

Merging Counter-Propagation and Back-Propagation Algorithms: Overcoming the Limitations of Counter-Propagation Neural Network Models



Abstract
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Datasets
3.1.1. Water Solubility
3.1.2. Acute Fish Toxicity
3.1.3. Bio-Concentration Factor
3.2. Molecular Descriptors
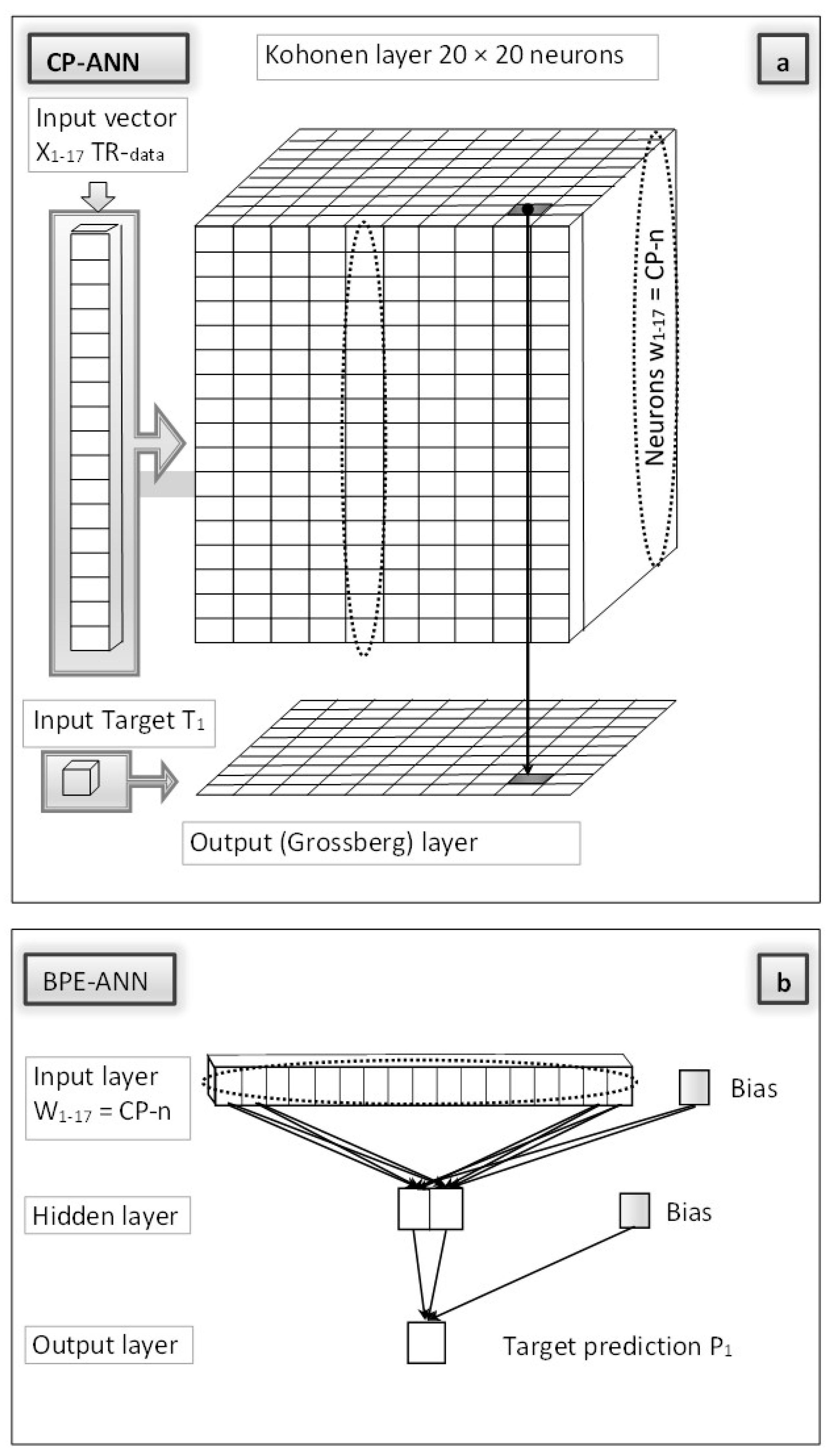
3.3. Counter-Propagation–Back-Propagation Artificial Neural Network (CP-BPE-ANN)
3.3.1. CP-BPE-ANN “Stage I”
3.3.2. CP-BPE-ANN “Stage II”
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schmeisser, S.; Miccoli, A.; von Bergen, M.; Berggren, E.; Braeuning, A.; Busch, W.; Desaintes, C.; Gourmelon, A.; Grafström, R.; Harrill, J.; et al. New approach methodologies in human regulatory toxicology-Not if, but how and when! Environ. Int. 2023, 178, 108082. [Google Scholar] [CrossRef] [PubMed]
- Agatonovic-Kustrin, S.; Morton, D.W.; Razic, S. Modelling of Pesticide Aquatic Toxicity. Comb. Chem. High Throughput Screen. 2014, 17, 808–818. [Google Scholar] [CrossRef]
- In, Y.; Lee, S.K.; Kim, P.J.; No, K.T. Prediction of Acute Toxicity to Fathead Minnow by Local Model Based QSAR and Global QSAR Approaches. Bull. Korean Chem. Soc. 2012, 33, 613–619. [Google Scholar] [CrossRef][Green Version]
- Mazzatorta, P.; Benfenati, E.; Neagu, C.D.; Gini, G. Tuning neural and fuzzy-neural networks for toxicity modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.P.; Gupta, S.; Rai, P. Predicting acute aquatic toxicity of structurally diverse chemicals in fish using artificial intelligence approaches. Ecotoxicol. Environ. Saf. 2013, 95, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Drgan, V.; Župerl, Š.; Vračko, M.; Cappelli, C.I.; Novič, M. CPANNatNIC software for counter-propagation neural network to assist in read-across. J. Cheminform. 2017, 9, 30. [Google Scholar] [CrossRef]
- OECD. Guideline no. ENV/JM/MONO 2, Chapter 5. In Guidance on the Principle of Measure of Goodness-of-Fit, Robustness and Predictivity; OECD: Paris, France, 2007; p. 103. [Google Scholar]
- Lovrić, M.; Pavlović, K.; Žuvela, P.; Spataru, A.; Lučić, B.; Kern, R.; Wong, M.W. Machine learning in prediction of intrinsic aqueous solubility of drug-like compounds: Generalization, complexity, or predictive ability? J. Chemometr. 2021, 35, e3349. [Google Scholar] [CrossRef]
- 9Sluga, J.; Venko, K.; Drgan, V.; Novič, M. QSPR Models for Prediction of Aqueous Solubility: Exploring the Potency of Randic-type Indices. Croat. Chem. Acta 2020, 93, 311–319. [Google Scholar] [CrossRef]
- Kim, T.; You, B.H.; Han, S.; Shin, H.C.; Chung, K.C.; Park, H. Quantum Artificial Neural Network Approach to Derive a Highly Predictive 3D-QSAR Model for Blood-Brain Barrier Passage. Int. J. Mol. Sci. 2021, 22, 10995. [Google Scholar] [CrossRef]
- Alexander, D.L.J.; Tropsha, A.; Winkler, D.A. Beware of R2: Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [Google Scholar] [CrossRef]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, But Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P. Principles of QSAR Modeling: Comments and Suggestions from Personal Experience. Int. J. Quant. Struct. -Prop. Relatsh. 2020, 5, 37. [Google Scholar] [CrossRef]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A.; Gramatica, P.; Gombar, V.K. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Muratov, E.N.; Bajorath, J.; Sheridan, R.P.; Tetko, I.V.; Filimonov, D.; Poroikov, V.; Oprea, T.I.; Baskin, I.I.; Varnek, A.; Roitberg, A.; et al. QSAR without borders. Chem. Soc. Rev. 2020, 10, 531, Correction in Chem. Soc. Rev. 2020, 49, 3716–3716. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.; Goel, A.K.; Kumar, A. The role of artificial neural network and machine learning in utilizing spatial information. Spat. Inf. Res. 2023, 31, 275–285. [Google Scholar] [CrossRef]
- Emmert-Streib, F.; Yang, Z.; Feng, H.; Tripathi, S.; Dehmer, M. An Introductory Review of Deep Learning for Prediction Models With Big Data. Front. Artif. Intell. 2020, 3, 4. [Google Scholar] [CrossRef]
- Emmert-Streib, F.; Yli-Harja, O.; Dehmer, M. Explainable artificial intelligence and machine learning: A reality rooted perspective. Wires Data Min. Knowl. 2020, 10, e1368. [Google Scholar] [CrossRef]
- Emmert-Streib, F.; Yli-Harja, O.; Dehmer, M. Artificial Intelligence: A Clarification of Misconceptions, Myths and Desired Status. Front. Artif. Intell. 2020, 3, 524339. [Google Scholar] [CrossRef]
- Alzubaidi, L.; Zhang, J.L.; Humaidi, A.J.; Al-Dujaili, A.; Duan, Y.; Al-Shamma, O.; Santamaria, J.; Fadhel, M.A.; Al-Amidie, M.; Farhan, L. Review of deep learning: Concepts, CNN architectures, challenges, applications, future directions. J. Big Data-Ger. 2021, 8, 53. [Google Scholar] [CrossRef]
- Jimenez-Luna, J.; Cuzzolin, A.; Bolcato, G.; Sturlese, M.; Moro, S. A Deep-Learning Approach toward Rational Molecular Docking Protocol Selection. Molecules 2020, 25, 2487. [Google Scholar] [CrossRef] [PubMed]
- Baskin, I.I.; Palyulin, V.A.; Zefirov, N.S. Neural networks in building QSAR models. Methods Mol. Biol. 2008, 458, 137–158. [Google Scholar] [PubMed]
- Dayhoff, J. Neural Network Architectures, An Introduction; Van Nostrand Reinhold: New York, NY, USA, 1990. [Google Scholar]
- Hecht-Nielsen, R. Counterpropagation networks. Appl. Opt. 1987, 26, 4979–4983. [Google Scholar] [CrossRef] [PubMed]
- Zupan, J.; Novič, M.; Ruisanchez, I. Kohonen and counterpropagation artificial neural networks in analytical chemistry. Chemom. Intell. Lab. Syst. 1997, 38, 1–23. [Google Scholar] [CrossRef]
- Kuzmanovski, I.; Novič, M. Counter-propagation neural networks in Matlab. Chemom. Intell. Lab. Syst. 2008, 90, 84–91. [Google Scholar] [CrossRef]
- Rumelhart, D.E.; Hinton, G.E.; Williams, R.J. Learning Representations by Back-Propagating Errors. Nature 1986, 323, 533–536. [Google Scholar] [CrossRef]
- Zupan, J.; Gasteiger, J. Neural Networks in Chemistry and Drug Design; Wiley: Weinheim, Germany, 1999. [Google Scholar]
- Borišek, J.; Drgan, V.; Minovski, N.; Novič, M. Mechanistic interpretation of artificial neural network-based QSAR model for prediction of cathepsin K inhibition potency. J. Chemometr. 2014, 28, 272–281. [Google Scholar] [CrossRef]
- Drgan, V.; Župerl, Š.; Vračko, M.; Como, F.; Novič, M. Robust modelling of acute toxicity towards fathead minnow (Pimephales promelas) using counter-propagation artificial neural networks and genetic algorithm. SAR QSAR Environ. Res. 2016, 27, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Sorkun, M.C.; Khetan, A.; Er, S. AqSolDB, a curated reference set of aqueous solubility and 2D descriptors for a diverse set of compounds. Sci. Data 2019, 6, 143. [Google Scholar] [CrossRef]
- Sorkun, M.C.; Koelman, J.M.V.A.; Er, S. Pushing the limits of solubility prediction via quality-oriented data selection. iScience 2021, 24, 101961. [Google Scholar] [CrossRef]
- Lusci, A.; Pollastri, G.; Baldi, P. Deep Architectures and Deep Learning in Chemoinformatics: The Prediction of Aqueous Solubility for Drug-Like Molecules. J. Chem. Inf. Model. 2013, 53, 1563–1575. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemometr. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Jaworska, J.; Nikolova-Jeliazkova, N.; Aldenberg, T. QSAR applicability domain estimation by projection of the training set in descriptor space: A review. Atla-Altern. Lab. Anim. 2005, 33, 445–459. [Google Scholar] [CrossRef]
- Minovski, N.; Župerl, Š.; Drgan, V.; Novič, M. Assessment of applicability domain for multivariate counter-propagation artificial neural network predictive models by minimum Euclidean distance space analysis: A case study. Anal. Chim. Acta 2013, 759, 28–42. [Google Scholar] [CrossRef]
- Russom, C.L.; Bradbury, S.P.; Broderius, S.J.; Hammermeister, D.E.; Drummond, R.A. Predicting modes of toxic action from chemical structure: Acute toxicity in the fathead minnow (Pimephales promelas). Environ. Toxicol. Chem. 1997, 16, 948–967. [Google Scholar] [CrossRef]
- Gissi, A.; Gadaleta, D.; Floris, M.; Olla, S.; Carotti, A.; Novellino, E.; Benfenati, E.; Nicolotti, O. An Alternative QSAR-Based Approach for Predicting the Bioconcentration Factor for Regulatory Purposes. ALTEX-Altern. Anim. Exp. 2014, 31, 23–36. [Google Scholar]
- Gissi, A.; Nicolotti, O.; Carotti, A.; Gadaleta, D.; Lombardo, A.; Benfenati, E. Integration of QSAR models for bioconcentration suitable for REACH. Sci. Total Environ. 2013, 456, 325–332. [Google Scholar] [CrossRef]
- Landrum, G.; Tosco, P.; Kelley, B.; Ric; Cosgrove, D.; sriniker; gedeck; Vianello, R.; NadineSchneider; Kawashima, E.; et al. rdkit/rdkit: 2023_09_4 (Q3 2023). 2024. Available online: https://doi.org/10.5281/zenodo.10460537 (accessed on 4 April 2024).
- Kode. DRAGON (Software for Molecular Descriptor Calculation), Version 7.0; Kode: Pisa, Italy, 2016. [Google Scholar]
- Willighagen, E.L.; Mayfield, J.W.; Alvarsson, J.; Berg, A.; Carlsson, L.; Jeliazkova, N.; Kuhn, S.; Pluskal, T.; Rojas-Chertó, M.; Spjuth, O.; et al. The Chemistry Development Kit (CDK) v2.0: Atom typing, depiction, molecular formulas, and substructure searching. J. Cheminform. 2017, 9, 33, Erratum in J. Cheminform. 2017, 9, 53. [Google Scholar] [CrossRef]


{kind=link}
| Model | Number of Descriptors | 3 Size | 2 RMSE | |||
|---|---|---|---|---|---|---|
| 4 Training (Neurons) | Training (Compounds) | Test | Validation | |||
| CP-ANN 1 (AqSol) | 17 | 20 × 20 | / | 0.68 | 0.88 | 0.92 |
| CP-BPE-ANN 1-1 | 5 | 0.88 | 0.99 | 0.86 | 0.83 | |
| CP-BPE-ANN 1-2 | 10 | 0.71 | 0.90 | 0.86 | 0.83 | |
| CP-BPE-ANN 1-3 | 12 | 0.73 | 0.89 | 0.86 | 0.80 | |
| CP-ANN 2 (AqSol) | 17 | 20 × 20 | / | 0.68 | 0.90 | 0.91 |
| CP-BPE-ANN 2-1 | 5 | 0.84 | 0.98 | 0.89 | 0.84 | |
| CP-BPE-ANN 2-2 | 10 | 0.74 | 0.92 | 0.84 | 0.88 | |
| CP-BPE-ANN 2-3 | 12 | 0.74 | 0.92 | 0.98 | 0.88 | |
| CP-ANN 3 (AqSol) | 6 | 15 × 15 | / | 0.79 | 0.80 | 0.80 |
| CP-BPE-ANN 3-1 | 5 | 0.95 | 1.10 | 0.93 | 0.90 | |
| CP-BPE-ANN 3-2 | 10 | 0.84 | 1.04 | 0.91 | 0.83 | |
| CP-BPE-ANN 3-3 | 12 | 0.78 | 1.00 | 0.89 | 0.83 | |
| CP-ANN 4 (AqSol) | 9 | 20 × 20 | / | 0.66 | 0.85 | 0.86 |
| CP-BPE-ANN 4-1 | 5 | 0.85 | 0.95 | 0.85 | 0.81 | |
| CP-BPE-ANN 4-2 | 10 | 0.82 | 0.93 | 0.93 | 0.80 | |
| CP-BPE-ANN 4-3 | 12 | 0.79 | 0.91 | 0.96 | 0.94 | |
| CP-ANN 5 (FHM 2) | 23 | 18 × 18 | / | 5 0.87 | / | 1.01 |
| CP-BPE-ANN 5-1 | 5 | 0.45 | 0.69 | / | 0.95 | |
| CP-BPE-ANN 5-2 | 10 | 0.38 | 0.65 | / | 0.94 | |
| CP-BPE-ANN 4-3 | 12 | 0.36 | 0.66 | / | 0.91 | |
| CP-ANN 6 (FHM 8) | 28 | 25 × 25 | / | 6 0.82 | / | 0.98 |
| CP-BPE-ANN 6-1 | 5 | 0.23 | 0.70 | / | 1.06 | |
| CP-BPE-ANN 6-2 | 10 | 0.21 | 0.68 | / | 1.06 | |
| CP-BPE-ANN 6-3 | 12 | 0.20 | 0.68 | / | 1.04 | |
| CP-ANN 7 (BCF) | 10 | 9 × 9 | / | 0.68 | 0.84 | 0.93 |
| CP-BPE-ANN 7-1 | 5 | 0.30 | 0.70 | 0.78 | 0.79 | |
| CP-BPE-ANN 7-2 | 10 | 0.24 | 0.68 | 0.83 | 0.88 | |
| CP-BPE-ANN 7-3 | 12 | 0.20 | 0.69 | 0.84 | 0.88 | |
| Model | IDoutlier | 1 LogS | ||
|---|---|---|---|---|
| 2 Exp. | 3 PredCP-ANN | 4 PredCP-BPE-ANN | ||
| CP-BPE-ANN 1 | 330 | −5.418 | −4.114 | −5.653 |
| CP-BPE-ANN 2 | 330 | −5.418 | 0.057 | −0.460 |
| 638 | −6.294 | −6.134 | −6.052 | |
| CP-BPE-ANN 3 | / | / | / | / |
| CP-BPE-ANN 4 | / | / | / | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drgan, V.; Venko, K.; Sluga, J.; Novič, M. Merging Counter-Propagation and Back-Propagation Algorithms: Overcoming the Limitations of Counter-Propagation Neural Network Models. Int. J. Mol. Sci. 2024, 25, 4156. https://doi.org/10.3390/ijms25084156
Drgan V, Venko K, Sluga J, Novič M. Merging Counter-Propagation and Back-Propagation Algorithms: Overcoming the Limitations of Counter-Propagation Neural Network Models. International Journal of Molecular Sciences. 2024; 25(8):4156. https://doi.org/10.3390/ijms25084156
Chicago/Turabian StyleDrgan, Viktor, Katja Venko, Janja Sluga, and Marjana Novič. 2024. "Merging Counter-Propagation and Back-Propagation Algorithms: Overcoming the Limitations of Counter-Propagation Neural Network Models" International Journal of Molecular Sciences 25, no. 8: 4156. https://doi.org/10.3390/ijms25084156
APA StyleDrgan, V., Venko, K., Sluga, J., & Novič, M. (2024). Merging Counter-Propagation and Back-Propagation Algorithms: Overcoming the Limitations of Counter-Propagation Neural Network Models. International Journal of Molecular Sciences, 25(8), 4156. https://doi.org/10.3390/ijms25084156