

The Role of Sirtuin-1 (SIRT1) in the Physiology and Pathophysiology of the Human Placenta



{kind=link}
{kind=link}
{kind=link}
Abstract
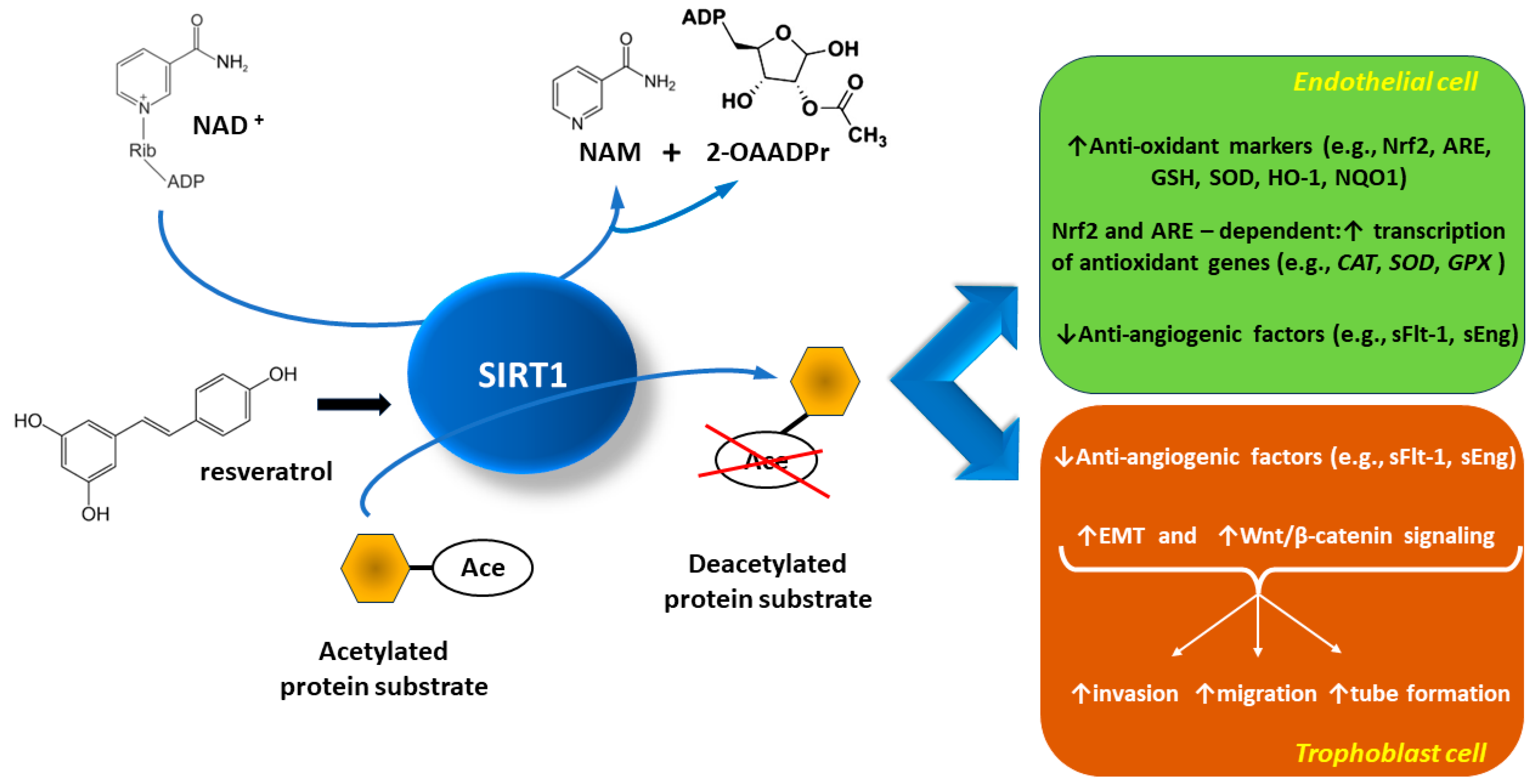
:1. Introduction
2. SIRT1 in the Regulation of Trophoblast Function
2.1. Effects towards Placental Development and Differentiation
2.2. Effects of SIRT1 on Autophagy within Trophoblast
2.3. Effects on Cell Senescence Phenotype Occurrence within the Placenta
3. SIRT1 and PPARγ
3.1. Role of SIRT1- and PPARγ-Dependent Signaling Pathways in Placental Pathology
3.1.1. Effects of Hypoxia on PPARγ Activity
3.1.2. Effects of Hypoxia on SIRT1 Activity
3.1.3. Effects of SIRT1 and PPARγ Action towards Placentas Exposed to Oxidative Stress
3.1.4. Effects of SIRT1 and PPARγ towards Placentas Affected by Inflammatory Response
3.1.5. Correlations between Hyperglycemia and Placental SIRT1/PPARγ Activity
4. SIRT1-Dependent Prevention of Pre-Eclampsia
4.1. SIRT1 Protective Actions towards Vascular Endothelial Cells
4.1.1. SIRT1 and the Protection of Endothelial Cells against Oxidative Stress and Inflammatory Response
4.1.2. SIRT1 May Protect Endothelial Cells through Autophagy Regulation
4.1.3. SIRT1 and Possible Protection of Endothelial Cells against Senescence
4.2. Anti-Inflammatory Action of SIRT1 within the Placenta in the Context of Pre-Eclampsia
4.3. SIRT1 Alleviates PE Course on Animal Models of PE
4.4. SIRT1 Induction Alleviates PE Manifestations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-OAADPr | 2′-O-acetyl-adenosine diphosphate(ADP)-ribose. |
| AMPK | adenosine monophosphate(AMP)-activated protein kinase. |
| ARE | antioxidant response element. |
| ATG5: ATG7, ATG8 | autophagy related-proteins 5, 7, 8. |
| Beclin-1 | the mammalian ortholog of yeast Atg6/Vps30, an essential autophagy protein that contains a Bcl-2 homology-3 domain. |
| BECN1 | Beclin-1 gene. |
| CAT: CAT | catalase gene, catalase (anti-oxidant enzyme), respectively. |
| CD40 | 50-kDa integral membrane protein of the tumor necrosis factor receptor (TNF-R) family. |
| CSP | cell senescence phenotype. |
| CTSD | cathepsin D. |
| EMT | epithelial–mesenchymal transition. |
| eNOS | endothelial nitric oxide synthase. |
| ETC | electron transport chain. |
| FEEL-1 | link domain-containing scavenger receptor-1. |
| FoxOs | forkhead O class box transcription factors. |
| FoxO1: FoxO3 | forkhead box protein O1 and O3, respectively. |
| GCM1 | glial cells missing-1 (transcription factor). |
| GLUT1 | glucose transporter 1. |
| GOT | glutamicoxaloacetic transaminase. |
| GPT | glutamic pyruvic transaminase. |
| GPX | glutathione peroxidase. |
| GPX1: GPX2, GPX3 | glutathione peroxidase isoforms 1, 2, and 3, respectively. |
| GSH | glutathione. |
| H2O2 | hydrogen peroxide. |
| HIF | hypoxia-inducible factor (HIF). |
| HIF-1α | hypoxia-inducible factor 1 subunit alpha. |
| HIF-2α | hypoxia-inducible factor 2 subunit alpha. |
| HIFα: HIFβ | domains that make up the (hypoxia-inducible factor) HIF molecule domain. |
| HMGB1 | high mobility group box 1 (non-histone nuclear protein). |
| HO-1 | heme oxygenase-1. |
| HSF1 | heat shock transcription factor 1. |
| HSP70 | 70-kDa heat shock proteins. |
| HSPs | heat shock proteins. |
| HUVEC | human umbilical vein endothelial cells. |
| ICAM-1 | inter-cellular adhesion molecule 1, also known as CD54 (cluster of differentiation 54). |
| IL-1: IL-1β, IL-6, IL-8, IL-10, IL-12 | interleukins: 1, 1 beta, 6, 8, 10, and 12. |
| IUFD | intra-uterine fetal death. |
| IL-12p40 | interleukin-12 subunit beta (p40). |
| KO | knockout. |
| L-NAME | L-NG-nitroarginine methyl ester. |
| LAMP1: LAMP2 | lysosomal associated membrane protein 1,2. |
| LC3 | microtubule-associated protein 1 light chain 3 (MAP1LC3), a human homologue of yeast Atg8, an essential component of autophagy. |
| LC3-II | membrane-bound: lipidated form of LC3. |
| LDH | lactate dehydrogenase. |
| LKB1 | liver kinase B1. |
| LOX-1 | lectin-like oxidized low-density lipoprotein-1. |
| LPL | placental lipoprotein lipase. |
| LPS | lipopolysaccharide. |
| MCP-1 | monocyte chemoattractant protein-1. |
| mi-R217: mi-R34a, mi-R155, mi-R22 | micro-RNA molecules. |
| MnSOD | manganese superoxide dismutase (antioxidant enzyme). |
| mTOR | mammalian target of rapamycin (an ubiquitous serine-threonine protein kinase). |
| NAD | nicotinamide adenine dinucleotide. |
| NAD+ | nicotinamide adenine dinucleotide (oxidized form). |
| NADH | nicotinamide adenine dinucleotide (reduced form, H for hydrogen). |
| NAM | nicotinamide. |
| NCoR1 | nuclear receptor co-repressor-1. |
| NDRG1 | N-myc downstream-regulated gene 1 (formerly known as Drg1, Cap43, Rit42, RTP, and PROXY-1). |
| Nampt | nicotinamide mononucleotide adenyltransferase. |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells. |
| NMN | nicotinamide mononucleotide. |
| NMNAT | nicotinamide-(mono)nucleotide adenylyltransferase. |
| NMNAT1: NMNAT2, NMNAT3 | nicotinamide-(mono)nucleotide adenylyltransferase isoforms: 1,2, and 3. |
| NO | nitric oxide. |
| NOS | nitric oxide synthase. |
| Nox | nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. |
| NQO1 | nicotinamide adenine dinucleotide phosphate (NADPH)-quinone oxidoreductase-1. |
| Nrf2 | nuclear factor erythroid 2-related factor 2. |
| p62 | ubiquitin-binding scaffold protein, also known as sequestosome 1 (SQSTM1). |
| p53 | protein encoded by the TP53 tumor suppressor gene, marker of cell senescence. |
| p21 | cyclin-dependent kinase inhibitor p21, protein marker of cell senescence. |
| PE | pre-eclampsia. |
| PlGF | placenta growth factor. |
| PPARγ | peroxisome proliferator-activated receptor γ. |
| PPARγ2 | an isoform of PPARγ typical for adipose tissue. |
| PPRE | PPARγ-reactive elements. |
| PRDM1 | positive regulatory (PR) domain zinc finger protein 1, a coactivator selectively activating PPARγ. |
| Prdm16 | positive regulatory domain containing 16 |
| qPCR | quantitative polymerase chain reaction. |
| Rab7 | a small GTPase: member of the Rab family that controls transport to late endocytic compartments such as late endosomes and lysosomes. |
| RAB7 | Rab7 gene. |
| RAGE | receptor for advanced glycation end-products. |
| ROS | reactive oxygen species. |
| RUPP | reduced uterine perfusion pressure. |
| RXR | retinoid X-receptor. |
| SASP | senescence-associated secretory phenotype. |
| sEng | soluble endoglin: the extracellular domain of membrane endoglin. |
| sFlt-1 | soluble fms-like tyrosine kinase 1, also known as soluble vascular endothelial growth factor (VEGF) receptor-1. |
| siRNA | small interfering RNA. |
| SIRT1 | silent information regulator 2 homolog 1 or sirtuin-1. |
| SIRT7 | sirtuins 1 to 7. |
| SMAD2: SMAD3 | small mothers against decapentaplegic proteins 2 and 3 (transcription factors). |
| SMRT | silencing mediator of retinoid and thyroid hormone receptors. |
| SOD | superoxide dismutase. |
| SQSTM1 | sequestosome 1: also known as ubiquitin-binding scaffold protein p62. |
| SREC-1 | scavenger receptor expressed by endothelial cell-1. |
| SRT2104 | experimental drug, a selective small molecule activator of SIRT1. |
| STAT | signal transducer and activator of transcription (transcription factor). |
| STAT3 | signal transducer and activator of transcription 3 (transcription factor). |
| TCA | tricarboxylic acid cycle, also known as the Krebs cycle or the citric acid cycle. |
| TFEB | transcription factor EB (TFEB), a member of the MiT/TFE family of basic helix-loop-helix leucine zipper transcription factors, a key regulator of the autophagy/lysosomal-to-nucleus signaling pathway. |
| TLR2: TLR4 | toll-like receptor 2 and 4. |
| TNF-α | tumor necrosis factor alpha. |
| TRX | thioredoxin (anti-oxidant protein). |
| TSC | trophoblast stem cells. |
| TZD | thiazolidinediones: synthetic activators of PPARγ. |
| WT TSC | wild-type trophoblast stem cells (TSC). |
| VCAM-1 | vascular cell adhesion molecule 1, also known as CD106 (cluster of differentiation 106). |
| VEGF | vascular endothelial growth factor. |
References
- Pham, J.; Arul Nambi Rajan, K.; Li, P.; Parast, M.M. The role of Sirtuin1-PPARγ axis in placental development and function. J. Mol. Endocrinol. 2018, 60, R201–R212. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. What is the placenta? Am. J. Obstet. Gynecol. 2015, 213 (Suppl. S4), S6.E1–S6.E4. [Google Scholar] [CrossRef] [PubMed]
- James, J.L.; Carter, A.M.; Chamley, L.W. Human placentation from nidation to 5 weeks of gestation. Part I: What do we know about formative placental development following implantation? Placenta 2012, 33, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Soncin, F.; Natale, D.; Parast, M.M. Signaling pathways in mouse and human trophoblast differentiation: A comparative review. Cell. Mol. Life Sci. 2015, 72 (Suppl. S4), 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.J. Why is placentation abnormal in preeclampsia? Am. J. Obstet. Gynecol. 2015, 213, S115–S122. [Google Scholar] [CrossRef]
- Thornburg, K.L.; Marshall, N. The placenta is the center of the chronic disease universe. Am. J. Obstet. Gynecol. 2015, 213 (Suppl. S4), S14–S20. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, C.; Pei, J.; Li, M.; Gu, W. SIRT1: A Novel Protective Molecule in Pre-eclampsia. Int. J. Med. Sci. 2022, 19, 993–1002. [Google Scholar] [CrossRef]
- Kojima, J.; Dai, Y.; Suzuki, T.; Ono, M.; Nishi, H. Sirtuin 1 is a potential therapeutic candidate gene for fetal growth restriction via insulin-like 4. J. Matern. Fetal Neonatal Med. 2023, 36, 2253486. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Hannan, N.J.; Beard, S.; Binder, N.K.; Onda, K.; Kaitu’u-Lino, T.J.; Chen, Q.; Tuohey, L.; De Silva, M.; Tong, S. Key players of the necroptosis pathway RIPK1 and SIRT2 are altered in placenta from preeclampsia and fetal growth restriction. Placenta 2017, 51, 1–9. [Google Scholar] [CrossRef]
- Yu, Y.; An, X.; Fan, D. Histone Deacetylase Sirtuin 2 Enhances Viability of Trophoblasts Through p65-Mediated MicroRNA-146a/ACKR2 Axis. Reprod. Sci. 2021, 28, 1370–1381. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, Y.; Liu, M.; Liao, L.; Wei, X.; Zhou, R. SIRT3 deficiency affects the migration, invasion, tube formation and necroptosis of trophoblast and is implicated in the pathogenesis of preeclampsia. Placenta 2022, 120, 1–9. [Google Scholar] [CrossRef]
- Castex, J.; Willmann, D.; Kanouni, T.; Arrigoni, L.; Li, Y.; Friedrich, M.; Schleicher, M.; Wöhrle, S.; Pearson, M.; Kraut, N.; et al. Inactivation of Lsd1 triggers senescence in trophoblast stem cells by induction of Sirt4. Cell Death Dis. 2017, 8, e2631. [Google Scholar] [CrossRef] [PubMed]
- Sandvoß, M.; Potthast, A.B.; von Versen-Höynck, F.; Das, A.M. HELLP Syndrome: Altered Hypoxic Response of the Fatty Acid Oxidation Regulator SIRT 4. Reprod. Sci. 2017, 24, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Bartho, L.A.; O’Callaghan, J.L.; Fisher, J.J.; Cuffe, J.S.M.; Kaitu’u-Lino, T.J.; Hannan, N.J.; Clifton, V.L.; Perkins, A.V. Analysis of mitochondrial regulatory transcripts in publicly available datasets with validation in placentae from pre-term, post-term and fetal growth restriction pregnancies. Placenta 2021, 112, 162–171. [Google Scholar] [CrossRef]
- Huynh, J.; Dawson, D.; Roberts, D.; Bentley-Lewis, R. A systematic review of placental pathology in maternal diabetes mellitus. Placenta 2015, 36, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Kwak-Kim, J.; Bao, S.; Lee, S.K.; Kim, J.W.; Gilman-Sachs, A. Immunological modes of pregnancy loss: Inflammation, immune effectors, and stress. Am. J. Reprod. Immunol. 2014, 72, 129–140. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Piani, F.; Crescimanno, C.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Modulation of NRF2/KEAP1 signaling in preeclampsia. Cells 2023, 12, 1545. [Google Scholar] [CrossRef]
- Singh, V.; Singh, V.; Ubaid, S. Role of silent information regulator 1 (SIRT1) in regulating oxidative stress and inflammation. Inflammation 2020, 43, 1589–1598. [Google Scholar] [CrossRef]
- Arul Nambi Rajan, K.; Khater, M.; Soncin, F.; Pizzo, D.; Moretto-Zita, M.; Pham, J.; Stus, O.; Iyer, P.; Tache, V.; Laurent, L.C.; et al. Sirtuin1 is required for proper trophoblast differentiation and placental development in mice. Placenta 2018, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Mitton, A.; Lim, R.; Barker, G.; Riley, C.; Permezel, M. SIRT1 is a novel regulator of key pathways of human labor. Biol. Reprod. 2011, 84, 167–178. [Google Scholar] [CrossRef]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Borg, A.J.; Yong, H.E.; Lappas, M.; Degrelle, S.A.; Keogh, R.J.; Da Silva-Costa, F.; Fournier, T.; Abumaree, M.; Keelan, J.A.; Kalionis, B.; et al. Decreased STAT3 in human idiopathic fetal growth restriction contributes to trophoblast dysfunction. Reproduction 2015, 149, 523–532. [Google Scholar] [CrossRef]
- Erlebacher, A.; Price, K.A.; Glimcher, L.H. Maintenance of mouse trophoblast stem cell proliferation by TGF-beta/activin. Dev. Biol. 2004, 275, 158–169. [Google Scholar] [CrossRef]
- Tang, S.; Huang, G.; Fan, W.; Chen, Y.; Ward, J.M.; Xu, X.; Xu, Q.; Kang, A.; McBurney, M.W.; Fargo, D.C.; et al. SIRT1-mediated deacetylation of CRABPII regulates cellular retinoic acid signaling and modulates embryonic stem cell differentiation. Mol. Cell 2014, 55, 843–855. [Google Scholar] [CrossRef]
- Takahashi, Y.; Takahashi, M.; Carpino, N.; Jou, S.T.; Chao, J.R.; Tanaka, S.; Shigeyoshi, Y.; Parganas, E.; Ihle, J.N. Leukemia inhibitory factor regulates trophoblast giant cell differentiation via Janus kinase 1-signal transducer and activator of transcription 3-suppressor of cytokine signaling 3 pathway. Mol. Endocrinol. 2008, 22, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, F.; Xu, Y.; Wei, J.; Zhang, Y.; Yang, H.; Gao, B.; Yu, G.; Fang, D. JAK1-mediated Sirt1 phosphorylation functions as a negative feedback of the J.A.K1-STAT3 pathway. J. Biol. Chem. 2018, 293, 11067–11075. [Google Scholar] [CrossRef] [PubMed]
- Psilopatis, I.; Vrettou, K.; Fleckenstein, F.N.; Theocharis, S. The Role of Peroxisome Proliferator-Activated Receptors in Preeclampsia. Cells 2023, 12, 647. [Google Scholar] [CrossRef]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Kobayashi, M.; Kitagishi, Y. Expression and Function of PPARs in Placenta. PPAR Res. 2013, 2013, 256508. [Google Scholar] [CrossRef]
- Pei, J.; Liu, Z.; Wang, C.; Chu, N.; Liu, L.; Tang, Y.; Liu, H.; Xiang, Q.; Cheng, H.; Li, M.; et al. Progesterone Attenuates SIRT1-Deficiency-Mediated Pre-Eclampsia. Biomolecules 2022, 12, 422. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Dröge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Klionsky, D.J. Autophagy contributes to lysosomal storage disorders. Autophagy 2012, 8, 715–716. [Google Scholar] [CrossRef]
- Yoshimori, T. Autophagy: Paying Charon’s toll. Cell 2007, 128, 833–836. [Google Scholar] [CrossRef]
- Nakashima, A.; Cheng, S.B.; Ikawa, M.; Yoshimori, T.; Huber, W.J.; Menon, R.; Huang, Z.; Fierce, J.; Padbury, J.F.; Sadovsky, Y.; et al. Evidence for lysosomal biogenesis proteome defect and impaired autophagy in preeclampsia. Autophagy 2020, 16, 1771–1785. [Google Scholar] [CrossRef]
- Wang, P.; Huang, C.X.; Gao, J.J.; Shi, Y.; Li, H.; Yan, H.; Yan, S.J.; Zhang, Z. Resveratrol induces SIRT1-Dependent autophagy to prevent H2O2-Induced oxidative stress and apoptosis in HTR8/SVneo cells. Placenta 2020, 91, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xia, B.; Tang, L.; Wu, W.; Tang, J.; Liang, Y.; Yang, H.; Zhang, Z.; Lu, Y.; Chen, G.; et al. Echinacoside protects against MPTP/MPP+-induced neurotoxicity via regulating autophagy pathway mediated by Sirt1. Metab. Brain Dis. 2019, 34, 203–212. [Google Scholar] [CrossRef]
- Jiang, Q.; Hao, R.; Wang, W.; Gao, H.; Wang, C. SIRT1/Atg5/autophagy are involved in the antiatherosclerosis effects of ursolic acid. Mol. Cell. Biochem. 2016, 420, 171–184. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signaling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Napolitano, G.; Esposito, A.; Choi, H.; Matarese, M.; Benedetti, V.; Di Malta, C.; Monfregola, J.; Medina, D.L.; Lippincott-Schwartz, J.; Ballabio, A. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 2018, 9, 3312. [Google Scholar] [CrossRef] [PubMed]
- Akaishi, R.; Yamada, T.; Nakabayashi, K.; Nishihara, H.; Furuta, I.; Kojima, T.; Morikawa, M.; Yamada, T.; Fujita, N.; Minakami, H. Autophagy in the placenta of women with hypertensive disorders in pregnancy. Placenta 2014, 35, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Qi, H.B.; Kamana, K.C.; Zhang, X.M.; Zhang, H.; Baker, P.N. Excessive autophagy induces the failure of trophoblast invasion and vasculature: Possible relevance to the pathogenesis of preeclampsia. J. Hypertens. 2015, 33, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Choi, S.J.; Kim, K.H.; Cho, E.Y.; Kim, J.H.; Roh, C.R. Autophagy-related proteins, LC3 and Beclin-1, in placentas from pregnancies complicated by preeclampsia. Reprod. Sci. 2008, 15, 912–920. [Google Scholar] [CrossRef]
- Feng, L.; Chen, M.; Li, Y.; Li, M.; Hu, S.; Zhou, B.; Zhu, L.; Yu, L.; Zhou, Q.; Tan, L.; et al. Sirt1 deacetylates and stabilizes p62 to promote hepato-carcinogenesis. Cell Death Dis. 2021, 12, 405. [Google Scholar] [CrossRef]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; et al. Deacetylation of Nuclear LC3 drives autophagy initiation under starvation. Mol. Cell. 2015, 57, 456–466. [Google Scholar] [CrossRef]
- Pi, Q.Z.; Wang, X.W.; Jian, Z.L.; Chen, D.; Zhang, C.; Wu, Q.C. Melatonin Alleviates Cardiac Dysfunction Via Increasing Sirt1-Mediated Beclin-1 Deacetylation and Autophagy During Sepsis. Inflammation 2021, 44, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef]
- Sultana, Z.; Maiti, K.; Dedman, L.; Smith, R. Is there a role for placental senescence in the genesis of obstetric complications and fetal growth restriction? Am. J. Obstet. Gynecol. 2018, 218, S762–S773. [Google Scholar] [CrossRef]
- Biron-Shental, T.; Sukenik-Halevy, R.; Sharon, Y.; Goldberg-Bittman, L.; Kidron, D.; Fejgin, M.D.; Amiel, A. Short telomeres may play a role in placental dysfunction in preeclampsia and intrauterine growth restriction. Am. J. Obstet. Gynecol. 2010, 202, 381.e1–381.e7. [Google Scholar] [CrossRef]
- Tasta, O.; Swiader, A.; Grazide, M.H.; Rouahi, M.; Parant, O.; Vayssière, C.; Bujold, E.; Salvayre, R.; Guerby, P.; Negre-Salvayre, A. A role for 4-hydroxy-2-nonenal in premature placental senescence in preeclampsia and intrauterine growth restriction. Free Radic. Biol. Med. 2021, 164, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Halperin, D.S.; Tontonoz, P. Before they were fat: Adipocyte progenitors. Cell Metab. 2008, 8, 454–457. [Google Scholar] [CrossRef]
- Koppen, A.; Kalkhoven, E. Brown vs white adipocytes: The PPARγ coregulator story. FEBS Lett. 2010, 584, 3250–3259. [Google Scholar] [CrossRef]
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–286. [Google Scholar] [CrossRef]
- Feng, S.; Reuss, L.; Wang, Y. Potential of Natural Products in the Inhibition of Adipogenesis through Regulation of PPARγ Expression and/or Its Transcriptional Activity. Molecules 2016, 21, 1278. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.R.; Milner, J. SIRT1, metabolism and cancer. Curr. Opin. Oncol. 2012, 24, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.E., Jr.; Pruitt, W.M.; Pruitt, K. Diverse roles of SIRT1 in cancer biology and lipid metabolism. Int. J. Mol. Sci. 2015, 16, 950–965. [Google Scholar] [CrossRef] [PubMed]
- Farghali, H.; Kutinová Canová, N.; Lekić, N. Resveratrol and related compounds as antioxidants with an allosteric mechanism of action in epigenetic drug targets. Physiol. Res. 2013, 62, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. SIRT1 is regulated by a PPARγ-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010, 38, 7458–7471. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kulp, S.K.; Chen, C.S. Energy restriction as an antitumor target of thiazolidinediones. J. Biol. Chem. 2010, 285, 9780–9791. [Google Scholar] [CrossRef] [PubMed]
- Calleri, E.; Pochetti, G.; Dossou, K.S.S.; Laghezza, A.; Montanari, R.; Capelli, D.; Prada, E.; Loiodice, F.; Massolini, G.; Bernier, M.; et al. Resveratrol and its metabolites bind to PPARs. Chembiochem 2014, 15, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, D.M.; Cobankent Aytekin, E.; Mitranovic, M.I.; Turdean, S.G.; Moharer, M.S.; Cotoi, O.S.; Toru, H.S. Human Placenta and Evolving Insights into Pathological Changes of Preeclampsia: A Comprehensive Review of the Last Decade. Fetal Pediatr. Pathol. 2023, 31, 1–14. [Google Scholar] [CrossRef]
- Racicot, K.; Kwon, J.Y.; Aldo, P.; Silasi, M.; Mor, G. Understanding the complexity of the immune system during pregnancy. Am. J. Reprod. Immunol. 2014, 72, 107–116. [Google Scholar] [CrossRef]
- Huynh, J.; Yamada, J.; Beauharnais, C.; Wenger, J.B.; Thadhani, R.I.; Wexler, D.; Roberts, D.J.; Bentley-Lewis, R. Type 1, type 2 and gestational diabetes mellitus differentially impact placental pathologic characteristics of uteroplacental malperfusion. Placenta 2015, 36, 1161–1166. [Google Scholar] [CrossRef]
- Chang, C.W.; Wakeland, A.K.; Parast, M.M. Trophoblast lineage specification, differentiation and their regulation by oxygen tension. J. Endocrinol. 2018, 236, R43–R56. [Google Scholar] [CrossRef]
- Lee, J.W.; Bae, S.H.; Jeong, J.W.; Kim, S.H.; Kim, K.W. Hypoxia-inducible factor (HIF-1)alpha: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef]
- Adelman, D.M.; Gertsenstein, M.; Nagy, A.; Simon, M.C.; Maltepe, E. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000, 14, 3191–3203. [Google Scholar] [CrossRef]
- Maltepe, E.; Krampitz, G.W.; Okazaki, K.M.; Red-Horse, K.; Mak, W.; Simon, M.C.; Fisher, S.J. Hypoxia-inducible factor-dependent histone deacetylase activity determines stem cell fate in the placenta. Development 2005, 132, 3393–3403. [Google Scholar] [CrossRef]
- Wakeland, A.K.; Soncin, F.; Moretto-Zita, M.; Chang, C.W.; Horii, M.; Pizzo, D.; Nelson, K.K.; Laurent, L.C.; Parast, M.M. Hypoxia Directs Human Extravillous Trophoblast Differentiation in a Hypoxia-Inducible Factor-Dependent Manner. Am. J. Pathol. 2017, 187, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Yun, Z.; Maecker, H.L.; Johnson, R.S.; Giaccia, A.J. Inhibition of PPARγ2 gene expression by the HIF-1-regulated gene, D.E.C1/Stra13: A mechanism for regulation of adipogenesis by hypoxia. Dev. Cell. 2002, 2, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Tache, V.; Ciric, A.; Moretto-Zita, M.; Li, Y.; Peng, J.; Maltepe, E.; Milstone, D.S.; Parast, M.M. Hypoxia and trophoblast differentiation: A key role for PPARγ. Stem Cells Dev. 2013, 22, 2815–2824. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Chen, Z.; Sun, Q.; Li, Y.; Gu, H.; Ni, X. Reduced expression of 11β-hydroxysteroid dehydrogenase type 2 in preeclamptic placentas is associated with Decreased PPARγ but Increased PPARα Expression. Endocrinology 2014, 155, 299–309. [Google Scholar] [CrossRef]
- Chen, C.P.; Chen, C.Y.; Yang, Y.C.; Su, T.H.; Chen, H. Decreased Placental GCM1 (glial cells missing) gene expression in pre-eclampsia. Placenta 2004, 25, 413–421. [Google Scholar] [CrossRef]
- Langbein, M.; Strick, R.; Strissel, P.L.; Vogt, N.; Parsch, H.; Beckmann, M.W.; Schild, R.L. Impaired cytotrophoblast cell-cell fusion is associated with reduced Syncytin and increased apoptosis in patients with placental dysfunction. Mol. Reprod. Dev. 2008, 75, 175–183. [Google Scholar] [CrossRef]
- Baczyk, D.; Drewlo, S.; Proctor, L.; Dunk, C.; Lye, S.; Kingdom, J. Glial cell missing-1 transcription factor is required for the differentiation of the human trophoblast. Cell Death Differ. 2009, 16, 719–727. [Google Scholar] [CrossRef]
- Karumanchi, S.A.; Epstein, F.H. Placental ischemia and soluble fms-like tyrosine kinase 1: Cause or consequence of preeclampsia? Kidney Int. 2007, 71, 959–961. [Google Scholar] [CrossRef]
- Nagamatsu, T.; Fujii, T.; Kusumi, M.; Zou, L.; Yamashita, T.; Osuga, Y.; Momoeda, M.; Kozuma, S.; Taketani, Y. Cytotrophoblasts up-regulate soluble fms-like tyrosine kinase-1 expression under reduced oxygen: An implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology 2004, 145, 4838–4845. [Google Scholar] [CrossRef]
- Li, H.; Gu, B.; Zhang, Y.; Lewis, D.F.; Wang, Y. Hypoxia-induced increase in soluble Flt-1 production correlates with enhanced oxidative stress in trophoblast cells from the human placenta. Placenta 2005, 26, 210–217. [Google Scholar] [CrossRef]
- Nevo, O.; Soleymanlou, N.; Wu, Y.; Xu, J.; Kingdom, J.; Many, A.; Zamudio, S.; Caniggia, I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by H.I.F-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1085–R1093. [Google Scholar] [CrossRef]
- Munaut, C.; Lorquet, S.; Pequeux, C.; Blacher, S.; Berndt, S.; Frankenne, F.; Foidart, J.M. Hypoxia is responsible for soluble vascular endothelial growth factor receptor-1 (VEGFR-1) but not for soluble endoglin induction in villous trophoblast. Hum. Reprod. 2008, 23, 1407–1415. [Google Scholar] [CrossRef]
- Taché, V.; LaCoursiere, D.Y.; Saleemuddin, A.; Parast, M.M. Placental expression of vascular endothelial growth factor receptor-1/soluble vascular endothelial growth factor receptor-1 correlates with severity of clinical preeclampsia and villous hypermaturity. Hum. Pathol. 2011, 42, 1283–1288. [Google Scholar] [CrossRef]
- Huppertz, B.; Abe, E.; Murthi, P.; Nagamatsu, T.; Szukiewicz, D.; Salafia, C. Placental angiogenesis, maternal and fetal vessels—A workshop report. Placenta 2007, 28 (Suppl. A), S94–S96. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, F.P.; Drewlo, S.; English, F.A.; Kingdom, J.; Johns, E.J.; Kenny, L.C.; Walsh, S.K. Evidence implicating peroxisome proliferator-activated receptor-γ in the pathogenesis of preeclampsia. Hypertension 2011, 58, 882–887. [Google Scholar] [CrossRef]
- Bainbridge, S.A.; Minhas, A.; Whiteley, K.J.; Qu, D.; Sled, J.G.; Kingdom, J.C.; Adamson, S.L. Effects of reduced Gcm1 expression on trophoblast morphology, fetoplacental vascularity, and pregnancy outcomes in mice. Hypertension 2012, 59, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.N.; Smith, S.C.; To, K.F.; Sahota, D.S.; Baker, P.N. Increased placental apoptosis in pregnancies complicated by preeclampsia. Am. J. Obstet. Gynecol. 2001, 184, 1249–1250. [Google Scholar] [CrossRef]
- Nelson, D.M.; Johnson, R.D.; Smith, S.D.; Anteby, E.Y.; Sadovsky, Y. Hypoxia limits differentiation and up-regulates expression and activity of prostaglandin H synthase 2 in cultured trophoblast from term human placenta. Am. J. Obstet. Gynecol. 1999, 180, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Elchalal, U.; Humphrey, R.G.; Smith, S.D.; Hu, C.; Sadovsky, Y.; Nelson, D.M. Troglitazone attenuates hypoxia-induced injury in cultured term human trophoblasts. Am. J. Obstet. Gynecol. 2004, 191, 2154–2159. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Dioum, E.M.; Chen, R.; Alexander, M.S.; Zhang, Q.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 2009, 324, 1289–1293. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Dioum, E.M.; Hogg, R.T.; Gerard, R.D.; Garcia, J.A. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J. Biol. Chem. 2011, 286, 13869–13878. [Google Scholar] [CrossRef]
- Chen, B.; Nelson, D.M.; Sadovsky, Y. N-myc down-regulated gene 1 modulates the response of term human trophoblasts to hypoxic injury. J. Biol. Chem. 2006, 281, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Broady, A.J.; Loichinger, M.H.; Ahn, H.J.; Davy, P.M.; Allsopp, R.C.; Bryant-Greenwood, G.D. Protective proteins and telomere length in placentas from patients with pre-eclampsia in the last trimester of gestation. Placenta 2017, 50, 44–52. [Google Scholar] [CrossRef]
- Cudmore, M.J.; Ramma, W.; Cai, M.; Fujisawa, T.; Ahmad, S.; Al-Ani, B.; Ahmed, A. Resveratrol inhibits the release of soluble fms-like tyrosine kinase (sFlt-1) from human placenta. Am. J. Obstet. Gynecol. 2012, 206, 253.e10–253.e15. [Google Scholar] [CrossRef]
- Hannan, N.J.; Brownfoot, F.C.; Cannon, P.; Deo, M.; Beard, S.; Nguyen, T.V.; Palmer, K.R.; Tong, S.; Kaitu’u-Lino, T.J. Resveratrol inhibits release of soluble fms-like tyrosine kinase (sFlt-1) and soluble endoglin and improves vascular dysfunction—Implications as a preeclampsia treatment. Sci. Rep. 2017, 7, 1819. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zuo, Q.; Huang, S.; Yu, X.; Jiang, Z.; Zou, S.; Fan, M.; Sun, L. Resveratrol inhibits trophoblast apoptosis through oxidative stress in preeclampsia-model rats. Molecules 2014, 19, 20570–20579. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef]
- Meher, A.P.; Joshi, A.A.; Joshi, S.R. Maternal micronutrients, omega-3 fatty acids, and placental PPARγ expression. Appl. Physiol. Nutr. Metab. 2014, 39, 793–800. [Google Scholar] [CrossRef]
- Matsubara, K.; Higaki, T.; Matsubara, Y.; Nawa, A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int. J. Mol. Sci. 2015, 16, 4600–4614. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.R.; Tong, S.; Kaitu’u-Lino, T.J. Placental-specific sFLT-1: Role in pre-eclamptic pathophysiology and its translational possibilities for clinical prediction and diagnosis. Mol. Hum. Reprod. 2017, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Piragine, E.; Brogi, S.; Camodeca, C.; Fucci, R.; Calderone, V.; Nencetti, S.; Martelli, A.; Orlandini, E. Resveratrol-like Compounds as SIRT1 Activators. Int. J. Mol. Sci. 2022, 23, 15105. [Google Scholar] [CrossRef] [PubMed]
- Bokuda, K.; Ichihara, A. Preeclampsia up to date—What’s going on? Hypertens. Res. 2023, 46, 1900–1907. [Google Scholar] [CrossRef]
- Yagel, S.; Verlohren, S. Role of placenta in development of pre-eclampsia: Revisited. Ultrasound Obstet. Gynecol. 2020, 56, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, J.; Baker, P.N.; Tong, C. Revisiting preeclampsia: A metabolic disorder of the placenta. FEBS J. 2022, 289, 336–354. [Google Scholar] [CrossRef]
- Hadley, E.E.; Richardson, L.S.; Torloni, M.R.; Menon, R. Gestational tissue inflammatory biomarkers at term labor: A systematic review of literature. Am. J. Reprod. Immunol. 2018, 79, e12776. [Google Scholar] [CrossRef]
- Kim, S.H.; Shim, S.H.; Sung, S.R.; Lee, K.A.; Shim, J.Y.; Cha, D.H.; Lee, K.J. Gene expression analysis of the microdissected trophoblast layer of human placenta after the spontaneous onset of labor. PLoS ONE 2013, 8, e77648. [Google Scholar] [CrossRef]
- Tsai, P.J.; Davis, J.; Thompson, K.; Bryant-Greenwood, G. Visfatin/Nampt and SIRT1: Roles in Postterm Delivery in Pregnancies Associated with Obesity. Reprod. Sci. 2015, 22, 1028–1036. [Google Scholar] [CrossRef]
- Harmon, A.C.; Cornelius, D.C.; Amaral, L.M.; Faulkner, J.L.; Cunningham, M.W., Jr.; Wallace, K.; LaMarca, B. The role of inflammation in the pathology of preeclampsia. Clin. Sci. 2016, 130, 409–419. [Google Scholar] [CrossRef]
- Doshani, A.; Konje, J.C. Placental dysfunction in obese women and antenatal surveillance. Best Pract. Res. Clin. Obstet. Gynaecol. 2023, 91, 102407. [Google Scholar] [CrossRef]
- Baker, B.C.; Hayes, D.J.; Jones, R.L. Effects of micronutrients on placental function: Evidence from clinical studies to animal models. Reproduction 2018, 156, R69–R82. [Google Scholar] [CrossRef] [PubMed]
- Bo, Q.L.; Chen, Y.H.; Yu, Z.; Fu, L.; Zhou, Y.; Zhang, G.B.; Wang, H.; Zhang, Z.H.; Xu, D.X. Rosiglitazone pretreatment protects against lipopolysaccharide-induced fetal demise through inhibiting placental inflammation. Mol. Cell. Endocrinol. 2016, 423, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Yu, Y.; Guan, H.B.; Qiao, C. Effect of Human Umbilical Cord Mesenchymal Stem Cell Transplantation in a Rat Model of Preeclampsia. Reprod. Sci. 2016, 23, 1058–1070. [Google Scholar] [CrossRef]
- Leon-Garcia, S.M.; Roeder, H.A.; Nelson, K.K.; Liao, X.; Pizzo, D.P.; Laurent, L.C.; Parast, M.M.; LaCoursiere, D.Y. Maternal obesity and sex-specific differences in placental pathology. Placenta 2016, 38, 33–40. [Google Scholar] [CrossRef]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef]
- Qiao, L.; Guo, Z.; Bosco, C.; Guidotti, S.; Wang, Y.; Wang, M.; Parast, M.; Schaack, J.; Hay, W.W., Jr.; Moore, T.R.; et al. Maternal High-Fat Feeding Increases Placental Lipoprotein Lipase Activity by Reducing SIRT1 Expression in Mice. Diabetes 2015, 64, 3111–3120. [Google Scholar] [CrossRef]
- Cawyer, C.R.; Horvat, D.; Leonard, D.; Allen, S.R.; Jones, R.O.; Zawieja, D.C.; Kuehl, T.J.; Uddin, M.N. Hyperglycemia impairs cytotrophoblast function via stress signaling. Am. J. Obstet. Gynecol. 2014, 211, 541.e1–541.e8. [Google Scholar] [CrossRef] [PubMed]
- Suwaki, N.; Masuyama, H.; Masumoto, A.; Takamoto, N.; Hiramatsu, Y. Expression and potential role of peroxisome proliferator-activated receptor gamma in the placenta of diabetic pregnancy. Placenta 2007, 28, 315–323. [Google Scholar] [CrossRef]
- Jawerbaum, A.; Capobianco, E.; Pustovrh, C.; White, V.; Baier, M.; Salzberg, S.; Pesaresi, M.; Gonzalez, E. Influence of peroxisome proliferator-activated receptor gamma activation by its endogenous ligand 15-deoxy Delta12,14 prostaglandin J2 on nitric oxide production in term placental tissues from diabetic women. Mol. Hum. Reprod. 2004, 10, 671–676. [Google Scholar] [CrossRef]
- Holdsworth-Carson, S.J.; Lim, R.; Mitton, A.; Whitehead, C.; Rice, G.E.; Permezel, M.; Lappas, M. Peroxisome proliferator-activated receptors are altered in pathologies of the human placenta: Gestational diabetes mellitus, intrauterine growth restriction and preeclampsia. Placenta 2010, 31, 222–229. [Google Scholar] [CrossRef]
- Knabl, J.; Hüttenbrenner, R.; Hutter, S.; Günthner-Biller, M.; Vrekoussis, T.; Karl, K.; Friese, K.; Kainer, F.; Jeschke, U. Peroxisome proliferator-activated receptor-gamma (PPARγ) is down-regulated in trophoblast cells of gestational diabetes mellitus (GDM) and in trophoblast tumour cells BeWo in vitro after stimulation with PPARγ agonists. J. Perinat. Med. 2014, 42, 179–187. [Google Scholar] [CrossRef]
- Capobianco, E.; Fornes, D.; Linenberg, I.; Powell, T.L.; Jansson, T.; Jawerbaum, A. A novel rat model of gestational diabetes induced by intrauterine programming is associated with alterations in placental signaling and fetal overgrowth. Mol. Cell. Endocrinol. 2016, 422, 221–232. [Google Scholar] [CrossRef]
- Fantone, S.; Giannubilo, S.R.; Marzioni, D.; Tossetta, G. HTRA family proteins in pregnancy outcome. Tissue Cell 2021, 72, 101549. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Gesuita, R.; Di Renzo, G.C.; Meyyazhagan, A.; Tersigni, C.; Scambia, G.; Di Simone, N.; Marzioni, D. HtrA1 in Gestational Diabetes Mellitus: A Possible Biomarker? Diagnostics 2022, 12, 2705. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Viana-Mattioli, S.; Nunes, P.; Cavalli, R.; Sandrim, V. Analysis of, S.I.RT1 Expression in Plasma and in an In Vitro Model of Preeclampsia. Oxid. Med. Cell. Longev. 2020, 2020, 4561083. [Google Scholar] [CrossRef]
- Yin, Y.; Feng, Y.; Zhao, H.; Zhao, Z.; Yua, H.; Xu, J.; Che, H. SIRT1 inhibits releases of HMGB1 and HSP70 from human umbilical vein endothelial cells caused by IL-6 and the serum from a preeclampsia patient and protects the cells from death. Biomed. Pharmacother. 2017, 88, 449–458. [Google Scholar] [CrossRef]
- Majeed, Y.; Halabi, N.; Madani, A.Y.; Engelke, R.; Bhagwat, A.M.; Abdesselem, H.; Agha, M.V.; Vakayil, M.; Courjaret, R.; Goswami, N.; et al. SIRT1 promotes lipid metabolism and mitochondrial biogenesis in adipocytes and coordinates adipogenesis by targeting key enzymatic pathways. Sci. Rep. 2021, 11, 8177. [Google Scholar] [CrossRef]
- Hopp, A.K.; Grüter, P.; Hottiger, M.O. Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation. Cells 2019, 8, 1371, Erratum in Cells 2019, 8, 1371. [Google Scholar] [CrossRef]
- Yanagisawa, S.; Baker, J.R.; Vuppusetty, C.; Koga, T.; Colley, T.; Fenwick, P.; Donnelly, L.E.; Barnes, P.J.; Ito, K. The dynamic shuttling of SIRT1 between cytoplasm and nuclei in bronchial epithelial cells by single and repeated cigarette smoke exposure. PLoS ONE 2018, 13, e0193921. [Google Scholar] [CrossRef]
- Choudhury, F.K. Mitochondrial Redox Metabolism: The Epicenter of Metabolism during Cancer Progression. Antioxidants 2021, 10, 1838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Huang, Q.; Zeng, Z.; Wu, J.; Zhang, Y.; Chen, Z. Sirt1 Inhibits Oxidative Stress in Vascular Endothelial Cells. Oxid. Med. Cell. Longev. 2017, 2017, 7543973. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Giannakou, M.E.; Partridge, L. The interaction between FOXO and SIRT1: Tipping the balance towards survival. Trends Cell Biol. 2004, 14, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Furukawa-Hibi, Y.; Chen, C.; Horio, Y.; Isobe, K.; Ikeda, K.; Motoyama, N. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int. J. Mol. Med. 2005, 16, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Zhang, H.; Zhang, B.; Zhang, E.; Wu, Y. Tumor Necrosis Factor-alpha (TNF-α) Enhances miR-155-Mediated Endothelial Senescence by Targeting Sirtuin1 (SIRT1). Med. Sci. Monit. 2019, 25, 8820–8835. [Google Scholar] [CrossRef]
- Guo, Y.; Chao, L.; Chao, J. Kallistatin attenuates endothelial senescence by modulating Let-7g-mediated miR-34a-SIRT1-eNOS pathway. J. Cell. Mol. Med. 2018, 22, 4387–4398. [Google Scholar] [CrossRef]
- Ming, G.F.; Wu, K.; Hu, K.; Chen, Y.; Xiao, J. NAMPT regulates senescence, proliferation, and migration of endothelial progenitor cells through the SIRT1 AS lncRNA/miR-22/SIRT1 pathway. Biochem. Biophys. Res. Commun. 2016, 478, 1382–1388. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, D.; Zhang, N.; Yuan, T.; Tao, H. MiR-217 promotes endothelial cell senescence through the SIRT1/p53 signaling pathway. J. Mol. Histol. 2021, 52, 257–267. [Google Scholar] [CrossRef]
- Chen, L.; Holder, R.; Porter, C.; Shah, Z. Vitamin D3 attenuates doxorubicin-induced senescence of human aortic endothelial cells by upregulation of IL-10 via the pAMPKα/Sirt1/Foxo3a signaling pathway. PLoS ONE 2021, 16, e0252816. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, J.; Fu, M.; Dong, R.; Yang, Y.; Luo, J.; Hu, S.; Li, W.; Xu, X.; Tu, L. Dipeptidyl peptidase-4 inhibition improves endothelial senescence by activating AMPK/SIRT1/Nrf2 signaling pathway. Biochem. Pharmacol. 2020, 177, 113951. [Google Scholar] [CrossRef]
- Duan, J.L.; Ruan, B.; Song, P.; Fang, Z.Q.; Yue, Z.S.; Liu, J.J.; Dou, G.R.; Han, H.; Wang, L. Shear stress-induced cellular senescence blunts liver regeneration through Notch-sirtuin 1-P21/P16 axis. Hepatology 2022, 75, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Diabetes, oxidative stress and therapeutic strategies. Biochim. Biophys. Acta 2014, 1840, 2709–2729. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Di Lisa, F. Mitochondrial, R.O.S Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; de Bittencourt, P.I., Jr.; Newsholme, P. Molecular Events Linking Oxidative Stress and Inflammation to Insulin Resistance and β-Cell Dysfunction. Oxid. Med. Cell. Longev. 2015, 2015, 181643. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Duraisamy, A.J.; Kowluru, R.A. Sirt1: A Guardian of the Development of Diabetic Retinopathy. Diabetes 2018, 67, 745–754. [Google Scholar] [CrossRef]
- Karbasforooshan, H.; Karimi, G. The role of SIRT1 in diabetic retinopathy. Biomed. Pharmacother. 2018, 97, 190–194. [Google Scholar] [CrossRef]
- Kume, S.; Uzu, T.; Kashiwagi, A.; Koya, D. SIRT1, a calorie restriction mimetic, in a new therapeutic approach for type 2 diabetes mellitus and diabetic vascular complications. Endocr. Metab. Immune Disord. Drug Targets 2010, 10, 16–24. [Google Scholar] [CrossRef]
- Orimo, M.; Minamino, T.; Miyauchi, H.; Tateno, K.; Okada, S.; Moriya, J.; Komuro, I. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 889–894. [Google Scholar] [CrossRef]
- Ota, H.; Eto, M.; Kano, M.R.; Kahyo, T.; Setou, M.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2205–2211. [Google Scholar] [CrossRef]
- Pal, P.B.; Sonowal, H.; Shukla, K.; Srivastava, S.K.; Ramana, K.V. Aldose reductase regulates hyperglycemia-induced HUVEC death via SIRT1/AMPK-α1/mTOR pathway. J. Mol. Endocrinol. 2019, 63, 11–25. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in A.M.P-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Sun, J.; Chen, G.; Niu, C.; Wang, Y.; Zhao, C.; Sun, J.; Huang, H.; Huang, S.; Liang, Y.; et al. Resveratrol Promotes Diabetic Wound Healing via SIRT1-FOXO1-c-Myc Signaling Pathway-Mediated Angiogenesis. Front. Pharmacol. 2019, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Peng, I.C.; Sun, W.; Su, M.I.; Hsu, P.H.; Fu, Y.; Zhu, Y.; DeFea, K.; Pan, S.; Tsai, M.D.; et al. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circ. Res. 2009, 104, 496–505. [Google Scholar] [CrossRef]
- Cornelius, D.C.; Wallace, K. Autophagy in preeclampsia: A new target? EBioMedicine 2020, 57, 102864. [Google Scholar] [CrossRef]
- Wu, Q.; Hu, Y.; Jiang, M.; Wang, F.; Gong, G. Effect of Autophagy Regulated by Sirt1/FoxO1 Pathway on the Release of Factors Promoting Thrombosis from Vascular Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 4132. [Google Scholar] [CrossRef]
- Daitoku, H.; Hatta, M.; Matsuzaki, H.; Aratani, S.; Ohshima, T.; Miyagishi, M.; Nakajima, T.; Fukamizu, A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10042–10047. [Google Scholar] [CrossRef]
- Liu, J.; Bi, X.; Chen, T.; Zhang, Q.; Wang, S.X.; Chiu, J.J.; Liu, G.S.; Zhang, Y.; Bu, P.; Jiang, F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015, 6, e1827. [Google Scholar] [CrossRef] [PubMed]
- Kuchitsu, Y.; Fukuda, M. Revisiting Rab7 Functions in Mammalian Autophagy: Rab7 Knockout Studies. Cells 2018, 7, 215. [Google Scholar] [CrossRef]
- Takeda-Watanabe, A.; Kitada, M.; Kanasaki, K.; Koya, D. SIRT1 inactivation induces inflammation through the dysregulation of autophagy in human THP-1 cells. Biochem. Biophys. Res. Commun. 2012, 427, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.N.; Jiang, X.; Tang, W.; Song, P. Influence of intermittent fasting on autophagy in the liver. Biosci. Trends. 2023. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.I.; Kim, K.I.; Woo, S.K.; Kim, J.S.; Choi, K.J.; Lee, H.J.; Kim, K.S. Stromal Cell-Derived Factor 1 Protects Brain Vascular Endothelial Cells from Radiation-Induced Brain Damage. Cells 2019, 8, 1230. [Google Scholar] [CrossRef]
- Tian, X.L.; Li, Y. Endothelial cell senescence and age-related vascular diseases. J. Genet. Genom. 2014, 41, 485–495. [Google Scholar] [CrossRef]
- Hwang, H.S.; Maeng, Y.S.; Park, Y.W.; Koos, B.J.; Kwon, Y.G.; Kim, Y.H. Increased senescence and reduced functional ability of fetal endothelial progenitor cells in pregnancies complicated by preeclampsia without intrauterine growth restriction. Am. J. Obstet. Gynecol. 2008, 199, 259.e1–259.e7. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, J.; Mitsui-Saito, M.; Hayashi, C.; Hoshiai, T.; Senoo, M.; Chisaka, H.; Yaegashi, N.; Okamura, K. Decrease and senescence of endothelial progenitor cells in patients with preeclampsia. J. Clin. Endocrinol. Metab. 2005, 90, 5329–5332. [Google Scholar] [CrossRef]
- Hwang, J.S.; Choi, H.S.; Ham, S.A.; Yoo, T.; Lee, W.J.; Paek, K.S.; Seo, H.G. Deacetylation-mediated interaction of SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci. Rep. 2015, 5, 15971. [Google Scholar] [CrossRef]
- Holmlund, U.; Wähämaa, H.; Bachmayer, N.; Bremme, K.; Sverremark-Ekström, E.; Palmblad, K. The novel inflammatory cytokine high mobility group box protein 1 (HMGB1) is expressed by human term placenta. Immunology 2007, 122, 430–437. [Google Scholar] [CrossRef]
- Jiang, R.; Cai, J.; Zhu, Z.; Chen, D.; Wang, J.; Wang, Q.; Teng, Y.; Huang, Y.; Tao, M.; Xia, A.; et al. Hypoxic trophoblast HMGB1 induces endothelial cell hyperpermeability via the TRL-4/caveolin-1 pathway. J. Immunol. 2014, 193, 5000–5012. [Google Scholar] [CrossRef]
- Lai, H.; Nie, L.; Zeng, X.; Xin, S.; Wu, M.; Yang, B.; Luo, Y.; Liu, B.; Zheng, J.; Liu, H. Enhancement of heat shock protein 70 attenuates inducible nitric oxide synthase in preeclampsia complicated with fetal growth restriction. J. Matern. Fetal Neonatal Med. 2022, 35, 2555–2563. [Google Scholar] [CrossRef]
- Peraçoli, J.C.; Bannwart-Castro, C.F.; Romao, M.; Weel, I.C.; Ribeiro, V.R.; Borges, V.T.; Rudge, M.V.; Witkin, S.S.; Peraçoli, M.T. High levels of heat shock protein 70 are associated with pro-inflammatory cytokines and may differentiate early- from late-onset preeclampsia. J. Reprod. Immunol. 2013, 100, 129–134. [Google Scholar] [CrossRef]
- Molvarec, A.; Szarka, A.; Walentin, S.; Beko, G.; Karádi, I.; Prohászka, Z.; Rigó, J., Jr. Serum heat shock protein 70 levels in relation to circulating cytokines, chemokines, adhesion molecules and angiogenic factors in women with preeclampsia. Clin. Chim. Acta 2011, 412, 1957–1962. [Google Scholar] [CrossRef]
- Watanabe, S.; Ageta-Ishihara, N.; Nagatsu, S.; Takao, K.; Komine, O.; Endo, F.; Miyakawa, T.; Misawa, H.; Takahashi, R.; Kinoshita, M.; et al. SIRT1 overexpression ameliorates a mouse model of S.O.D1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol. Brain 2014, 7, 62. [Google Scholar] [CrossRef]
- Gu, X.; Gu, B.; Lv, X.; Yu, Z.; Wang, R.; Zhou, X.; Qiao, W.; Mao, Z.; Zuo, G.; Li, Q.; et al. 1, 25-dihydroxy-vitamin D3 with tumor necrosis factor-alpha protects against rheumatoid arthritis by promoting p53 acetylation-mediated apoptosis via Sirt1 in synoviocytes. Cell Death Dis. 2016, 7, e2423. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, X.D.; Li, H. Protective role of SIRT1-mediated Sonic Hedgehog signaling pathway in the preeclampsia rat models. J. Assist. Reprod. Genet. 2021, 38, 1843–1851. [Google Scholar] [CrossRef]
- ACOG. Practice Bulletin No. 202: Gestational Hypertension and Preeclampsia. Obstet. Gynecol. 2019, 133, 1. [Google Scholar] [CrossRef]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef]
- McBurney, M.W.; Yang, X.; Jardine, K.; Hixon, M.; Boekelheide, K.; Webb, J.R.; Lansdorp, P.M.; Lemieux, M. The Mammalian SIR2α protein has a role in embryogenesis and gametogenesis. Mol. Cell Biol. 2003, 23, 38–54. [Google Scholar] [CrossRef]
- Wang, R.H.; Sengupta, K.; Li, C.; Kim, H.S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef]
- Xiong, L.; Ye, X.; Chen, Z.; Fu, H.; Li, S.; Xu, P.; Yu, J.; Wen, L.; Gao, R.; Fu, Y.; et al. Advanced Maternal Age-associated SIRT1 Deficiency Compromises Trophoblast Epithelial-Mesenchymal Transition through an Increase in Vimentin Acetylation. Aging Cell 2021, 20, e13491. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef] [PubMed]
- Taskin, I.I.; Gurbuz, S.; Icen, M.S.; Derin, D.C.; Findik, F.M. Expression of sirtuin 2 and 7 in placenta accreta spectrum. Rev. Assoc. Med. Bras 2023, 69, e20230360. [Google Scholar] [CrossRef]
- Zhang, Q.; Ye, X.; Xu, X.; Yan, J. Placenta-derived exosomal miR-135a-5p promotes gestational diabetes mellitus pathogenesis by activating PI3K/AKT signaling pathway via SIRT1. J. Cell. Mol. Med. 2023. [Google Scholar] [CrossRef]
- Su, S.; Zhong, L.; Huang, S.; Deng, L.; Pang, L. MiRNA-494 induces trophoblast senescence by targeting SIRT1. Hypertens. Pregnancy 2023, 42, 2219774. [Google Scholar] [CrossRef]
- Liu, Z.; Pei, J.; Zhang, X.; Wang, C.; Tang, Y.; Liu, H.; Yu, Y.; Luo, S.; Gu, W. CD74 deficiency reduces trophoblast invasion and proliferation mediated by SIRT1 in preeclampsia. Reproduction 2023, 166, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Deodati, A.; Inzaghi, E.; Cianfarani, S. Epigenetics and In Utero Acquired Predisposition to Metabolic Disease. Front. Genet. 2020, 10, 1270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kraus, W.L. SIRT1-dependent regulation of chromatin and transcription: Linking NAD+ metabolism and signaling to the control of cellular functions. Biochim. Biophys. Acta 2010, 1804, 1666–1675. [Google Scholar] [CrossRef]
- Fortuny, L.; Sebastián, C. Sirtuins as Metabolic Regulators of Immune Cells Phenotype and Function. Genes 2021, 12, 1698. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Wang, Y.; Chao, Y.; Zhang, J.; Jia, Y.; Tie, J.; Hu, D. Regulation of SIRT1 and Its Roles in Inflammation. Front. Immunol. 2022, 13, 831168. [Google Scholar] [CrossRef]
- Tamura, N.; Heidari, N.; Faragher, R.G.A.; Smith, R.K.W.; Dudhia, J. Effects of resveratrol and its analogues on the cell cycle of equine mesenchymal stem/stromal cells. J. Equine Sci. 2023, 34, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Watroba, M.; Szukiewicz, D. Sirtuins at the Service of Healthy Longevity. Front. Physiol. 2021, 12, 724506. [Google Scholar] [CrossRef] [PubMed]
- Aksu, K.; Golal, E.; Aslan, M.A.; Ustunel, I.; Acar, N. The investigation of the role of sirtuin-1 on embryo implantation in oxidative stress-induced mice. J. Assist. Reprod. Genet. 2021, 38, 2349–2361. [Google Scholar] [CrossRef] [PubMed]
- Sande, A.K.; Dalen, I.; Torkildsen, E.A.; Sande, R.K.; Morken, N.H. Pregestational maternal risk factors for preterm and term preeclampsia: A population-based cohort study. Acta Obstet. Gynecol. Scand. 2023, 102, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xie, X.; Yuan, T.; Wang, Y.; Zhao, F.; Zhou, Z.; Zhang, H. Epidemiological trends of maternal hypertensive disorders of pregnancy at the global, regional, and national levels: A population-based study. BMC Pregnancy Childbirth 2021, 21, 364. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wątroba, M.; Szewczyk, G.; Szukiewicz, D. The Role of Sirtuin-1 (SIRT1) in the Physiology and Pathophysiology of the Human Placenta. Int. J. Mol. Sci. 2023, 24, 16210. https://doi.org/10.3390/ijms242216210
Wątroba M, Szewczyk G, Szukiewicz D. The Role of Sirtuin-1 (SIRT1) in the Physiology and Pathophysiology of the Human Placenta. International Journal of Molecular Sciences. 2023; 24(22):16210. https://doi.org/10.3390/ijms242216210
Chicago/Turabian StyleWątroba, Mateusz, Grzegorz Szewczyk, and Dariusz Szukiewicz. 2023. "The Role of Sirtuin-1 (SIRT1) in the Physiology and Pathophysiology of the Human Placenta" International Journal of Molecular Sciences 24, no. 22: 16210. https://doi.org/10.3390/ijms242216210
APA StyleWątroba, M., Szewczyk, G., & Szukiewicz, D. (2023). The Role of Sirtuin-1 (SIRT1) in the Physiology and Pathophysiology of the Human Placenta. International Journal of Molecular Sciences, 24(22), 16210. https://doi.org/10.3390/ijms242216210