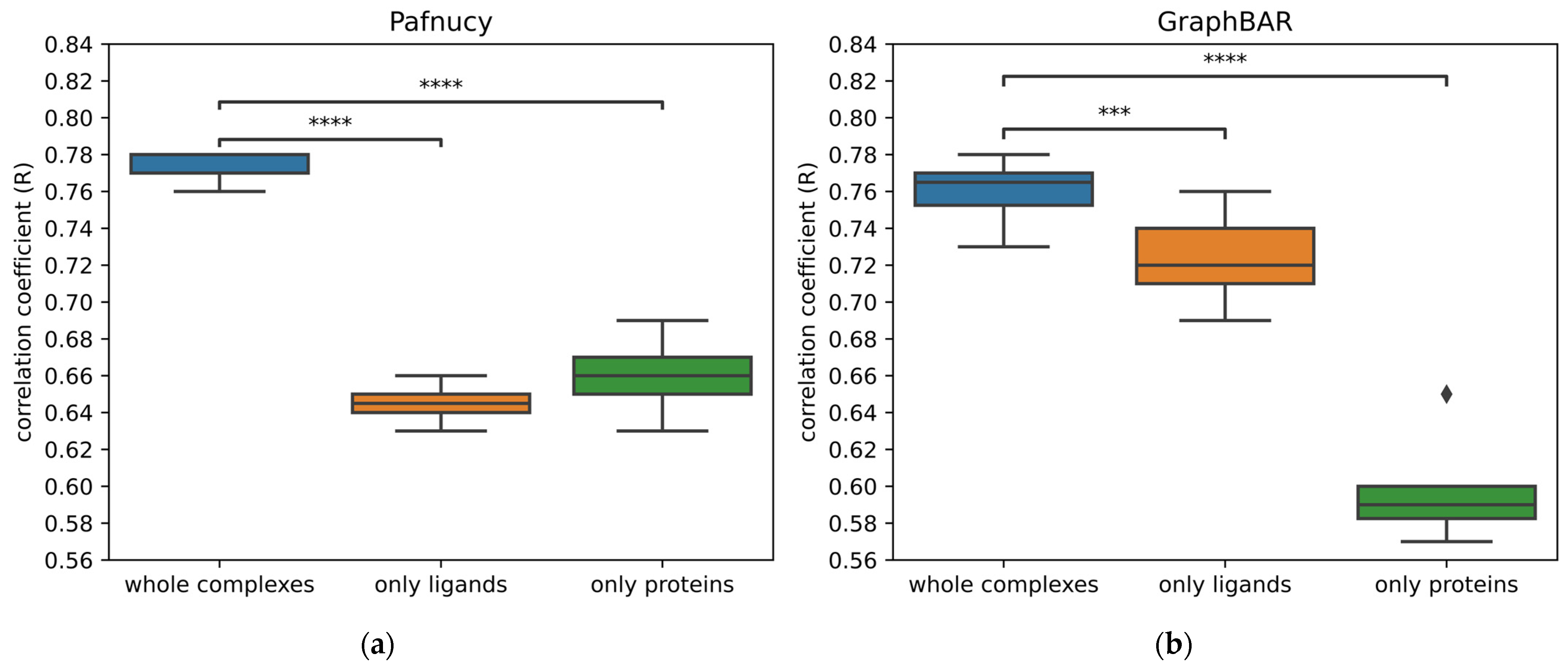
2.5. Learning from Ligand Only, Protein Only, or Interactions
2.6. Other Test Sets
- An example of such a dataset can be found in Volkov et al. [7], where a modular MPNN and Pafnucy were trained on the PDBbind 2016 and were evaluated by predicting on a 2019 holdout set. To create this test set, they selected 3,386 complexes from the PDBbind 2019 that are not in the PDBbind 2016. Instead of using the files provided by the PDBbind, they downloaded the structures from the Protein Data Bank [51]. The complexes were curated and processed with Protoss v.4.0 [52] and IChem [53], e.g., protonation was optimized. Subsequently, Isert et al. [54] reused these data to train models with electron density-based geometric neural networks, and they validated their binding affinity predictions on the same 2019 holdout set.
- Another 2019 holdout set of 4,366 complexes was used to evaluate GIGN [21]. They compared their results against a dozen neural networks, including OnionNet [55], Pafnucy, and GNN-DTI [56]. It is worth mentioning that the protein overlap rate between test and training sets is 69% instead of 100% for the core set 2016. As for the ligand overlap rate, it goes down to 25%, while it was at 38% for the core set 2016.
- Due to similar considerations, Deep Fusion [13] was evaluated on a test set of 222 complexes that was developed from the 2019 holdout set by removing complexes with ligands or proteins already present in the PDBbind 2016. Deep Fusion, KDEEP, and Pafnucy were trained on the PDBbind 2016 and evaluated on this test set.
- AK-score [57] was trained on the refined set of the PDBbind 2016, and it was evaluated by predicting the binding affinity of 534 complexes newly released in the refined set of the PDBbind 2018. For comparison purposes, they also evaluated the performance of other scoring functions, namely X-score [58] and ChemPLP [59].
- The atomic convolutional neural network (ACNN) [60] was trained and tested on several different splits of the PDBbind dataset. On top of a temporal split, they used a stratified split based on the pKi value of complexes and a ligand scaffold split. The stratified split allowed the selection of complexes covering all binding affinities in the training and test sets. In the case of the scaffold split, ligands with unusual scaffold were placed in the test set, therefore preventing the effect of QSAR in the prediction.
- In a similar way, MoleculeNet [61] has been trained and tested on the PDBbind dataset with a temporal split. As for PotentialNet [62], they performed cross-validation by performing a pairwise structural homology split and a sequence similarity split. Both splits are explained in detail in Li and Yang [63]. They were carried out via an agglomerative hierarchical clustering on the PDBbind 2007 refined set, resulting in a test set of 118 and 101 samples, respectively.
- The DUD-E was used to train AtomNet [68] and to evaluate its virtual screening power. AtomNet is the first CNN applied on 3D grids to predict protein–ligand binding affinities. Thirty targets from DUD-E were used as the test set, while the remaining seventy-two targets were used as the training set. On top of using the DUD-E dataset, a derived dataset called “ChEMBL-20 PMD” has been compiled to further benchmark AtomNet. It was created based on several quality criteria and is composed of 78,904 actives, 2,367,120 property-matched decoys (PMD), and 290 targets. This dataset is composed of decoys structurally different from the active molecules to prevent the false-negative bias issue, which, on the other hand, results in an artificial enrichment issue. Therefore, another dataset called “ChEMBL-20 inactives” was developed in order to evaluate AtomNet’s ability to classify experimentally verified active and inactive molecules. ChEMBL-20 inactives were obtained by replacing the PMD with 363,187 molecules known to be inactive.
- Lim et al. [56] used the DUD-E and the PDBbind in order to constitute a training set and a test set. Molecules were docked with Smina [69], resulting in a dataset of docked poses for DUD-E’s 21,705 active molecules and 1,337,409 decoys. As for PDBbind, the molecules were redocked with Smina. If the pose had an RMSD < 2 Å from the crystallographic pose, it was classified as a positive sample, and if the pose was at >4 Å from the crystallographic pose, it was classified as a negative sample. Therefore, 2094 positive and 12,246 negative samples were obtained. The training set was subsequently created with the docked poses of 72 proteins from the DUDE and 70% of the PDBbind redocked dataset. The test set consisted of the docked poses from the remaining 25 proteins from the DUDE and 30% of the PDBbind redocked dataset. The PDBbind split of data was based on a split of the targets, so no proteins would be in the training and test sets. Thereafter, another test set was developed by selecting from the CHEMBL molecules with known binding affinity for the 25 proteins from the DUDE test set. The affinity threshold was put to an IC50 of 1.0 μM, splitting the test set into 27,389 active and 26,939 inactive molecules.
- To assess the docking power, decoy poses were generated by redocking PDBbind’s ligands in their binding site. For each complex, up to 100 decoy poses were selected by setting up 10 bins of 1 Å based on their RMSD values (0–10 Å) to the initial pose. For each bin, ligand poses were clustered based on their conformation, and up to 10 poses were selected. This led to a dataset composed of 22,492 decoy poses.
- In order to evaluate virtual screening power, the ligands were crossdocked. The dataset is composed of 16,245 protein–ligand interaction pairs by docking 285 ligands into 57 proteins. The docking was performed on the protein structure with the highest affinity for each cluster. One hundred poses were selected for each protein–ligand interaction pair. Overall, 1,624,500 decoy poses make up this dataset.
3. Materials and Methods
3.1. Datasets
- The version 2016 that contains 13,308 protein–ligand complexes
- The version 2018 that contains 16,151 protein–ligand complexes
- The version 2019 that contains 17,679 protein–ligand complexes
- Crystallographic structures, with a resolution of 2.5 Å maximum
- Complete ligands/pockets (without missing atoms) and without steric clash with the protein
- Noncovalently bound complexes, no nonstandard residues at a distance <5 Å from the ligand
- No other ligands are present in the binding site, e.g., cofactors or substrates
- Binding affinity evaluated in Ki or KD and with a pKi between 2 and 12
- Ligands with a molecular weight of less than 1000 and less than 10 residues for peptides
- With ligands made only of the following atoms: C, N, O, P, S, F, Cl, Br, I, and H
- The buried surface area of the ligand is higher than 15% of the total surface area of the complex
- The dataset sizes (general set or refined set)
- The types of ligands (peptide or nonpeptide)
- Using only ligands or only proteins
- All the atoms of the ligands
- The center of geometry (CoG) of the ligands (Figure 6)
3.2. Neural Networks
- 9 bits (one-hot or all null) encoding atom types: B, C, N, O, P, S, Se, halogen, and metal
- 1 integer (1, 2, or 3) with atom hybridization: hyb
- 1 integer counting the numbers of bonds with other heavyatoms: heavy_valence
- 1 integer counting the numbers of bonds with other heteroatoms: hetero_valence
- 5 bits (1 if present) encoding properties defined with SMARTS patterns: hydrophobic, aromatic, acceptor, donor, and ring
- 1 float with partial charge: partial charge
- 1 integer (1 for ligand, −1 for protein) to distinguish between the two molecules: moltype
- The hydrogen atom type
- van der Waals atomic radius
- A normal vector with three coordinate directions describing surface curvature and shape complementarity
3.3. Metrics
- ns: 5.00 × 10−2 < p ≤ 1.00 × 100
- *: 1.00 × 10−2 < p ≤ 5.00 × 10−2
- **: 1.00 × 10−3 < p ≤ 1.00 × 10−2
- ***: 1.00 × 10−4 < p ≤ 1.00 × 10−3
- ****: p ≤ 1.00 × 10−4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neural Network | CSAR NRC-HiQ set1 | CSAR NRC-HiQ set2 |
|---|---|---|
| KDEEP [46] | 55 | 49 |
| RosENet [44] | 33 | 10 |
| OnionNet-2 [47] | 55 | 49 |
| graphDelta [48] | 53 | 49 |
| GraphBAR [35] | 51 | 36 |
| PIGNet [37] | 48 & 37 | 37 & 22 |
| BAPA [49] | 50 | 44 |
| CAPLA [50] | 51 | 36 |
| GIGN [21] | 47 | |
| Target | PDB ID | Number of Ligands | Affinity Range (kcal/mol) |
|---|---|---|---|
| MCL1 | 4HW3 | 42 | 4.2 |
| BACE | 4DJW | 36 | 3.5 |
| p38 | 3FLY | 34 | 3.8 |
| PTP1B | 2QBS | 23 | 5.1 |
| JNK1 | 2GMX | 21 | 3.4 |
| CDK2 | 1H1Q | 16 | 4.2 |
| Tyk2 | 4GIH | 16 | 4.3 |
| Thrombin | 2ZFF | 11 | 1.7 |
References
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provazník, I.; Choi, I. Computer Aided Drug Design: Success and Limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef]
- Meli, R.; Morris, G.; Biggin, P. Scoring functions for protein-ligand binding affinity prediction using structure-based deep learning: A review. Front. Bioinform. 2022, 2, 885983. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, X.; Hsieh, C.-Y.; Deng, Y.; Wang, D.; Xu, L.; Wu, J.; Li, D.; Kang, Y.; Hou, T.; et al. A generalized protein–ligand scoring framework with balanced scoring, docking, ranking and screening powers. Chem. Sci. 2023, 14, 8129–8146. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Jukič, M.; Janežič, D.; Bren, U. Potential Novel Thioether-Amide or Guanidine-Linker Class of SARS-CoV-2 Virus RNA-Dependent RNA Polymerase Inhibitors Identified by High-Throughput Virtual Screening Coupled to Free-Energy Calculations. Int. J. Mol. Sci. 2021, 22, 11143. [Google Scholar] [CrossRef]
- Gapsys, V.; Pérez-Benito, L.; Aldeghi, M.; Seeliger, D.; van Vlijmen, H.; Tresadern, G.; de Groot, B.L. Large scale relative protein ligand binding affinities using non-equilibrium alchemy. Chem. Sci. 2020, 11, 1140–1152. [Google Scholar] [CrossRef]
- Volkov, M.; Turk, J.-A.; Drizard, N.; Martin, N.; Hoffmann, B.; Gaston-Mathé, Y.; Rognan, D. On the Frustration to Predict Binding Affinities from Protein–Ligand Structures with Deep Neural Networks. J. Med. Chem. 2022, 65, 7946–7958. [Google Scholar] [CrossRef]
- Deng, J.; Dong, W.; Socher, R.; Li, L.J.; Kai, L.; Li, F.-F. ImageNet: A large-scale hierarchical image database. In Proceedings of the 2009 IEEE Conference on Computer Vision and Pattern Recognition, Miami, FL, USA, 20–25 June 2009; pp. 248–255. [Google Scholar]
- Wang, R.; Fang, X.; Lu, Y.; Wang, S. The PDBbind database: Collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem. 2004, 47, 2977–2980. [Google Scholar] [CrossRef]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and evaluation of a deep learning model for protein-ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef]
- Braka, A.; Garnier, N.; Bonnet, P.; Aci-Sèche, S. Residence Time Prediction of Type 1 and 2 Kinase Inhibitors from Unbinding Simulations. J. Chem. Inf. Model. 2020, 60, 342–348. [Google Scholar] [CrossRef]
- Ziada, S.; Diharce, J.; Raimbaud, E.; Aci-Sèche, S.; Ducrot, P.; Bonnet, P. Estimation of Drug-Target Residence Time by Targeted Molecular Dynamics Simulations. J. Chem. Inf. Model. 2022, 62, 5536–5549. [Google Scholar] [CrossRef]
- Jones, D.; Kim, H.; Zhang, X.; Zemla, A.; Stevenson, G.; Bennett, W.F.D.; Kirshner, D.; Wong, S.E.; Lightstone, F.C.; Allen, J.E. Improved Protein–Ligand Binding Affinity Prediction with Structure-Based Deep Fusion Inference. J. Chem. Inf. Model. 2021, 61, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Unarta, I.C.; Xu, J.; Shang, Y.; Cheung, C.H.P.; Zhu, R.; Chen, X.; Cao, S.; Cheung, P.P.; Bierer, D.; Zhang, M.; et al. Entropy of stapled peptide inhibitors in free state is the major contributor to the improvement of binding affinity with the GK domain. RSC Chem. Biol. 2021, 2, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Mam, B.; Sowdhamini, R. DEELIG: A Deep Learning Approach to Predict Protein-Ligand Binding Affinity. Bioinform. Biol. Insights 2021, 15, 11779322211030364. [Google Scholar] [CrossRef] [PubMed]
- Jukič, M.; Bren, U. Machine Learning in Antibacterial Drug Design. Front. Pharmacol. 2022, 13, 864412. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shen, C.; Huang, N. Predicting or Pretending: Artificial Intelligence for Protein-Ligand Interactions Lack of Sufficiently Large and Unbiased Datasets. Front. Pharmacol. 2020, 11, 69. [Google Scholar] [CrossRef]
- Li, S.; Zhou, J.; Xu, T.; Huang, L.; Wang, F.; Xiong, H.; Huang, W.; Dou, D.; Xiong, H. Structure-Aware Interactive Graph Neural Networks for the Prediction of Protein-Ligand Binding Affinity. In Proceedings of the 27th ACM SIGKDD Conference on Knowledge Discovery & Data Mining, Singapore, 14–18 August 2021; pp. 975–985. [Google Scholar]
- Wang, Y.; Wu, S.; Duan, Y.; Huang, Y. A point cloud-based deep learning strategy for protein-ligand binding affinity prediction. Brief. Bioinform. 2022, 23, bbab474. [Google Scholar] [CrossRef]
- Li, Y.; Rezaei, M.A.; Li, C.; Li, X. DeepAtom: A Framework for Protein-Ligand Binding Affinity Prediction. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019; pp. 303–310. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, W.; Lv, Q.; Dong, T.; Yu-Chian Chen, C. Geometric Interaction Graph Neural Network for Predicting Protein–Ligand Binding Affinities from 3D Structures (GIGN). J. Phys. Chem. Lett. 2023, 14, 2020–2033. [Google Scholar] [CrossRef]
- Francoeur, P.G.; Masuda, T.; Sunseri, J.; Jia, A.; Iovanisci, R.B.; Snyder, I.; Koes, D.R. Three-Dimensional Convolutional Neural Networks and a Cross-Docked Data Set for Structure-Based Drug Design. J. Chem. Inf. Model. 2020, 60, 4200–4215. [Google Scholar] [CrossRef]
- Wang, R.; Fang, X.; Lu, Y.; Yang, C.-Y.; Wang, S. The PDBbind Database: Methodologies and Updates. J. Med. Chem. 2005, 48, 4111–4119. [Google Scholar] [CrossRef]
- Hu, L.; Benson, M.L.; Smith, R.D.; Lerner, M.G.; Carlson, H.A. Binding MOAD (Mother of All Databases). Proteins Struct. Funct. Bioinform. 2005, 60, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, P.-S.; Zhu, C.; Gaines, B.B.; Zhu, T.; Bi, J.; Song, M. OctSurf: Efficient hierarchical voxel-based molecular surface representation for protein-ligand affinity prediction. J. Mol. Graph. Model. 2021, 105, 107865. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Shen, C.; Yang, Z.; Jiang, D.; Liu, S.; Lu, A.; Chen, X.; Hou, T.; Cao, D. Featurization strategies for protein–ligand interactions and their applications in scoring function development. WIREs Comput. Mol. Sci. 2022, 12, e1567. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Montavon, G.; Binder, A.; Lapuschkin, S.; Samek, W.; Müller, K.-R. Layer-Wise Relevance Propagation: An Overview. In Explainable AI: Interpreting, Explaining and Visualizing Deep Learning; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Karpov, P.; Godin, G.; Tetko, I.V. Transformer-CNN: Swiss knife for QSAR modeling and interpretation. J. Cheminform. 2020, 12, 17. [Google Scholar] [CrossRef]
- Nielsen, I.E.; Dera, D.; Rasool, G.; Ramachandran, R.P.; Bouaynaya, N.C. Robust Explainability: A tutorial on gradient-based attribution methods for deep neural networks. IEEE Signal Process. Mag. 2022, 39, 73–84. [Google Scholar] [CrossRef]
- Hochuli, J.; Helbling, A.; Skaist, T.; Ragoza, M.; Koes, D.R. Visualizing convolutional neural network protein-ligand scoring. J. Mol. Graph. Model. 2018, 84, 96–108. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Y.; Han, L.; Li, J.; Liu, J.; Zhao, Z.; Nie, W.; Liu, Y.; Wang, R. PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics 2014, 31, 405–412. [Google Scholar] [CrossRef]
- Bournez, C.; Carles, F.; Peyrat, G.; Aci-Sèche, S.; Bourg, S.; Meyer, C.; Bonnet, P. Comparative Assessment of Protein Kinase Inhibitors in Public Databases and in PKIDB. Molecules 2020, 25, 3226. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2020, 2, 573–584. [Google Scholar] [CrossRef]
- Son, J.; Kim, D. Development of a graph convolutional neural network model for efficient prediction of protein-ligand binding affinities. PLoS ONE 2021, 16, e0249404. [Google Scholar] [CrossRef]
- Lakshminarayanan, B.; Pritzel, A.; Blundell, C. Simple and scalable predictive uncertainty estimation using deep ensembles. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 6405–6416. [Google Scholar]
- Moon, S.; Zhung, W.; Yang, S.; Lim, J.; Kim, W.Y. PIGNet: A physics-informed deep learning model toward generalized drug–target interaction predictions. Chem. Sci. 2022, 13, 3661–3673. [Google Scholar] [CrossRef] [PubMed]
- Sieg, J.; Flachsenberg, F.; Rarey, M. In Need of Bias Control: Evaluating Chemical Data for Machine Learning in Structure-Based Virtual Screening. J. Chem. Inf. Model. 2019, 59, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Scantlebury, J.; Brown, N.; Von Delft, F.; Deane, C.M. Data Set Augmentation Allows Deep Learning-Based Virtual Screening to Better Generalize to Unseen Target Classes and Highlight Important Binding Interactions. J. Chem. Inf. Model. 2020, 60, 3722–3730. [Google Scholar] [CrossRef]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein-Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef]
- Li, H.; Leung, K.-S.; Wong, M.-H.; Ballester, P.J. Correcting the impact of docking pose generation error on binding affinity prediction. BMC Bioinform. 2016, 17, 308. [Google Scholar] [CrossRef] [PubMed]
- Boyles, F.; Deane, C.M.; Morris, G.M. Learning from Docked Ligands: Ligand-Based Features Rescue Structure-Based Scoring Functions When Trained on Docked Poses. J. Chem. Inf. Model. 2022, 62, 5329–5341. [Google Scholar] [CrossRef]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.M.; Mortenson, P.N.; Murray, C.W. Diverse, High-Quality Test Set for the Validation of Protein−Ligand Docking Performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef]
- Hassan-Harrirou, H.; Zhang, C.; Lemmin, T. RosENet: Improving Binding Affinity Prediction by Leveraging Molecular Mechanics Energies with an Ensemble of 3D Convolutional Neural Networks. J. Chem. Inf. Model. 2020, 60, 2791–2802. [Google Scholar] [CrossRef]
- Dunbar, J.B., Jr.; Smith, R.D.; Damm-Ganamet, K.L.; Ahmed, A.; Esposito, E.X.; Delproposto, J.; Chinnaswamy, K.; Kang, Y.-N.; Kubish, G.; Gestwicki, J.E.; et al. CSAR Data Set Release 2012: Ligands, Affinities, Complexes, and Docking Decoys. J. Chem. Inf. Model. 2013, 53, 1842–1852. [Google Scholar] [CrossRef]
- Jiménez, J.; Škalič, M.; Martínez-Rosell, G.; De Fabritiis, G. KDEEP: Protein–Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zheng, L.; Liu, Y.; Qu, Y.; Li, Y.-Q.; Zhao, M.; Mu, Y.; Li, W. OnionNet-2: A convolutional neural network model for predicting protein-ligand binding affinity based on residue-atom contacting shells. Front. Chem. 2021, 9, 753002. [Google Scholar] [CrossRef] [PubMed]
- Karlov, D.S.; Sosnin, S.; Fedorov, M.V.; Popov, P. graphDelta: MPNN Scoring Function for the Affinity Prediction of Protein–Ligand Complexes. ACS Omega 2020, 5, 5150–5159. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Choi, J.; Park, S.; Ahn, J. Binding affinity prediction for protein–ligand complex using deep attention mechanism based on intermolecular interactions. BMC Bioinform. 2021, 22, 542. [Google Scholar] [CrossRef]
- Jin, Z.; Wu, T.; Chen, T.; Pan, D.; Wang, X.; Xie, J.; Quan, L.; Lyu, Q. CAPLA: Improved prediction of protein–ligand binding affinity by a deep learning approach based on a cross-attention mechanism. Bioinformatics 2023, 39, btad049. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Bietz, S.; Urbaczek, S.; Schulz, B.; Rarey, M. Protoss: A holistic approach to predict tautomers and protonation states in protein-ligand complexes. J. Cheminform. 2014, 6, 12. [Google Scholar] [CrossRef]
- Da Silva, F.; Desaphy, J.; Rognan, D. IChem: A Versatile Toolkit for Detecting, Comparing, and Predicting Protein–Ligand Interactions. ChemMedChem 2018, 13, 507–510. [Google Scholar] [CrossRef]
- Isert, C.; Atz, K.; Riniker, S.; Schneider, G. Exploring protein-ligand binding affinity prediction with electron density-based geometric deep learning. ChemRxiv 2023. [Google Scholar] [CrossRef]
- Zheng, L.; Fan, J.; Mu, Y. OnionNet: A Multiple-Layer Intermolecular-Contact-Based Convolutional Neural Network for Protein–Ligand Binding Affinity Prediction. ACS Omega 2019, 4, 15956–15965. [Google Scholar] [CrossRef]
- Lim, J.; Ryu, S.; Park, K.; Choe, Y.J.; Ham, J.; Kim, W.Y. Predicting Drug–Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation. J. Chem. Inf. Model. 2019, 59, 3981–3988. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Shin, W.-H.; Ko, J.; Lee, J. AK-Score: Accurate Protein-Ligand Binding Affinity Prediction Using an Ensemble of 3D-Convolutional Neural Networks. Int. J. Mol. Sci. 2020, 21, 8424. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput.-Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Gomes, J.; Ramsundar, B.; Feinberg, E.N.; Pande, V.S. Atomic convolutional networks for predicting protein-ligand binding affinity. arXiv 2017, arXiv:1703.10603. [Google Scholar]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef]
- Feinberg, E.N.; Sur, D.; Wu, Z.; Husic, B.E.; Mai, H.; Li, Y.; Sun, S.; Yang, J.; Ramsundar, B.; Pande, V.S. PotentialNet for Molecular Property Prediction. ACS Cent. Sci. 2018, 4, 1520–1530. [Google Scholar] [CrossRef]
- Li, Y.; Yang, J. Structural and Sequence Similarity Makes a Significant Impact on Machine-Learning-Based Scoring Functions for Protein–Ligand Interactions. J. Chem. Inf. Model. 2017, 57, 1007–1012. [Google Scholar] [CrossRef]
- Tosstorff, A.; Rudolph, M.G.; Cole, J.C.; Reutlinger, M.; Kramer, C.; Schaffhauser, H.; Nilly, A.; Flohr, A.; Kuhn, B. A high quality, industrial data set for binding affinity prediction: Performance comparison in different early drug discovery scenarios. J. Comput.-Aided Mol. Des. 2022, 36, 753–765. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking Sets for Molecular Docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Chen, L.; Cruz, A.; Ramsey, S.; Dickson, C.J.; Duca, J.S.; Hornak, V.; Koes, D.R.; Kurtzman, T. Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLoS ONE 2019, 14, e0220113. [Google Scholar] [CrossRef] [PubMed]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. arXiv 2015, arXiv:1510.02855. [Google Scholar]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.R.; Ibrahim, T.M.; Vogel, S.M.; Boeckler, F.M. Evaluation and Optimization of Virtual Screening Workflows with DEKOIS 2.0—A Public Library of Challenging Docking Benchmark Sets. J. Chem. Inf. Model. 2013, 53, 1447–1462. [Google Scholar] [CrossRef]
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep. 2017, 7, 46710. [Google Scholar] [CrossRef]
- Chen, P.; Ke, Y.; Lu, Y.; Du, Y.; Li, J.; Yan, H.; Zhao, H.; Zhou, Y.; Yang, Y. DLIGAND2: An improved knowledge-based energy function for protein–ligand interactions using the distance-scaled, finite, ideal-gas reference state. J. Cheminform. 2019, 11, 52. [Google Scholar] [CrossRef]
- Ballester, P.J. Selecting machine-learning scoring functions for structure-based virtual screening. Drug Discov. Today Technol. 2019, 32–33, 81–87. [Google Scholar] [CrossRef]
- Yasuo, N.; Sekijima, M. Improved Method of Structure-Based Virtual Screening via Interaction-Energy-Based Learning. J. Chem. Inf. Model. 2019, 59, 1050–1061. [Google Scholar] [CrossRef]
- Riniker, S.; Landrum, G.A. Open-source platform to benchmark fingerprints for ligand-based virtual screening. J. Cheminform. 2013, 5, 26. [Google Scholar] [CrossRef]
- Imrie, F.; Bradley, A.R.; van der Schaar, M.; Deane, C.M. Protein Family-Specific Models Using Deep Neural Networks and Transfer Learning Improve Virtual Screening and Highlight the Need for More Data. J. Chem. Inf. Model. 2018, 58, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2019, 59, 895–913. [Google Scholar] [CrossRef] [PubMed]
- Brocidiacono, M.; Francoeur, P.; Aggarwal, R.; Popov, K.; Koes, D.; Tropsha, A. BigBind: Learning from Nonstructural Data for Structure-Based Virtual Screening. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Z.; Li, J.; Han, L.; Liu, J.; Zhao, Z.; Wang, R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 1. Compilation of the Test Set. J. Chem. Inf. Model. 2014, 54, 1700–1716. [Google Scholar] [CrossRef]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 2. Evaluation Methods and General Results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef]
- Li, Y.; Su, M.; Liu, Z.; Li, J.; Liu, J.; Han, L.; Wang, R. Assessing protein–ligand interaction scoring functions with the CASF-2013 benchmark. Nat. Protoc. 2018, 13, 666–680. [Google Scholar] [CrossRef]
- Özçelik, R.; van Tilborg, D.; Jiménez-Luna, J.; Grisoni, F. Structure-Based Drug Discovery with Deep Learning. ChemBioChem 2023, 24, e202200776. [Google Scholar] [CrossRef]
- Isert, C.; Atz, K.; Schneider, G. Structure-based drug design with geometric deep learning. arXiv 2022, arXiv:2210.11250. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, H.; Hu, Q.; Liu, H.; Liu, L.; Bennamoun, M. Deep learning for 3d point clouds: A survey. IEEE Trans. Pattern Anal. Mach. Intell. 2020, 43, 4338–4364. [Google Scholar] [CrossRef]
- Meagher, D. Octree Encoding: A New Technique for the Representation, Manipulation and Display of Arbitrary 3-D Objects by Computer; Stanford University: Stanford, CA, USA, 1980. [Google Scholar]
- Fan, F.J.; Shi, Y. Effects of data quality and quantity on deep learning for protein-ligand binding affinity prediction. Bioorg. Med. Chem. 2022, 72, 117003. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, S.G.; Baumann, K. Maximum Unbiased Validation (MUV) Data Sets for Virtual Screening Based on PubChem Bioactivity Data. J. Chem. Inf. Model. 2009, 49, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Pérez, A.; Martínez-Rosell, G.; De Fabritiis, G. Simulations meet machine learning in structural biology. Curr. Opin. Struct. Biol. 2018, 49, 139–144. [Google Scholar] [CrossRef] [PubMed]
| Neural Network | Results from Publication | Results from Replication | ||
|---|---|---|---|---|
| Pafnucy | R = 0.78 1 | RMSE = 1.42 1 | R = 0.77 SD = 0.01 1 | RMSE = 1.41 SD = 0.01 1 |
| GraphBAR | R = 0.76 1 | RMSE = 1.44 1 | R = 0.76 SD = 0.02 3 | RMSE = 1.43 SD = 0.03 3 |
| OctSurf | R = 0.79 ± 0.01 2 | RMSE = 1.45 ± 0.02 2 | R = 0.79 SD = 0.01 2 | RMSE = 1.46 SD = 0.03 2 |
| Neural Network | Whole Complex (R, RMSE) | Only Ligand (R, RMSE) | Only Protein (R, RMSE) |
|---|---|---|---|
| Pafnucy 1 | 0.77, 1.41 | 0.65, 1.67 | 0.66, 1.64 |
| GraphBAR 2 | 0.76, 1.43 | 0.73, 1.51 | 0.59, 1.77 |
| OctSurf 1 | 0.79, 1.45 | 0.73 (n.a.) | 0.64 (n.a.) |
| Modular MPNN 2 [7] | 0.81, 1.51 | 0.75, 1.57 | 0.73, 1.57 |
| Deep Fusion 3 [13] | 0.81, 1.31 | 0.49, 3.01 | 0.5, 4.00 |
| PointTransformer 4 [19] | 0.86, 1.19 | 0.45 (n.a.) | 0.2 (n.a.) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Libouban, P.-Y.; Aci-Sèche, S.; Gómez-Tamayo, J.C.; Tresadern, G.; Bonnet, P. The Impact of Data on Structure-Based Binding Affinity Predictions Using Deep Neural Networks. Int. J. Mol. Sci. 2023, 24, 16120. https://doi.org/10.3390/ijms242216120
Libouban P-Y, Aci-Sèche S, Gómez-Tamayo JC, Tresadern G, Bonnet P. The Impact of Data on Structure-Based Binding Affinity Predictions Using Deep Neural Networks. International Journal of Molecular Sciences. 2023; 24(22):16120. https://doi.org/10.3390/ijms242216120
Chicago/Turabian StyleLibouban, Pierre-Yves, Samia Aci-Sèche, Jose Carlos Gómez-Tamayo, Gary Tresadern, and Pascal Bonnet. 2023. "The Impact of Data on Structure-Based Binding Affinity Predictions Using Deep Neural Networks" International Journal of Molecular Sciences 24, no. 22: 16120. https://doi.org/10.3390/ijms242216120
APA StyleLibouban, P.-Y., Aci-Sèche, S., Gómez-Tamayo, J. C., Tresadern, G., & Bonnet, P. (2023). The Impact of Data on Structure-Based Binding Affinity Predictions Using Deep Neural Networks. International Journal of Molecular Sciences, 24(22), 16120. https://doi.org/10.3390/ijms242216120