
Modulating Stress Proteins in Response to Therapeutic Interventions for Parkinson’s Disease

Abstract
1. Introduction
2. Methods
3. Heat Shock Proteins (HSPs) in PD: An Overview of Mechanisms and Implications
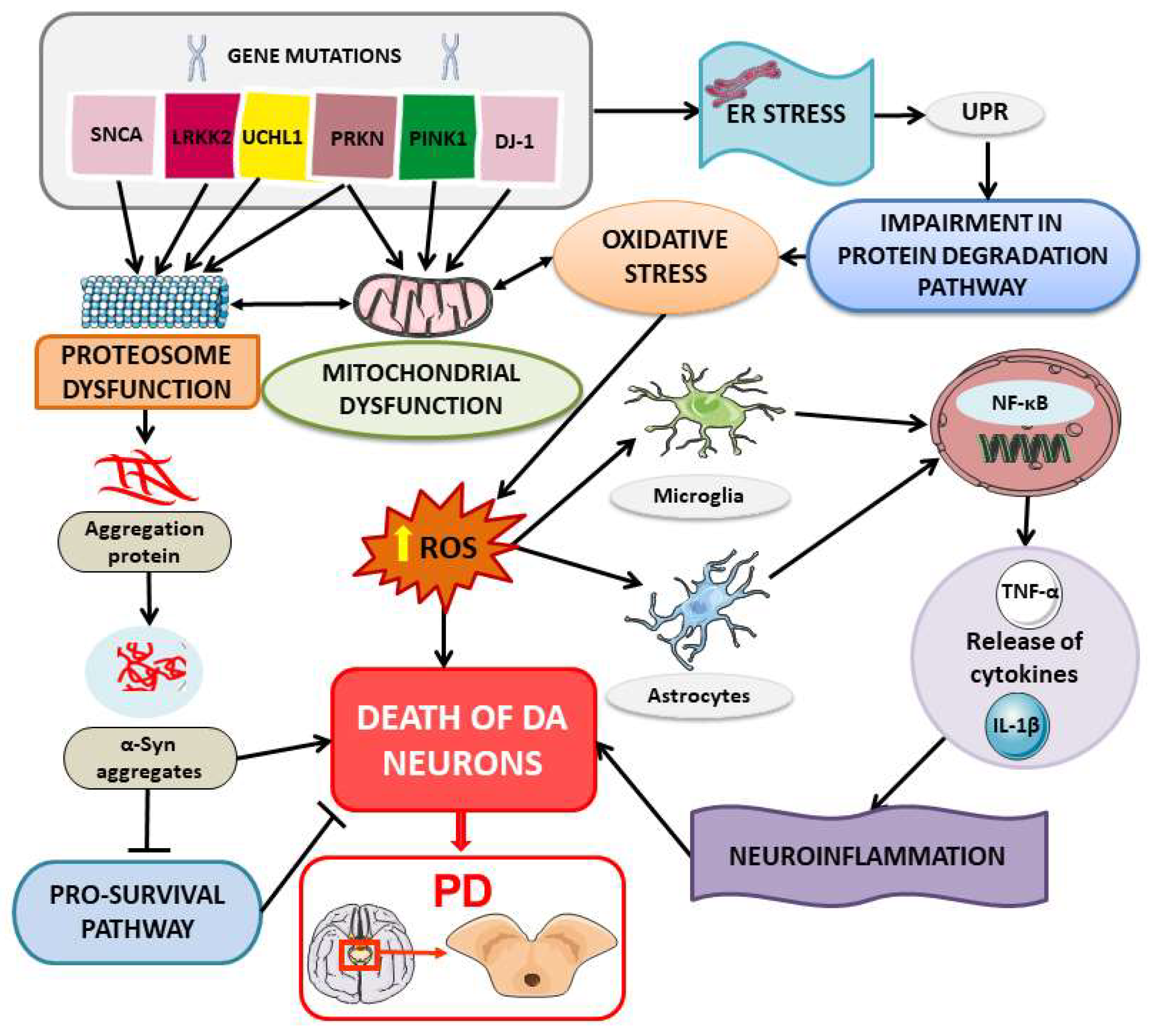
3.1. Protein Misfolding and Aggregation
3.2. ER Stress in PD Pathogenesis
3.3. Immune Response in PD Pathogenesis and Possible Contribution of HSP in Peripheral Immune Processes
4. Therapeutic Approaches for Modulating Stress Proteins in PD
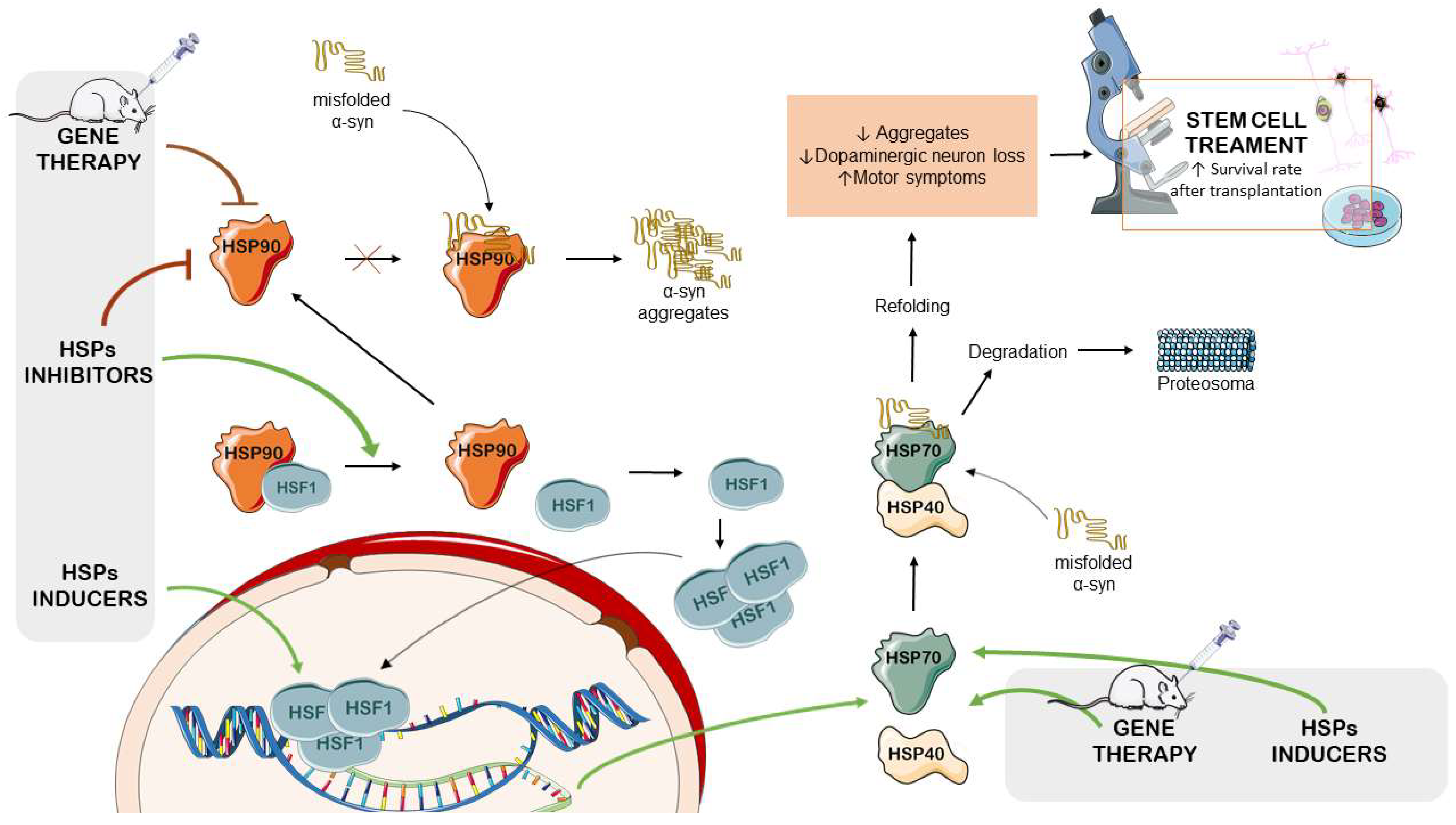
4.1. HSP Inducers
4.2. HSP90 Inhibitors
4.3. HSP-Modulation-Based Gene Therapy for PD Management
4.4. The Therapeutic Potential of HSPs in Stem-Cell-Based PD Treatments
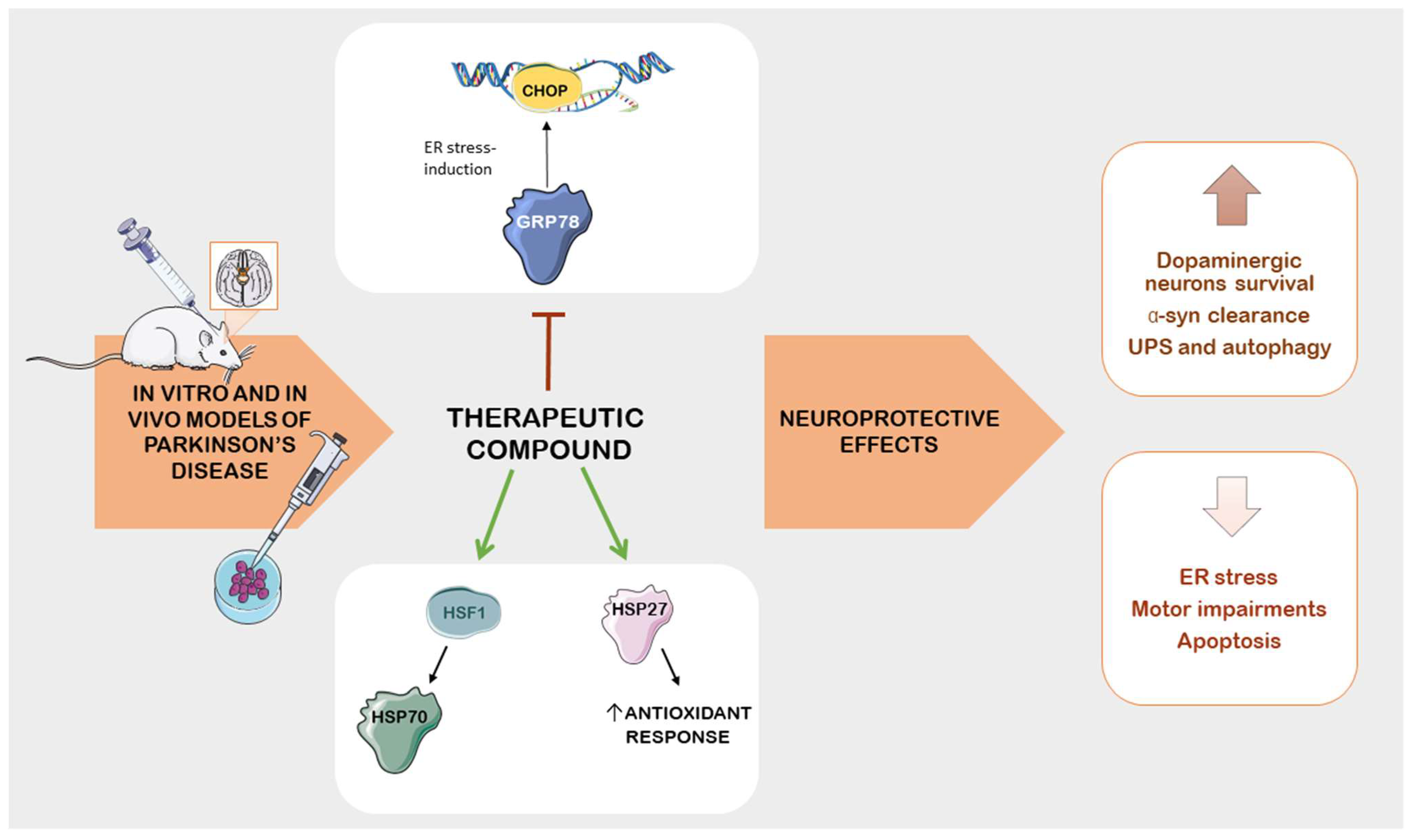
5. Therapeutic Compounds as Potential Treatment in PD: Insights from Preclinical Studies on Stress Protein Modulation
5.1. Pharmacological Modulation of HSPs and Neuroprotective Effects in PD Models
5.2. Natural Remedies Modulating HSPs for Neuroprotection in PD Models
5.2.1. The Activation of HSF1/HSP70 by Natural Compounds
5.2.2. The Activation of HSP27 by Natural Compounds
5.2.3. Impact of Natural Compounds on ER Stress

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Model | HSP Interaction | Effect | Ref. |
|---|---|---|---|---|
| Effects of commonly used compounds on HSP modulation in PD models | ||||
| Selegiline | Primary cultures of hippocampal-derived NSCs were pretreated with various concentrations of selegiline (0, 10, 20, 30, and 40 µM) for 48 h, followed by treatment with H2O2 (125 µM) for 30 min. The positive control cells were cultured for 48 h at 37 °C, then treated with H2O2 for 30 min. | HSPA4 | Selegiline increased HSPA4 and Bcl-2 mRNA expressions, improving cell viability and reducing oxidative-stress-induced cell death. | [171] |
| Lapatinib | Wistar albino rats received subcutaneous injections of the vehicle, ROT (2 mg/kg), or lapatinib (100 mg/kg/day) orally administered 1 h after ROT injection. The experiment was conducted for a total of 21 days. | HSP90/CDC37 chaperone complex | Lapatinib improved motor deficits in ROT-induced rats and reduced nigrostriatal dopaminergic depletions. Reduction in HSP90, CDC37, and c-SRC levels resulted in neuroprotective effects in ROT-induced rats by suppressing the main proteins involved in PD pathogenesis (α-syn, LRRK2, and c-ABL). | [177] |
| Rifampicin | PC12 cells were differentiated with 50 ng/mL of nerve growth factor for 6 days and then treated with rifampicin at 150 μM for 24 h. Gene silencing was performed using GRP78-specific siRNA or control siRNAs for 24 h. Then, rifampicin (150 μM) was administered for 2 h, followed by 1 μM ROT for 24 h. | GRP78 | Pretreatment with rifampicin provided neuroprotection by inducing GRP78 in a time- and dose-dependent manner. This upregulation occurred via the PERK-eIF2α-ATF4 pathway. | [180] |
| Empagliflozin | Wistar rats received 11 subcutaneous injections with the vehicle or ROT (1.5 mg/kg in 1% DMSO). The other animals were treated with empagliflozin alone (10 mg/kg/day orally) or empagliflozin with ROT starting from the 11th day, for 21 days. | GRP78 | Empagliflozin improved histopathological alterations and motor deficit in ROT-induced rats by inhibiting GRP78/PERK/eIF4α/CHOP. Empagliflozin enhanced α-syn clearance by improving autophagy and UPS impairments. | [182] |
| Metformin | C57BL/6 mice received a stereotaxic injection of ROT (2 mg/kg), metformin (50 mg/kg), or metformin for 3 days followed by ROT treatment. | GRP78 | Metformin pretreatment attenuated ER stress via inhibition of ATF4, ATF6, XBP1, GRP78, and CHOP mRNA levels, ameliorating dopaminergic neuron degeneration. | [183] |
| Metformin | Fibroblasts isolated from the skin biopsies of PD patients with R47X mutations or healthy patient controls and HeLa, SH-SY5Y, and HEK293 cells were cultured at suitable conditions. Metformin (10 mM) was used as a treatment for 24 h. Fibroblasts from TRAP1 or HTRA2 knockout mice were also used for the experiments. Other analysis required vectors containing TRAP1 and HTRA2 cDNA and specific siRNA for TRAP1, HTRA2, and controls. | TRAP1 | R47X TRAP1 mutations could lead to upregulation in mitochondrial UPR and enhancement of mitochondrial membrane potential as a response to an imbalance of proteins, including HSP90, HSP60, and HSP70. Metformin was able to reverse the effect of mutation. | [185] |
| Sodium salicylate (HSF-1 inducer) | Sprague Dawley rats were treated with subcutaneous injection of vehicle, i.p. injection of ROT (2 mg/kg suspended), ROT + sodium salicylate (100 mg/kg; i.p. injection), or sodium salicylate alone for five weeks. | HSF-1, HSP-40, and HSP-27 | Sodium salicylate protects the proteasome from oxidative stress and induces the expression of HSF-1, HSP-40, and HSP27, thereby also reducing α-syn aggregation. | [179] |
| Natural therapies modulating HSPs for neuroprotection in PD models | ||||
| Melatonin | SH-SY5Y cells were differentiated by using 10 μM retinoic acid for 6 days and subsequently exposed to melatonin (0.1, 1, 10, and 100 µM/mL) for 24 h. MPP+ (400 μM) was used to induce the PD model. For gene silencing, cells were transfected with HSF1-specific siRNA or control siRNAs for 24 h, then the cells were incubated with melatonin and MPP+ for 24 h. | HSF1 and MSP70 | Melatonin increased HSF1 and HSP70 compared with the MPP+ group. HSF1 silencing resulted in HSP70 downregulation and lower protection, whereas pro-apoptotic proteins and oxidative stress were increased. | [188] |
| Glutamine | SH-SY5Y cells overexpressing α-syn were obtained using transfection with recombinant plasmid. Cultured cells were treated with glutamine (0, 2, 4, 8, or 16 mM) for 6, 12, 24, and 48 h. Gene silencing was performed using HSF1-specific siRNA or control siRNA. | HSF1 and HSP70 | Glutamine increased both HSP70 mRNA and protein expression. This upregulation was dependent on HSF1 activation. This activation resulted in increased α-syn degradation. | [190] |
| Glutamine | PC12 cells overexpressing α-syn were achieved using transfection with recombinant plasmid. Cultured cells were treated with glutamine (0, 5, 10, or 20 mM) for 0, 4, 8, 12, 24, or 48 h. Gene silencing was performed using HSF1-specific siRNA or control siRNA. | HSF1 and HSP70 | Glutamine increased both HSP70 mRNA and protein expression. This upregulation was dependent on HSF1 activation. This activation resulted in increased α-syn degradation. | [191] |
| Ethyl acetate fraction from Holothuria leucospilota | Transgenic BY250, NL5901, CF1553, CL2166, TJ356, and wild-type N2 strains were used as C. elegans models for the study. The worms were exposed to 6-OHDA followed by compounds isolated from the ethyl acetate fraction from sea cucumber (1, 5, 25, and 50 μg/mL). Compounds were mixed with E. coli OP50 and then administered to worms as a food source. | HSP27 homologs (HSP16.1, HSP16.2, and HSP12.6) | Decanoic acid from the ethyl acetate fraction at low doses protected dopaminergic neurons against α-syn aggregation and improved motor deficits in worms. These effects occurred via activating DAF16 and its downstream genes sod-3, HSP16.1, HSP16.2, and HSP12.6. | [201] |
| Frondoside A | Frondoside A was administered at doses of 0.1, 0.5, 1, 5, and 10 μM to C. elegans strains exposed to 50 mM 6-OHDA. BZ555 and NL5901 were used as mutant strains of C. elegans. N2 was employed as wild-type control strain. | HSF1, HSP27 homologs (HSP16.1, HPS16.2) | Frondoside A (1 µM) reduced α-syn aggregation by enhancing the UPS and HPSs expression. | [202] |
| Nicotine | PC12 cells were pretreated with nicotine (100 nM) or inhibitors (LY294002 and methyllycaconitine) 30 min prior to exposure to MPP+ treatment (0.3 mM). Then, cells were incubated for 24 h. For the PD animal model, C57BL/6 mice were i.p. injected with 20 mg/kg MPTP-HCl twice daily for 7 days. The nicotine (0.25 mg/kg twice daily for 7 days) was i.p. injected alone or 30 min prior MPTP administration. | GPR78 and CHOP | Nicotine enhanced cell viability and attenuated MPP+-induced neurotoxicity by reducing the GRP78 and CHOP ER stress proteins. | [205] |
| Sesamol and Naringerin | Male Wistar rats received i.p. injection of vehicle or ROT (3 mg/kg). Sesamol (15 mg/kg) and naringenin (10 mg/kg) was administered orally. Flavonoids were administered for 10 days after the 11 days of ROT treatment. | HSP70 and HSP90 | Sesamol and narigerin promoted neuronal survival and improved muscle health by restoring protective proteins and increasing HSP70 and HSP90. | [192] |
| Quercitin | Dopaminergic SN4741 cells were pretreated with quercetin (10, 20, or 40 µM) 1 h prior the treatment with dieldrin (20 µM) for a total of 48 h. Gene silencing was performed using CHOP-specific siRNA or control siRNA. | GRP78, CHOP | Quercitin suppressed dieldrin-induced apoptosis in a dose-dependent manner, restoring CHOP and GRP78 levels and the p- eIF4α/eIF4α ratio induced by dieldrin. | [208] |
| Resveratrol liposomes (Polygonum cuspidatum) | The experiments were performed in Wistar rats through intracerebroventricular injection of 6-OHDA (15 μg) or vehicle or without injection. Resveratrol liposome was administered orally at a concentration of 20 mg/kg once daily for 2 weeks. | TRAP1 (HSP75) | Resveratrol liposome protected mitochondrial respiratory chain function and inhibited apoptosis in the substantia nigra of PD rats by increasing the phosphorylated TRAP1/TRAP ratio and PINK1 levels. | [211] |
| Resveratrol | Primary skin fibroblasts from a PD patient with a heterozygous parkin mutation and a healthy subject from the same family (control) were isolated from skin biopsy and cultured under suitable conditions. Then, the cells were treated with resveratrol (25 µM) or vehicle alone (0.2% DMSO). Proteomic analysis was performed using 2-DE, MALDI-TOF-MS, and Western blot. | HSP90B1, GPR78 (HSPA5), HSPA8 | Resveratrol improved protein folding regulation by modulating chaperones levels, including HSPA8 and HSP90, as well as SIRT1 deacetylase, resulting in improved chaperone-mediated autophagy. | [213] |
| Spirulina/C-phycocyanin (Arthrospira platensis) | Dried spirulina powder was added in fly food media to obtain 5% and 10% w/v concentrations. Wild-type Oregon R+ and transgenic DJ-1βΔ93 strains of Drosophila melanogaster were used as the control and PD models, respectively. | HSP70 | Spirulina supplementation increased the survival of flies and improved antioxidant defenses against cellular stress by reducing the HSP70 and JNK signaling pathways. | [193] |
| Amalaki rasayana | Amalaki rasayana was mixed with fly food media to obtain a 0.5% w/v concentration. Wild-type Oregon R+ and the transgenic DJ-1βΔ93 and Park13 strains of Drosophila melanogaster were used as the control and PD models, respectively. | HSP27 | Amalaki rasayana enhanced the tolerance to cellular stresses. This improvement was attributed to reduced ROS levels and lipid peroxidation, as well as an increase in SOD activity and HSP27 levels. | [203] |
| FLZ, a synthetic derivative of squamosamide (HSP70 inducer) | SH-SY5Y cells were transfected to produce a mutated α-syn (A53T) and then were exposed to 10 μM FLZ for 24 h. In the animal model, C3H mice expressing mutant α-syn received oral administration of 75 mg/kg FLZ for 7 weeks. However, gene silencing was performed using specific siRNAs for HSF1, HSP70, and co-chaperone genes. | HSP70 and its co-chaperone Hip | FLZ increased the expression of the co-chaperone Hip, enhancing HSP70 activity. FLZ directly bound to Hip and promoted its interaction with HSP70, which in turn reduced cytotoxic α-syn aggregates. | [197] |
| Wuzi Yanzong prescription | C57BL/6 mice were divided into a control group, a PD group (i.p. injection of MPTP for 1 week), and a PD + Wuzi Yanzong prescription group (16g/kg twice a day for 14 days). | GRP78, CHOP | Wuzi Yanzong prescription inhibited the UPR and ER-stress-induced apoptosis, possibly leading to an improvement in both PD symptoms and lesions. Specifically, the GRP78, p-PERK, p-eIF2α, ATF4, p-IRE1α, XBP1, ATF6, and CHOP ER related proteins were decreased after pretreatment in PD mice. | [214] |
| Uncaria rhynchophylla extract | Dried Uncharia rhynchophylla was used at concentrations 5, 10, or 20 µg/mL to treat SH-SY5Y cells for 6 h. Then, cells were exposed to MPP+ (1 mM) for 24 h. In the animal experiment, C57BL/6 mice were treated with vehicle, MPTP (30 mg/kg), or Uncharia rhynchophylla (20, 40, or 80 mg/kg orally) with or without MPTP. The experiment occurred for 19 days, with treatment from the 8th to the 12th day. | HSP90 | Uncaria rhynchophylla extract enhanced cell viability in vitro by modulating apoptotic and autophagic pathways through the inhibition of HSP90 expression. Thus, the compound improved behavioral deficits and increased DA concentrations. | [215] |
| Andrographolide | Male Swiss albino mice were pretreated with MPTP (25 mg/kg), followed by andrographolide (10 mg/kg). MPTP (5 applications) and andrographolide (10 applications) were administered on alternate days for 20 days. The study included control mice and mice treated with andrographoline alone. HCT116, HEK293, and Neuro-2A cells were used for in vitro studies. | HSF1, HSP70 | Andrographolide reduced the α-syn aggregation induced by MPTP via HSF1/HSP70, enhancing the protein quality control machinery. Additionally, andrographolide stimulated CHIP and ATG7 activity, increasing UPS activity and the autophagy process. | [200] |
6. Challenges and Future Prospects
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Schneider, S.A.; Obeso, J.A. Clinical and pathological features of parkinson’s disease. Behav. Neurobiol. Huntington’s Dis. Park. Dis. 2015, 22, 205–220. [Google Scholar]
- Sveinbjornsdottir, S. The clinical symptoms of parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 318–324. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef]
- Jellinger, K.A. Neuropathology and pathogenesis of extrapyramidal movement disorders: A critical update—I. Hypokinetic-rigid movement disorders. J. Neural Transm. 2019, 126, 933–995. [Google Scholar] [CrossRef]
- Xu, D.; Chen, Y.; Xu, Y.; ShenTu, C.; Peng, L. Signaling pathways in parkinson’s disease: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 73. [Google Scholar]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S.-i.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K. Familial parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M. Mutations in the dj-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Ohta, E.; Hasegawa, K.; Gasser, T.; Obata, F. Independent occurrence of i2020t mutation in the kinase domain of the leucine rich repeat kinase 2 gene in japanese and german parkinson’s disease families. Neurosci. Lett. 2007, 417, 21–23. [Google Scholar] [CrossRef]
- Funayama, M.; Hasegawa, K.; Ohta, E.; Kawashima, N.; Komiyama, M.; Kowa, H.; Tsuji, S.; Obata, F. An lrrk2 mutation as a cause for the parkinsonism in the original park8 family. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2005, 57, 918–921. [Google Scholar] [CrossRef]
- Hu, S.; Tan, J.; Qin, L.; Lv, L.; Yan, W.; Zhang, H.; Tang, B.; Wang, C. Molecular chaperones and parkinson’s disease. Neurobiol. Dis. 2021, 160, 105527. [Google Scholar] [CrossRef]
- Neef, D.W.; Turski, M.L.; Thiele, D.J. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010, 8, e1000291. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Group, P. Preferred reporting items for systematic reviews and meta-analyses: The prisma statement. BMJ 2009, 339, b2535. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Song, D.; Li, H.; He, M.L. Stress proteins: The biological functions in virus infection, present and challenges for target-based antiviral drug development. Signal Transduct. Target. Ther. 2020, 5, 125. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Licker, V.; Kovari, E.; Hochstrasser, D.F.; Burkhard, P.R. Proteomics in human parkinson’s disease research. J. Proteom. 2009, 73, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L. Parkinson’s disease: Etiology, neuropathology, and pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Exon Publication: Brisbane City, Australia, 2018; pp. 3–26. [Google Scholar]
- Voos, W.; Rottgers, K. Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim. Biophys. Acta 2002, 1592, 51–62. [Google Scholar] [CrossRef]
- Abeliovich, A. Mitochondrial damage control. Nature 2010, 463, 744–745. [Google Scholar] [CrossRef]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Orlacchio, A.; Maccarrone, M. Is modulation of oxidative stress an answer? The state of the art of redox therapeutic actions in neurodegenerative diseases. Oxidative Med. Cell. Longev. 2016, 2016, 7909380. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiu, V.; Maccarrone, M. Chronic inflammatory disorders and their redox control: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 2605–2641. [Google Scholar] [CrossRef] [PubMed]
- Bodner, R.A.; Outeiro, T.F.; Altmann, S.; Maxwell, M.M.; Cho, S.H.; Hyman, B.T.; McLean, P.J.; Young, A.B.; Housman, D.E.; Kazantsev, A.G. Pharmacological promotion of inclusion formation: A therapeutic approach for huntington’s and parkinson’s diseases. Proc. Natl. Acad. Sci. USA 2006, 103, 4246–4251. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.-J.; Rochet, J.-C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef]
- Liberek, K.; Lewandowska, A.; Zietkiewicz, S. Chaperones in control of protein disaggregation. EMBO J. 2008, 27, 328–335. [Google Scholar] [CrossRef]
- Tittelmeier, J.; Nachman, E.; Nussbaum-Krammer, C. Molecular chaperones: A double-edged sword in neurodegenerative diseases. Front. Aging Neurosci. 2020, 12, 581374. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Young, J.C.; Moarefi, I.; Hartl, F.U. Hsp90: A specialized but essential protein-folding tool. J. Cell Biol. 2001, 154, 267. [Google Scholar] [CrossRef] [PubMed]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; McLean, P.J. Hsp70 reduces α-synuclein aggregation and toxicity. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar] [CrossRef]
- Zhou, Y.; Gu, G.; Goodlett, D.R.; Zhang, T.; Pan, C.; Montine, T.J.; Montine, K.S.; Aebersold, R.H.; Zhang, J. Analysis of α-synuclein-associated proteins by quantitative proteomics. J. Biol. Chem. 2004, 279, 39155–39164. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.L.; Yang, Y.P.; Mao, C.J.; Zhang, X.Q.; Wang, C.T.; Yang, J.; Lv, D.J.; Wang, F.; Hu, L.F.; Liu, C.F. A role of bag3 in regulating snca/alpha-synuclein clearance via selective macroautophagy. Neurobiol. Aging 2017, 60, 104–115. [Google Scholar] [CrossRef]
- Bozaykut, P.; Sozen, E.; Kaga, E.; Ece, A.; Ozaltin, E.; Bergquist, J.; Kartal Ozer, N.; Karademir Yilmaz, B. Hsp70 inhibition leads to the activation of proteasomal system under mild hyperthermia conditions in young and senescent fibroblasts. Oxidative Med. Cell. Longev. 2020, 2020, 9369524. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The hsp70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- He, Y.; Wang, Z. The roles of hsp40/dnaj protein family in neurodegenerative diseases. Zhejiang Da Xue Xue Bao. Yi Xue Ban J. Zhejiang Univ. Med. Sci. 2022, 51, 640–646. [Google Scholar] [CrossRef]
- Asthana, A.; Bollapalli, M.; Tangirala, R.; Bakthisaran, R.; Mohan Rao, C. Hsp27 suppresses the cu(2+)-induced amyloidogenicity, redox activity, and cytotoxicity of alpha-synuclein by metal ion stripping. Free Radic. Biol. Med. 2014, 72, 176–190. [Google Scholar] [CrossRef]
- Rogalla, T.; Ehrnsperger, M.; Preville, X.; Kotlyarov, A.; Lutsch, G.; Ducasse, C.; Paul, C.; Wieske, M.; Arrigo, A.-P.; Buchner, J. Regulation of hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor α by phosphorylation. J. Biol. Chem. 1999, 274, 18947–18956. [Google Scholar] [CrossRef]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem. 2017, 86, 97–122. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M. Ubiquitin in chains. Trends Biochem. Sci. 2000, 25, 544–548. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Jenner, P. Proteasomal function is impaired in substantia nigra in parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Olanow, C.W.; Halliwell, B.; Isacson, O.; Jenner, P. Failure of the ubiquitin–proteasome system in parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 589–594. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; De Snoo, M.L.; Gondard, E.; Neudorfer, C.; Chau, H.; Ngana, S.G.; O’Hara, D.M.; Brotchie, J.M.; Koprich, J.B.; Lozano, A.M.; et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by alpha-synuclein. Acta Neuropathol. Commun. 2020, 8, 17. [Google Scholar] [CrossRef]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in parkinson’s disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Du, X.; Jiao, Q.; Chen, X.; Jiang, H. Expanding the role of proteasome homeostasis in parkinson’s disease: Beyond protein breakdown. Cell Death Dis. 2021, 12, 154. [Google Scholar] [CrossRef]
- Colla, E. Linking the endoplasmic reticulum to parkinson’s disease and alpha-synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Klappa, P.; Kietzmann, D.T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef]
- Smith, M.H.; Ploegh, H.L.; Weissman, J.S. Road to ruin: Targeting proteins for degradation in the endoplasmic reticulum. Science 2011, 334, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mrnas during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ni, M.; Lee, B.; Barron, E.; Hinton, D.; Lee, A. The unfolded protein response regulator grp78/bip is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008, 15, 1460–1471. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein kinase r-like er kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; LaVail, M.M.; Walter, P. Ire1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef]
- Ryu, E.J.; Harding, H.P.; Angelastro, J.M.; Vitolo, O.V.; Ron, D.; Greene, L.A. Endoplasmic reticulum stress and the unfolded protein response in cellular models of parkinson’s disease. J. Neurosci. 2002, 22, 10690–10698. [Google Scholar] [CrossRef]
- Hoozemans, J.; Van Haastert, E.; Eikelenboom, P.; De Vos, R.; Rozemuller, J.; Scheper, W. Activation of the unfolded protein response in parkinson’s disease. Biochem. Biophys. Res. Commun. 2007, 354, 707–711. [Google Scholar] [CrossRef]
- Silva, R.M.; Ries, V.; Oo, T.F.; Yarygina, O.; Jackson-Lewis, V.; Ryu, E.J.; Lu, P.D.; Marciniak, S.M.; Ron, D.; Przedborski, S. Chop/gadd153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J. Neurochem. 2005, 95, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, L.; Naik, I.; Braunstein, Z.; Zhong, J.; Ren, B. Transcription factor c/ebp homologous protein in health and diseases. Front. Immunol. 2017, 8, 1612. [Google Scholar] [CrossRef] [PubMed]
- Conn, K.J.; Gao, W.; McKee, A.; Lan, M.S.; Ullman, M.D.; Eisenhauer, P.B.; Fine, R.E.; Wells, J.M. Identification of the protein disulfide isomerase family member pdip in experimental parkinson’s disease and lewy body pathology. Brain Res. 2004, 1022, 164–172. [Google Scholar] [CrossRef]
- Bouman, L.; Schlierf, A.; Lutz, A.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D. Parkin is transcriptionally regulated by atf4: Evidence for an interconnection between mitochondrial stress and er stress. Cell Death Differ. 2011, 18, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.; Leung, C.T.; Liu, H.; Pang, S.Y.; Lam, C.S.; Xian, J.; Li, L.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Age-dependent accumulation of oligomeric snca/alpha-synuclein from impaired degradation in mutant lrrk2 knockin mouse model of parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (cma). Autophagy 2020, 16, 347–370. [Google Scholar] [CrossRef]
- Kanno, H.; Handa, K.; Murakami, T.; Aizawa, T.; Ozawa, H. Chaperone-mediated autophagy in neurodegenerative diseases and acute neurological insults in the central nervous system. Cells 2022, 11, 1205. [Google Scholar] [CrossRef]
- Abdi, I.Y.; Ghanem, S.S.; El-Agnaf, O.M. Immune-related biomarkers for parkinson’s disease. Neurobiol. Dis. 2022, 170, 105771. [Google Scholar] [CrossRef]
- Tang, G.; Minemoto, Y.; Dibling, B.; Purcell, N.H.; Li, Z.; Karin, M.; Lin, A. Inhibition of jnk activation through nf-κ b target genes. Nature 2001, 414, 313–317. [Google Scholar] [CrossRef]
- Flores-Morales, A.; Fernández, L.; Rico-Bautista, E.; Umana, A.; Negrín, C.; Zhang, J.G.; Norstedt, G. Endoplasmic reticulum stress prolongs gh-induced janus kinase (jak2)/signal transducer and activator of transcription (stat5) signaling pathway. Mol. Endocrinol. 2001, 15, 1471–1483. [Google Scholar] [CrossRef][Green Version]
- Meares, G.P.; Liu, Y.; Rajbhandari, R.; Qin, H.; Nozell, S.E.; Mobley, J.A.; Corbett, J.A.; Benveniste, E.N. Perk-dependent activation of jak1 and stat3 contributes to endoplasmic reticulum stress-induced inflammation. Mol. Cell. Biol. 2014, 34, 3911–3925. [Google Scholar] [CrossRef]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational repression mediates activation of nuclear factor kappa b by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef]
- Sprenkle, N.T.; Sims, S.G.; Sanchez, C.L.; Meares, G.P. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol. Neurodegener. 2017, 12, 42. [Google Scholar] [CrossRef]
- Zang, X.; Chen, S.; Zhu, J.; Ma, J.; Zhai, Y. The emerging role of central and peripheral immune systems in neurodegenerative diseases. Front. Aging Neurosci. 2022, 14, 872134. [Google Scholar] [CrossRef]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. Alpha-synuclein-specific t cell reactivity is associated with preclinical and early parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef]
- Fiszer, U.; Fredrikson, S.; Członkowska, A. Humoral response to hsp 65 and hsp 70 in cerebrospinal fluid in parkinson’s disease. J. Neurol. Sci. 1996, 139, 66–70. [Google Scholar] [CrossRef]
- Papuc, E.; Kurys-Denis, E.; Krupski, W.; Rejdak, K. Humoral response against small heat shock proteins in parkinson’s disease. PLoS ONE 2015, 10, e0115480. [Google Scholar] [CrossRef]
- Couch, Y.; Alvarez-Erviti, L.; Sibson, N.R.; Wood, M.J.; Anthony, D.C. The acute inflammatory response to intranigral alpha-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflamm. 2011, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guajardo, V.; Barnum, C.J.; Tansey, M.G.; Romero-Ramos, M. Neuroimmunological processes in parkinson’s disease and their relation to alpha-synuclein: Microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro 2013, 5, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Labrador-Garrido, A.; Cejudo-Guillen, M.; Klippstein, R.; De Genst, E.J.; Tomas-Gallardo, L.; Leal, M.M.; Villadiego, J.; Toledo-Aral, J.J.; Dobson, C.M.; Pozo, D.; et al. Chaperoned amyloid proteins for immune manipulation: Alpha-synuclein/hsp70 shifts immunity toward a modulatory phenotype. Immun. Inflamm. Dis. 2014, 2, 226–238. [Google Scholar] [CrossRef]
- Labrador-Garrido, A.; Cejudo-Guillen, M.; Daturpalli, S.; Leal, M.M.; Klippstein, R.; De Genst, E.J.; Villadiego, J.; Toledo-Aral, J.J.; Dobson, C.M.; Jackson, S.E.; et al. Chaperome screening leads to identification of grp94/gp96 and fkbp4/52 as modulators of the alpha-synuclein-elicited immune response. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 564–577. [Google Scholar]
- Villadiego, J.; Labrador-Garrido, A.; Franco, J.M.; Leal-Lasarte, M.; De Genst, E.J.; Dobson, C.M.; Pozo, D.; Toledo-Aral, J.J.; Roodveldt, C. Immunization with alpha-synuclein/grp94 reshapes peripheral immunity and suppresses microgliosis in a chronic parkinsonism model. Glia 2018, 66, 191–205. [Google Scholar] [CrossRef]
- San Gil, R.; Cox, D.; McAlary, L.; Berg, T.; Walker, A.K.; Yerbury, J.J.; Ooi, L.; Ecroyd, H. Neurodegenerative disease-associated protein aggregates are poor inducers of the heat shock response in neuronal cells. J. Cell Sci. 2020, 133, jcs243709. [Google Scholar]
- Nag, N.; Jelinek, G.A. A narrative review of lifestyle factors associated with parkinson’s disease risk and progression. Neuro-Degener. Dis. 2019, 19, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Bargues-Carot, A.; Riaz, Z.; Wickham, H.; Zenitsky, G.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Impact of environmental risk factors on mitochondrial dysfunction, neuroinflammation, protein misfolding, and oxidative stress in the etiopathogenesis of parkinson’s disease. Int. J. Mol. Sci. 2022, 23, 10808. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, G.; Marchese, R.; Avanzino, L.; Pelosin, E. Rehabilitation for parkinson’s disease: Current outlook and future challenges. Park. Relat. Disord. 2016, 22 (Suppl. 1), S60–S64. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef]
- Putcha, P.; Danzer, K.M.; Kranich, L.R.; Scott, A.; Silinski, M.; Mabbett, S.; Hicks, C.D.; Veal, J.M.; Steed, P.M.; Hyman, B.T. Brain-permeable small-molecule inhibitors of hsp90 prevent α-synuclein oligomer formation and rescue α-synuclein-induced toxicity. J. Pharmacol. Exp. Ther. 2010, 332, 849–857. [Google Scholar] [CrossRef]
- Kühn, K.; Wellen, J.; Link, N.; Maskri, L.; Lübbert, H.; Stichel, C.C. The mouse mptp model: Gene expression changes in dopaminergic neurons. Eur. J. Neurosci. 2003, 17, 1–12. [Google Scholar] [CrossRef]
- Sivanandy, P.; Leey, T.C.; Xiang, T.C.; Ling, T.C.; Wey Han, S.A.; Semilan, S.L.A.; Hong, P.K. Systematic review on parkinson’s disease medications, emphasizing on three recently approved drugs to control parkinson’s symptoms. Int. J. Environ. Res. Public Health 2021, 19, 364. [Google Scholar] [CrossRef]
- Li, H.; Yang, J.; Wang, Y.; Liu, Q.; Cheng, J.; Wang, F. Neuroprotective effects of increasing levels of hsp70 against neuroinflammation in parkinson’s disease model by inhibition of nf-κb and stat3. Life Sci. 2019, 234, 116747. [Google Scholar] [CrossRef]
- Yu, W.W.; Cao, S.N.; Zang, C.X.; Wang, L.; Yang, H.Y.; Bao, X.Q.; Zhang, D. Heat shock protein 70 suppresses neuroinflammation induced by alpha-synuclein in astrocytes. Mol. Cell. Neurosci. 2018, 86, 58–64. [Google Scholar] [CrossRef]
- Pastukhov, Y.F.; Plaksina, D.V.; Lapshina, K.V.; Guzhova, I.V.; Ekimova, I.V. Exogenous protein hsp70 blocks neurodegeneration in the rat model of the clinical stage of parkinson’s disease. Dokl. Biol. Sci. Proc. Acad. Sci. USSR Biol. Sci. Sect. 2014, 457, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Pragya, P.; Chaouhan, H.S.; Tiwari, A.K.; Patel, D.K.; Abdin, M.Z.; Chowdhuri, D.K. Heat shock protein-70 (hsp-70) suppresses paraquat-induced neurodegeneration by inhibiting jnk and caspase-3 activation in drosophila model of parkinson’s disease. PLoS ONE 2014, 9, e98886. [Google Scholar] [CrossRef]
- Luo, G.R.; Chen, S.; Le, W.D. Are heat shock proteins therapeutic target for parkinson’s disease? Int. J. Biol. Sci. 2006, 3, 20–26. [Google Scholar] [CrossRef]
- Ahmed, K.; Furusawa, Y.; Tabuchi, Y.; Emam, H.F.; Piao, J.L.; Hassan, M.A.; Yamamoto, T.; Kondo, T.; Kadowaki, M. Chemical inducers of heat shock proteins derived from medicinal plants and cytoprotective genes response. Int. J. Hyperth. Off. J. Eur. Soc. Hyperthermic Oncol. N. Am. Hyperth. Group 2012, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ekimova, I.V.; Plaksina, D.V.; Pastukhov, Y.F.; Lapshina, K.V.; Lazarev, V.F.; Mikhaylova, E.R.; Polonik, S.G.; Pani, B.; Margulis, B.A.; Guzhova, I.V.; et al. New hsf1 inducer as a therapeutic agent in a rodent model of parkinson’s disease. Exp. Neurol. 2018, 306, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, D.; Asai, M.; Katagiri, K.; Takeuchi, R.; Ohtsuka, K. Reinvestigation of the effect of carbenoxolone on the induction of heat shock proteins. Cell Stress Chaperones 2009, 14, 535–543. [Google Scholar] [CrossRef]
- Ali, Y.O.; Kitay, B.M.; Zhai, R.G. Dealing with misfolded proteins: Examining the neuroprotective role of molecular chaperones in neurodegeneration. Molecules 2010, 15, 6859–6887. [Google Scholar] [CrossRef]
- Thakur, P.; Nehru, B. Long-term heat shock proteins (hsps) induction by carbenoxolone improves hallmark features of parkinson’s disease in a rotenone-based model. Neuropharmacology 2014, 79, 190–200. [Google Scholar] [CrossRef]
- Chamberlain, L.H.; Burgoyne, R.D. Cysteine-string protein: The chaperone at the synapse. J. Neurochem. 2000, 74, 1781–1789. [Google Scholar] [CrossRef]
- Shirafuji, T.; Ueyama, T.; Adachi, N.; Yoshino, K.I.; Sotomaru, Y.; Uwada, J.; Kaneoka, A.; Ueda, T.; Tanaka, S.; Hide, I.; et al. The role of cysteine string protein α phosphorylation at serine 10 and 34 by protein kinase cγ for presynaptic maintenance. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Jing, H.; Akerblom, I.; Morgan, R.; Fisher, T.S.; Neveu, M. Identification of the human cdna for new survival/evasion peptide (dsep): Studies in vitro and in vivo of overexpression by neural cells. Exp. Neurol. 2002, 177, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Maciejewski, J.; Yao, L. Inhibition of secreted phospholipase a2 by neuron survival and anti-inflammatory peptide chec-9. J. Neuroinflamm. 2006, 3, 25. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Greenstein, J.I.; Loewenstern, J.; Degermentzidis, E.; Yao, L. Anti-inflammatory peptide regulates the supply of heat shock protein 70 monomers: Implications for aging and age-related disease. Rejuvenation Res. 2015, 18, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Amano, W.; Kubo, T.; Fukuhara, A.; Ihara, H.; Azuma, Y.-T.; Tajima, H.; Inui, T.; Sawa, A.; Takeuchi, T. Glyceraldehyde-3-phosphate dehydrogenase aggregate formation participates in oxidative stress-induced cell death. J. Biol. Chem. 2009, 284, 34331–34341. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, A.-M.; Selby, T.L.; Palkovits, M.; Lolait, S.J. Distribution of mrna encoding b78/apj, the rat homologue of the human apj receptor, and its endogenous ligand apelin in brain and peripheral tissues. Biochim. Biophys. Acta (BBA)-Gene Struct. Expr. 2000, 1492, 72–80. [Google Scholar] [CrossRef]
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and characterization of a novel endogenous peptide ligand for the human apj receptor. Biochem. Biophys. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef]
- Boal, F.; Timotin, A.; Roumegoux, J.; Alfarano, C.; Calise, D.; Anesia, R.; Parini, A.; Valet, P.; Tronchere, H.; Kunduzova, O. Apelin-13 administration protects against ischaemia/reperfusion-mediated apoptosis through the foxo1 pathway in high-fat diet-induced obesity. Br. J. Pharmacol. 2016, 173, 1850–1863. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, H.; Ji, B.; Wang, Z.; Wang, C.; Yang, C.; Pan, Y.; Chen, J.; Cheng, B.; Bai, B. Apelin-13 attenuates er stress-associated apoptosis induced by mpp+ in sh-sy5y cells. Int. J. Mol. Med. 2018, 42, 1732–1740. [Google Scholar] [CrossRef]
- Zhu, J.; Dou, S.; Jiang, Y.; Chen, J.; Wang, C.; Cheng, B. Apelin-13 protects dopaminergic neurons in mptp-induced parkinson’s disease model mice through inhibiting endoplasmic reticulum stress and promoting autophagy. Brain Res. 2019, 1715, 203–212. [Google Scholar] [CrossRef]
- Zhu, J.; Dou, S.; Wang, C.; Jiang, Y.; Wang, C.; Cheng, B. Apelin-36 mitigates mptp/mpp(+)-induced neurotoxicity: Involvement of α-synuclein and endoplasmic reticulum stress. Brain Res. 2019, 1721, 146334. [Google Scholar] [CrossRef]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator grp78/bip in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Jiang, M.; Yun, Q.; Shi, F.; Niu, G.; Gao, Y.; Xie, S.; Yu, S. Downregulation of mir-384-5p attenuates rotenone-induced neurotoxicity in dopaminergic sh-sy5y cells through inhibiting endoplasmic reticulum stress. Am. J. Physiol. Cell Physiol. 2016, 310, C755–C763. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, M.S.; Shabashvili, A.; Chen, W.; Meyers, C.; Sullivan, L.F.; Salganik, M.; Lin, J.H.; Lewin, A.S.; Muzyczka, N.; Gorbatyuk, O.S. Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of parkinson disease. Mol. Ther. 2012, 20, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, M.; Saarma, M.; Lindholm, P. Unconventional neurotrophic factors cdnf and manf: Structure, physiological functions and therapeutic potential. Neurobiol. Dis. 2017, 97, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Jiang, M.; Fu, X.; Cai, Q.; Zhang, J.; Yin, Y.; Guo, J.; Yu, L.; Jiang, Y.; Liu, Y.; et al. Mesencephalic astrocyte-derived neurotrophic factor reduces cell apoptosis via upregulating hsp70 in shsy-5y cells. Transl. Neurodegener. 2017, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, C.; Gu, H.; Li, C.; Fu, X.; Jiang, M.; Sun, H.; Xu, J.; Fang, J.; Jin, L. Mesencephalic astrocyte-derived neurotrophic factor reduces cell apoptosis via upregulating grp78 in sh-sy5y cells. Cell Biol. Int. 2016, 40, 803–811. [Google Scholar] [CrossRef]
- Pakarinen, E.; Lindholm, P.; Saarma, M.; Lindahl, M. Cdnf and manf regulate er stress in a tissue-specific manner. Cell. Mol. Life Sci. CMLS 2022, 79, 124. [Google Scholar] [CrossRef]
- Voutilainen, M.H.; De Lorenzo, F.; Stepanova, P.; Back, S.; Yu, L.Y.; Lindholm, P.; Porsti, E.; Saarma, M.; Mannisto, P.T.; Tuominen, R.K. Evidence for an additive neurorestorative effect of simultaneously administered cdnf and gdnf in hemiparkinsonian rats: Implications for different mechanism of action. eNeuro 2017, 4, 1–14. [Google Scholar] [CrossRef]
- Alam, Q.; Alam, M.Z.; Sait, K.H.W.; Anfinan, N.; Noorwali, A.W.; Kamal, M.A.; Khan, M.S.A.; Haque, A. Translational shift of hsp90 as a novel therapeutic target from cancer to neurodegenerative disorders: An emerging trend in the cure of alzheimer’s and parkinson’s diseases. Curr. Drug Metab. 2017, 18, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Fujikake, N.; Nagai, Y.; Popiel, H.A.; Okamoto, Y.; Yamaguchi, M.; Toda, T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 2008, 283, 26188–26197. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Saidi, L.-J.; Wahlster, L. Molecular chaperones and protein folding as therapeutic targets in parkinson’s disease and other synucleinopathies. Acta Neuropathol. Commun. 2013, 1, 79. [Google Scholar] [CrossRef]
- McFarland, N.R.; Dimant, H.; Kibuuka, L.; Ebrahimi-Fakhari, D.; Desjardins, C.A.; Danzer, K.M.; Danzer, M.; Fan, Z.; Schwarzschild, M.A.; Hirst, W. Chronic treatment with novel small molecule hsp90 inhibitors rescues striatal dopamine levels but not α-synuclein-induced neuronal cell loss. PLoS ONE 2014, 9, e86048. [Google Scholar] [CrossRef]
- Alani, B.; Salehi, R.; Sadeghi, P.; Zare, M.; Khodagholi, F.; Arefian, E.; Hakemi, M.G.; Digaleh, H. Silencing of hsp90 chaperone expression protects against 6-hydroxydopamine toxicity in pc12 cells. J. Mol. Neurosci. 2014, 52, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Yukitake, H.; Tajima, Y.; Suzuki, H.; Chikatsu, T.; Morimoto, S.; Funabashi, Y.; Omae, H.; Ito, T.; Yoneda, Y. Itz-1, a client-selective hsp90 inhibitor, efficiently induces heat shock factor 1 activation. Chem. Biol. 2010, 17, 18–27. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Ahn, Y.-H.; Benjamin, I.J.; Honda, T.; Hicks, R.J.; Calabrese, V.; Cole, P.A.; Dinkova-Kostova, A.T. Hsf1-dependent upregulation of hsp70 by sulfhydryl-reactive inducers of the keap1/nrf2/are pathway. Chem. Biol. 2011, 18, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Simões-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.-A.; Cuendet, M. Hdac6 as a target for neurodegenerative diseases: What makes it different from the other hdacs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef]
- Du, Y.; Wang, F.; Zou, J.; Le, W.; Dong, Q.; Wang, Z.; Shen, F.; Yu, L.; Li, Y. Histone deacetylase 6 regulates cytotoxic α-synuclein accumulation through induction of the heat shock response. Neurobiol. Aging 2014, 35, 2316–2328. [Google Scholar] [CrossRef] [PubMed]
- Leyk, J.; Goldbaum, O.; Noack, M.; Richter-Landsberg, C. Inhibition of hdac6 modifies tau inclusion body formation and impairs autophagic clearance. J. Mol. Neurosci. 2015, 55, 1031–1046. [Google Scholar] [CrossRef]
- Francelle, L.; Outeiro, T.F.; Rappold, G.A. Inhibition of hdac6 activity protects dopaminergic neurons from alpha-synuclein toxicity. Sci. Rep. 2020, 10, 6064. [Google Scholar] [CrossRef]
- Valdes, P.; Schneider, B.L. Gene therapy: A promising approach for neuroprotection in parkinson’s disease? Front. Neuroanat. 2016, 10, 123. [Google Scholar] [CrossRef]
- Zhang, C.W.; Adeline, H.B.; Chai, B.H.; Hong, E.T.; Ng, C.H.; Lim, K.L. Pharmacological or genetic activation of hsp70 protects against loss of parkin function. Neuro-Degener. Dis. 2016, 16, 304–316. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Chen, J.; Xue, J.; Ruan, J.; Zhao, J.; Tang, B.; Duan, R. Drosophila chip protects against mitochondrial dysfunction by acting downstream of pink1 in parallel with parkin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 5234–5245. [Google Scholar] [CrossRef]
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Höhfeld, J.; Patterson, C. The co-chaperone chip regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001, 3, 93–96. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, Z.W.; Mao, C.Y.; Shi, C.H.; Xu, Y.M. Chip as a therapeutic target for neurological diseases. Cell Death Dis. 2020, 11, 727. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Mao, C.; Wang, H.; Zhang, Z.; Zhang, S.; Luo, H.; Tang, M.; Yang, J.; Yuan, Y.; Wang, Y.; et al. Chip protects against mpp(+)/mptp-induced damage by regulating drp1 in two models of parkinson’s disease. Aging 2021, 13, 1458–1472. [Google Scholar] [CrossRef] [PubMed]
- Dimant, H.; Zhu, L.; Kibuuka, L.N.; Fan, Z.; Hyman, B.T.; McLean, P.J. Direct visualization of chip-mediated degradation of alpha-synuclein in vivo: Implications for pd therapeutics. PLoS ONE 2014, 9, e92098. [Google Scholar] [CrossRef]
- Rowland, N.C.; Kalia, S.K.; Kalia, L.V.; Larson, P.S.; Lim, D.A.; Bankiewicz, K.S. Merging dbs with viral vector or stem cell implantation:“Hybrid” stereotactic surgery as an evolution in the surgical treatment of parkinson’s disease. Mol. Ther. Methods Clin. Dev. 2016, 3, 15051. [Google Scholar] [CrossRef] [PubMed]
- Manfredsson, F.; Lewin, A.; Mandel, R. Rna knockdown as a potential therapeutic strategy in parkinson’s disease. Gene Ther. 2006, 13, 517–524. [Google Scholar] [CrossRef]
- Van Laar, A.D.; Van Laar, V.S.; San Sebastian, W.; Merola, A.; Elder, J.B.; Lonser, R.R.; Bankiewicz, K.S. An update on gene therapy approaches for parkinson’s disease: Restoration of dopaminergic function. J. Park. Dis. 2021, 11, S173–S182. [Google Scholar] [CrossRef]
- Parambi, D.G.T.; Alharbi, K.S.; Kumar, R.; Harilal, S.; Batiha, G.E.; Cruz-Martins, N.; Magdy, O.; Musa, A.; Panda, D.S.; Mathew, B. Gene therapy approach with an emphasis on growth factors: Theoretical and clinical outcomes in neurodegenerative diseases. Mol. Neurobiol. 2022, 59, 191–233. [Google Scholar] [CrossRef]
- Moloney, T.C.; Hyland, R.; O’Toole, D.; Paucard, A.; Kirik, D.; O’Doherty, A.; Gorman, A.M.; Dowd, E. Heat shock protein 70 reduces alpha-synuclein-induced predegenerative neuronal dystrophy in the alpha-synuclein viral gene transfer rat model of parkinson’s disease. CNS Neurosci. Ther. 2014, 20, 50–58. [Google Scholar] [CrossRef]
- Tiefensee Ribeiro, C.; Peixoto, D.O.; Santos, L.; Saibro-Girardi, C.; Brum, P.O.; Carazza-Kessler, F.G.; Somensi, N.; Behrens, L.M.P.; Bittencourt, R.R.; Soares, L.S.; et al. Intranasal hsp70 administration protects against dopaminergic denervation and modulates neuroinflammatory response in the 6-ohda rat model. Brain Behav. Immun. Health 2021, 14, 100253. [Google Scholar] [CrossRef] [PubMed]
- Salganik, M.; Sergeyev, V.G.; Shinde, V.; Meyers, C.A.; Gorbatyuk, M.S.; Lin, J.H.; Zolotukhin, S.; Gorbatyuk, O.S. The loss of glucose-regulated protein 78 (grp78) during normal aging or from sirna knockdown augments human alpha-synuclein (α-syn) toxicity to rat nigral neurons. Neurobiol. Aging 2015, 36, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Arkan, S.; Ljungberg, M.; Kirik, D.; Hansen, C. Dnajb6 suppresses alpha-synuclein induced pathology in an animal model of parkinson’s disease. Neurobiol. Dis. 2021, 158, 105477. [Google Scholar] [CrossRef] [PubMed]
- Koutras, C.; Braun, J.E. J protein mutations and resulting proteostasis collapse. Front. Cell. Neurosci. 2014, 8, 191. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A. New genes causing hereditary parkinson’s disease or parkinsonism. Curr. Neurol. Neurosci. Rep. 2017, 17, 66. [Google Scholar] [CrossRef]
- Mittal, S.O. Levodopa responsive-generalized dystonic spells and moaning in dnajc6 related juvenile parkinson’s disease. Park. Relat. Disord. 2020, 81, 188–189. [Google Scholar] [CrossRef]
- Calo, L.; Hidari, E.; Wegrzynowicz, M.; Dalley, J.W.; Schneider, B.L.; Podgajna, M.; Anichtchik, O.; Carlson, E.; Klenerman, D.; Spillantini, M.G. Cspalpha reduces aggregates and rescues striatal dopamine release in alpha-synuclein transgenic mice. Brain J. Neurol. 2021, 144, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, M.E.; Leung, E.H.; Sweeny, E.A.; Jackrel, M.E.; Cushman-Nick, M.; Neuhaus-Follini, A.; Vashist, S.; Sochor, M.A.; Knight, M.N.; Shorter, J. Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 2012, 151, 778–793. [Google Scholar] [CrossRef] [PubMed]
- Jackrel, M.E.; DeSantis, M.E.; Martinez, B.A.; Castellano, L.M.; Stewart, R.M.; Caldwell, K.A.; Caldwell, G.A.; Shorter, J. Potentiated hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 2014, 156, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Bouchez, G.; Sensebé, L.; Vourc’h, P.; Garreau, L.; Bodard, S.; Rico, A.; Guilloteau, D.; Charbord, P.; Besnard, J.C.; Chalon, S. Partial recovery of dopaminergic pathway after graft of adult mesenchymal stem cells in a rat model of parkinson’s disease. Neurochem. Int. 2008, 52, 1332–1342. [Google Scholar] [CrossRef]
- Shende, P.; Bhandarkar, S.; Prabhakar, B. Heat shock proteins and their protective roles in stem cell biology. Stem Cell Rev. Rep. 2019, 15, 637–651. [Google Scholar] [CrossRef]
- Venkataramana, N.K.; Kumar, S.K.; Balaraju, S.; Radhakrishnan, R.C.; Bansal, A.; Dixit, A.; Rao, D.K.; Das, M.; Jan, M.; Gupta, P.K.; et al. Open-labeled study of unilateral autologous bone-marrow-derived mesenchymal stem cell transplantation in parkinson’s disease. Transl. Res. J. Lab. Clin. Med. 2010, 155, 62–70. [Google Scholar] [CrossRef]
- Blandini, F.; Cova, L.; Armentero, M.T.; Zennaro, E.; Levandis, G.; Bossolasco, P.; Calzarossa, C.; Mellone, M.; Giuseppe, B.; Deliliers, G.L.; et al. Transplantation of undifferentiated human mesenchymal stem cells protects against 6-hydroxydopamine neurotoxicity in the rat. Cell Transplant 2010, 19, 203–217. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Carvalho, M.M.; Panchalingam, K.M.; Rodrigues, A.J.; Mendes-Pinheiro, B.; Anjo, S.; Manadas, B.; Behie, L.A.; Sousa, N.; Salgado, A.J. Impact of the secretome of human mesenchymal stem cells on brain structure and animal behavior in a rat model of parkinson’s disease. Stem Cells Transl. Med. 2017, 6, 634–646. [Google Scholar] [CrossRef]
- Teixeira, F.G.; Vilaca-Faria, H.; Domingues, A.V.; Campos, J.; Salgado, A.J. Preclinical comparison of stem cells secretome and levodopa application in a 6-hydroxydopamine rat model of parkinson’s disease. Cells 2020, 9, 315. [Google Scholar] [CrossRef]
- Cox, D.; Whiten, D.R.; Brown, J.W.P.; Horrocks, M.H.; San Gil, R.; Dobson, C.M.; Klenerman, D.; van Oijen, A.M.; Ecroyd, H. The small heat shock protein hsp27 binds alpha-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef]
- Mendes-Pinheiro, B.; Anjo, S.I.; Manadas, B.; Da Silva, J.D.; Marote, A.; Behie, L.A.; Teixeira, F.G.; Salgado, A.J. Bone marrow mesenchymal stem cells’ secretome exerts neuroprotective effects in a parkinson’s disease rat model. Front. Bioeng. Biotechnol. 2019, 7, 294. [Google Scholar] [CrossRef] [PubMed]
- Tome, D.; Fonseca, C.P.; Campos, F.L.; Baltazar, G. Role of neurotrophic factors in parkinson’s disease. Curr. Pharm. Des. 2017, 23, 809–838. [Google Scholar] [CrossRef] [PubMed]
- Rafi, M.A. Gene and stem cell therapy: Alone or in combination? BioImpacts BI 2011, 1, 213–218. [Google Scholar]
- Jiang, Z.; Wang, J.; Sun, G.; Feng, M. Bdnf-modified human umbilical cord mesenchymal stem cells-derived dopaminergic-like neurons improve rotation behavior of parkinson’s disease rats through neuroprotection and anti-neuroinflammation. Mol. Cell. Neurosci. 2022, 123, 103784. [Google Scholar] [CrossRef]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of parkinson’s disease. Lancet. Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.; Ho, P.W.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The interplay of aging, genetics and environmental factors in the pathogenesis of parkinson’s disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef]
- Sarkar, S.; Raymick, J.; Imam, S. Neuroprotective and therapeutic strategies against parkinson’s disease: Recent perspectives. Int. J. Mol. Sci. 2016, 17, 904. [Google Scholar] [CrossRef]
- Zeiss, C.J.; Allore, H.G.; Beck, A.P. Established patterns of animal study design undermine translation of disease-modifying therapies for parkinson’s disease. PLoS ONE 2017, 12, e0171790. [Google Scholar] [CrossRef]
- Alborghetti, M.; Bianchini, E.; De Carolis, L.; Galli, S.; Pontieri, F.E.; Rinaldi, D. Type-b monoamine oxidase inhibitors in neurological diseases: Clinical applications based on preclinical findings. Neural Regen. Res. 2024, 19, 16–21. [Google Scholar] [CrossRef]
- Abdanipour, A.; Jafari Anarkooli, I.; Shokri, S.; Ghorbanlou, M.; Bayati, V.; Nejatbakhsh, R. Neuroprotective effects of selegiline on rat neural stem cells treated with hydrogen peroxide. Biomed. Rep. 2018, 8, 41–46. [Google Scholar] [CrossRef]
- Elkenani, M.; Barakat, A.Z.; Held, T.; Rodrigues, D.M.; Mobarak, S.; Swarnka, S.; Adham, I.M.; Mohamed, B.A. Heat shock protein a4 ablation leads to skeletal muscle myopathy associated with dysregulated autophagy and induced apoptosis. J. Transl. Med. 2022, 20, 229. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three parkinson’s disease-causing genes: Parkin, pink1 and dj-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Duty, S.; Jenner, P. Animal models of parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Neuroprotective function of rasagiline and selegiline, inhibitors of type b monoamine oxidase, and role of monoamine oxidases in synucleinopathies. Int. J. Mol. Sci. 2022, 23, 11059. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, Z.H.; Li, X.Y.; Li, Y.F.; Li, J.R.; Hui, J.J.; Li, J.X.; Zhou, J.W.; Yi, Z.M. Efficacy and safety of selegiline for the treatment of parkinson’s disease: A systematic review and meta-analysis. Front. Aging Neurosci. 2023, 15, 1134472. [Google Scholar] [CrossRef] [PubMed]
- Mansour, H.M.; Mohamed, A.F.; Khattab, M.M.; El-Khatib, A.S. Lapatinib ditosylate rescues motor deficits in rotenone-intoxicated rats: Potential repurposing of anti-cancer drug as a disease-modifying agent in parkinson’s disease. Eur. J. Pharmacol. 2023, 954, 175875. [Google Scholar] [CrossRef]
- Choi, H.J.; Chen, T.X.; Hou, M.J.; Song, J.H.; Li, P.; Liu, C.F.; Wang, P.; Zhu, B.T. Protection against glutathione depletion-associated oxidative neuronal death by neurotransmitters norepinephrine and dopamine: Protein disulfide isomerase as a mechanistic target for neuroprotection. Acta Pharmacol. Sin. 2022, 43, 2527–2541. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Nehru, B. Modulatory effects of sodium salicylate on the factors affecting protein aggregation during rotenone induced parkinson’s disease pathology. Neurochem. Int. 2014, 75, 1–10. [Google Scholar] [CrossRef]
- Jing, X.; Shi, Q.; Bi, W.; Zeng, Z.; Liang, Y.; Wu, X.; Xiao, S.; Liu, J.; Yang, L.; Tao, E. Rifampicin protects pc12 cells from rotenone-induced cytotoxicity by activating grp78 via perk-eif2α-atf4 pathway. PLoS ONE 2014, 9, e92110. [Google Scholar] [CrossRef]
- Ghemrawi, R.; Khair, M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef]
- Motawi, T.K.; Al-Kady, R.H.; Abdelraouf, S.M.; Senousy, M.A. Empagliflozin alleviates endoplasmic reticulum stress and augments autophagy in rotenone-induced parkinson’s disease in rats: Targeting the grp78/perk/eif2alpha/chop pathway and mir-211-5p. Chem. Biol. Interact. 2022, 362, 110002. [Google Scholar] [CrossRef]
- Wang, D.X.; Chen, A.D.; Wang, Q.J.; Xin, Y.Y.; Yin, J.; Jing, Y.H. Protective effect of metformin against rotenone-induced parkinsonism in mice. Toxicol. Mech. Methods 2020, 30, 350–357. [Google Scholar] [CrossRef]
- Patil, S.P.; Jain, P.D.; Ghumatkar, P.J.; Tambe, R.; Sathaye, S. Neuroprotective effect of metformin in mptp-induced parkinson’s disease in mice. Neuroscience 2014, 277, 747–754. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Zimprich, A.; Carvajal Berrio, D.A.; Schindler, K.M.; Maurer, B.; Schulte, C.; Bus, C.; Hauser, A.K.; Kübler, M.; Lewin, R.; et al. Metformin reverses trap1 mutation-associated alterations in mitochondrial function in parkinson’s disease. Brain J. Neurol. 2017, 140, 2444–2459. [Google Scholar] [CrossRef] [PubMed]
- Nowell, J.; Blunt, E.; Gupta, D.; Edison, P. Antidiabetic agents as a novel treatment for alzheimer’s and parkinson’s disease. Ageing Res. Rev. 2023, 89, 101979. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.; Wang, X.; Xia, Y.; Huang, J.; Wang, T.; Lin, Z.; Xiong, N. Melatonin ameliorates parkinson’s disease via regulating microglia polarization in a roralpha-dependent pathway. NPJ Park. Dis. 2022, 8, 90. [Google Scholar] [CrossRef]
- Jung, Y.J.; Choi, H.; Oh, E. Melatonin attenuates mpp(+)-induced apoptosis via heat shock protein in a parkinson’s disease model. Biochem. Biophys. Res. Commun. 2022, 621, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P. Melatonin: Clinical perspectives in neurodegeneration. Front. Endocrinol. 2019, 10, 480. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y.; Zhao, S.; Zhang, Z.; Tong, X.; Wei, F.; Lu, Z. Heat shock protein 70 induction by glutamine increases the alpha-synuclein degradation in sh-sy5y neuroblastoma cells. Mol. Med. Rep. 2015, 12, 5524–5530. [Google Scholar] [CrossRef]
- Wang, H.; Tang, C.; Jiang, Z.; Zhou, X.; Chen, J.; Na, M.; Shen, H.; Lin, Z. Glutamine promotes hsp70 and inhibits alpha-synuclein accumulation in pheochromocytoma pc12 cells. Exp. Ther. Med. 2017, 14, 1253–1259. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sonia Angeline, M.; Sarkar, A.; Anand, K.; Ambasta, R.K.; Kumar, P. Sesamol and naringenin reverse the effect of rotenone-induced pd rat model. Neuroscience 2013, 254, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Christian, P.K.; Panchal, K.; Guruprasad, B.R.; Tiwari, A.K. Supplementation of spirulina (arthrospira platensis) improves lifespan and locomotor activity in paraquat-sensitive dj-1beta(delta93) flies, a parkinson’s disease model in drosophila melanogaster. J. Diet. Suppl. 2017, 14, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.Y. Effect and mechanism of chinese herbal medicine on parkinson’s disease. Int. Rev. Neurobiol. 2017, 135, 57–76. [Google Scholar]
- Westerheide, S.D.; Bosman, J.D.; Mbadugha, B.N.; Kawahara, T.L.; Matsumoto, G.; Kim, S.; Gu, W.; Devlin, J.P.; Silverman, R.B.; Morimoto, R.I. Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem. 2004, 279, 56053–56060. [Google Scholar] [CrossRef] [PubMed]
- McLean, P.J.; Klucken, J.; Shin, Y.; Hyman, B.T. Geldanamycin induces hsp70 and prevents α-synuclein aggregation and toxicity in vitro. Biochem. Biophys. Res. Commun. 2004, 321, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.Q.; Wang, X.L.; Zhang, D. Flz attenuates alpha-synuclein-induced neurotoxicity by activating heat shock protein 70. Mol. Neurobiol. 2017, 54, 349–361. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, J.-J.; Liu, G.-T. The novel squamosamide derivative (compound flz) attenuated 1-methyl, 4-phenyl-pyridinium ion (mpp+)-induced apoptosis and alternations of related signal transduction in sh-sy5y cells. Neuropharmacology 2007, 52, 423–429. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, J.-J.; Liu, G.-T. The novel squamosamide derivative flz protects against 6-hydroxydopamine-induced apoptosis through inhibition of related signal transduction in sh-sy5y cells. Eur. J. Pharmacol. 2007, 561, 1–6. [Google Scholar] [CrossRef]
- Dutta, N.; Ghosh, S.; Nelson, V.K.; Sareng, H.R.; Majumder, C.; Mandal, S.C.; Pal, M. Andrographolide upregulates protein quality control mechanisms in cell and mouse through upregulation of mtorc1 function. Biochim. Biophys. Acta. Gen. Subj. 2021, 1865, 129885. [Google Scholar] [CrossRef]
- Sanguanphun, T.; Sornkaew, N.; Malaiwong, N.; Chalorak, P.; Jattujan, P.; Niamnont, N.; Sobhon, P.; Meemon, K. Neuroprotective effects of a medium chain fatty acid, decanoic acid, isolated from h. Leucospilota against parkinsonism in c. Elegans pd model. Front. Pharmacol. 2022, 13, 1004568. [Google Scholar] [CrossRef]
- Chalorak, P.; Sanguanphun, T.; Limboonreung, T.; Meemon, K. Neurorescue effects of frondoside a and ginsenoside rg3 in c. Elegans model of parkinson’s disease. Molecules 2021, 26, 4843. [Google Scholar] [CrossRef]
- Dwivedi, V.; Lakhotia, S.C. Ayurvedic amalaki rasayana promotes improved stress tolerance and thus has anti-aging effects in drosophila melanogaster. J. Biosci. 2016, 41, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Khalid, R.; Afzal, M.; Anjum, F.; Fatima, H.; Zia, S.; Rasool, G.; Egbuna, C.; Mtewa, A.G.; Uche, C.Z.; et al. Phytobioactive compounds as therapeutic agents for human diseases: A review. Food Sci. Nutr. 2023, 11, 2500–2529. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, X.; Zhou, X.; Wu, X.; Li, Y.; Yao, J.; Bai, J. Nicotine suppresses the neurotoxicity by mpp(+)/mptp through activating alpha7nachr/pi3k/trx-1 and suppressing er stress. Neurotoxicology 2017, 59, 49–55. [Google Scholar] [CrossRef]
- Boyd, S.L.; Kuhn, N.C.; Patterson, J.R.; Stoll, A.C.; Zimmerman, S.A.; Kolanowski, M.R.; Neubecker, J.J.; Luk, K.C.; Ramsson, E.S.; Sortwell, C.E.; et al. Developmental exposure to the parkinson’s disease-associated organochlorine pesticide dieldrin alters dopamine neurotransmission in alpha-synuclein pre-formed fibril (pff)-injected mice. Toxicol. Sci. Off. J. Soc. Toxicol. 2023, 196, 99–111. [Google Scholar] [CrossRef]
- Ay, M.; Luo, J.; Langley, M.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Molecular mechanisms underlying protective effects of quercetin against mitochondrial dysfunction and progressive dopaminergic neurodegeneration in cell culture and mitopark transgenic mouse models of parkinson’s disease. J. Neurochem. 2017, 141, 766–782. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Chun, H.S. Protective effects of quercetin on dieldrin-induced endoplasmic reticulum stress and apoptosis in dopaminergic neuronal cells. Neuroreport 2016, 27, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A double-edged sword in health benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef]
- Putics, A.; Vegh, E.M.; Csermely, P.; Soti, C. Resveratrol induces the heat-shock response and protects human cells from severe heat stress. Antioxid. Redox Signal. 2008, 10, 65–75. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Q.; Wang, Y.; Guo, Y.; Xu, X.; Huang, P.; Lian, B.; Zhang, R.; Chen, Y.; Ha, Y. Protective effects of resveratrol liposomes on mitochondria in substantia nigra cells of parkinsonized rats. Ann. Palliat. Med. 2021, 10, 2458–2468. [Google Scholar] [CrossRef]
- Ramos Rego, I.; Santos Cruz, B.; Ambrosio, A.F.; Alves, C.H. TRAP1 in oxidative stress and neurodegeneration. Antioxidants 2021, 10, 1829. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Gaballo, A.; Signorile, A.; Ferretta, A.; Tanzarella, P.; Pacelli, C.; Di Paola, M.; Cocco, T.; Maffia, M. Resveratrol modulation of protein expression in parkin-mutant human skin fibroblasts: A proteomic approach. Oxid. Med. Cell. Longev. 2017, 2017, 2198243. [Google Scholar] [CrossRef]
- Li, Y.R.; Fan, H.J.; Sun, R.R.; Jia, L.; Yang, L.Y.; Zhang, H.F.; Jin, X.M.; Xiao, B.G.; Ma, C.G.; Chai, Z. Wuzi yanzong pill plays a neuroprotective role in parkinson’s disease mice via regulating unfolded protein response mediated by endoplasmic reticulum stress. Chin. J. Integr. Med. 2023, 29, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.L.; Zhou, J.J.; Liu, J.; Huo, X.K.; Wang, Y.L.; Liang, J.H.; Zhao, J.C.; Sun, C.P.; Yu, Z.L.; Fang, L.L.; et al. Uncaria rhynchophylla ameliorates parkinson’s disease by inhibiting hsp90 expression: Insights from quantitative proteomics. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 47, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
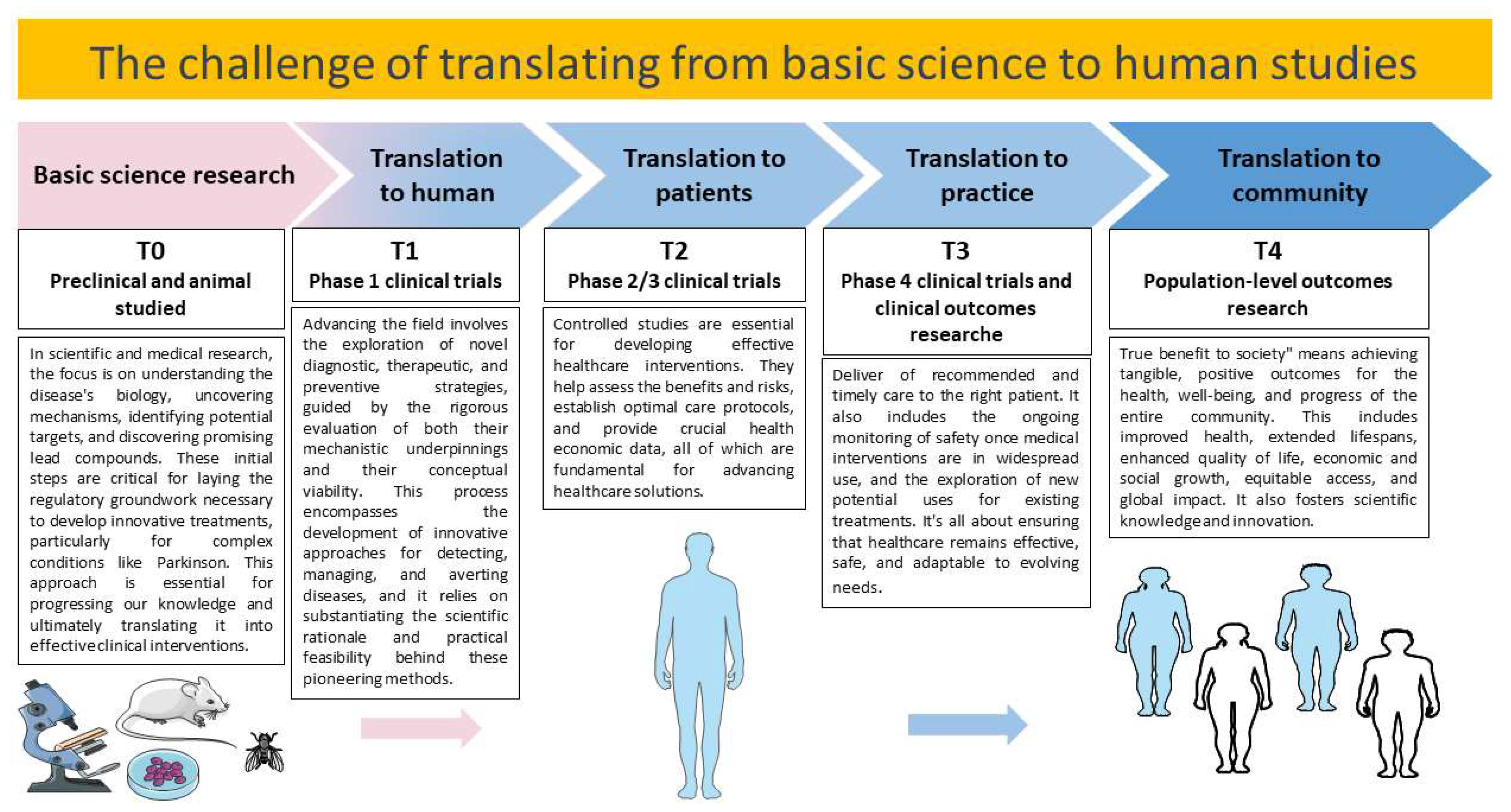
- Seyhan, A.A. Lost in translation: The valley of death across preclinical and clinical divide–identification of problems and overcoming obstacles. Transl. Med. Commun. 2019, 4, 1–19. [Google Scholar] [CrossRef]
- Cannon, J.R.; Greenamyre, J.T. Neurotoxic in vivo models of parkinson’s disease: Recent advances. Prog. Brain Res. 2010, 184, 17–33. [Google Scholar]
- Mahalmani, V.; Sinha, S.; Prakash, A.; Medhi, B. Translational research: Bridging the gap between preclinical and clinical research. Indian J. Pharmacol. 2022, 54, 393–396. [Google Scholar]
- Magen, I.; Chesselet, M.-F. Genetic mouse models of parkinson’s disease: The state of the art. Prog. Brain Res. 2010, 184, 53–87. [Google Scholar]
- Manning-BoĞ, A.B.; Langston, J.W. Model fusion: The next phase in developing animal models for parkinson’s disease. Neurotox. Res. 2007, 11, 219–240. [Google Scholar] [CrossRef]
- Chia, S.J.; Tan, E.K.; Chao, Y.X. Historical perspective: Models of parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef]
- Olanow, C.W. Can we achieve neuroprotection with currently available anti-parkinsonian interventions? Neurology 2009, 72, S59–S64. [Google Scholar] [CrossRef] [PubMed]





| Treatment | Model | HSP Interaction | Effect | Ref. |
|---|---|---|---|---|
| HSP inducers | ||||
| HSP70 exogenous | SH-SY5Y cells were exposed to ROT (3 μg/mL) for 1 h to induce the PD model and then treated with HSP70 (5, 10, 15, or 20 mg/L) for 72 h. | HSP70 | HSP70 reduced ROS and lipid peroxidation levels and protected cells from the apoptosis and mitochondrial dysfunction ROT-induced. Additionally, it reduced the levels of the NF-κB and STAT3 proteins. | [91] |
| HSP70 exogenous | Astrocytes were transfected with HSP70 cDNA and 4 h later were exposed to 100 nM of α-syn protein (A53T, human). For Hsp70 inhibition, 5 h before α-syn exposure, cells were exposed to VER155008 (10 μM). | HSP70 | HSP70 overexpression attenuated the neuroinflammatory and neurodegenerative response through inhibition of the JNK and NF-κB signaling pathways. | [92] |
| HSP70 exogenous | Male Wistar rats were subjected to several repeated bilateral microinjections of lactacystin (4 μg/μL) in the substantia nigra pars compacta every 7 days to induce PD. Then, 4 and 24 h after each lactacystin microinjection and a week after the last, the animals were treated the recombinant inducible human HSP70i (5 μg/10 μL) into each nostril of the rats. | HSP70 | HSP70i reduced the loss of dopaminergic neurons in the substantia nigra pars compacta that lactacystin induced, improved behavioral parameters in animals, and restored tyrosine hydroxylase levels. | [93] |
| HSP70 overexpression | Different fly strains were used, such as the control strain (w1118), the strain that did not express HSP70 (Df Hsp70), a dominant negative mutant strain of HSP70 (Hsp70K71E), a strain that overexpressed HSP70 (UAS-Hsp70), and a strain that overexpressed the human version of HSP70 (HSPA1L). All these mutants were exposed to paraquat (10 or 20 mM) for 12 and 24 h in order to induce PD-like symptoms. | HSP70 | The HSP70 overexpression in Drosophila mutants protected dopaminergic neuronal cells from oxidative stress and avoided the neuronal death that parquet induced. HSP70 also improved locomotor deficits in Drosophila and ameliorated survival. | [94] |
| Echinochrome derivative U-133 (HSP70 inducer) | A lactacystin-induced rodent PD model was treated with injections (i.p.) of echinochrome derivative U-133 at a dose of 5mg/kg 4 h and 24 h after each microinjection of irreversible proteasome inhibitor lactacystin and 7 days after. | HSP70 | HSP70 overexpression, via the U-133 HSP70 inducer, in dopaminergic neurons of the substantia nigra pars compacta protected the animals from lactacystin-induced PD-like symptoms. | [97] |
| Carbenoxolone (HSF-1 inducer) | The ROT model of PD was used for the study. The animals were divided into a control, ROT (ROT suspended in sunflower oil at a dose of 2 mg/kg; i.p.), ROT (ROT suspended in sunflower oil at a dose of 2 mg/kg; i.p.) + carbenoxolone, and carbenoxolone only (20 mg/kg; i.p.) groups for five weeks. | HSF-1 and HSP27 | Simultaneous treatment of carbenoxolone and ROT for five weeks slowed the neurodegenerative process and improved motor functions in the rat PD model via the elevation of HSF-1 and HSP-27 expression. | [100] |
| CSPα phosphorylation | For the in vivo study, PKCγ-KO mice and control mice (mice with the PKCγ gene intact) were used. To study the effect of phosphorylation on CSPα, the researchers would introduce CSPα mutants, such as CSPα(S10A/S34A) and CSPα(S10D/S34D), into cultured cells to evaluate how they affected function and interaction with other proteins such as HSP70 and SNAP25. | PKCγ–CSPα–HSC70/HSP70–SNAP25 axis | CSPα phosphorylation by PKCγ can protect the presynaptic terminal from neurodegeneration. | [102] |
| CHEC-9 peptide (HSF-1 inducer) | Sprague Dawley rats were fed a diluted strawberry gelatin solution containing the CHEC-9 peptide or not (1.0 mg/kg). For the in vitro study, human SY5Y neural cells were previously exposed to ROT at a concentration of 0.04 μM. After 10 min, a substance called CHEC-9 or a vehicle (a substance with no effects) was added to the cells for 24 h. | HSP70 | Oral treatment with the CHEC-9 peptide increased the level of active HSP70 monomers. In fact, it has been demonstrated in vitro that CHEC-9 also binds to HSP70 in the cytosol of the cerebral cortex. In the in vitro model of α-syn aggregation, CHEC-9 treatment induces HSP70-dependent dissolution of these aggregates in an HSP70-dependent manner. | [105] |
| Apelin-13 | SH-SY5Y cells were treated with MPP+ (0, 100, 250, 500, 750, or 1000 μM) with or without apelin-13 (100 nM) for 36 h. | GRP78 | Pretreatment with apelin-13 reduced ER stress through the inhibition of GRP78/CHOP/cleaved caspase-12 activation and the upregulation of phospho-ERK1/2, consequently reducing MPP+-induced apoptotic processes. | [110] |
| Apelin-13 | C57BL/6 male mice were treated with apelin-13 (0.3 μg/mice/day) or the same volume of saline into the substantia nigra pars compacta for 12 days and then were administered with MPTP (25 mg/kg/day) or saline intraperitoneally for 5 days. | GRP78, CHOP, and XBP1 | Apelin-13 exerted neuroprotective functions and improved motor impairments by inhibiting ER stress proteins and enhancing autophagy. It reduced levels of IRE1α, XBP1s, CHOP, and GRP78, which are stress proteins induced by MPTP. Furthermore, it decreased α-syn expression. | [111] |
| Apelin-36 | In order to induce the PD model, C57BL/6 mice were injected intraperitoneally with MPTP (25 mg/kg/day) or saline for 5 days. Afterwards, the animals were treated with apelin-36 (0.5 μg/mice/day) or the same volume of saline injected into the substantia nigra pars compacta for 7 days. | GRP78 and CHOP | Apelin-36 attenuated MPTP/MPP+-induced neurotoxicity in vitro and in vivo by inhibiting ER stress, apoptosis, and α-syn expression. It reduced levels of GRP78, CHOP, and cleaved caspase-12 in MPTP/MPP+-treated mice and cells. Thus, apelin-36 improved motor dysfunction and dopaminergic neurodegeneration. | [112] |
| miR-384-5p | SH-SY5Y cells were transfected with miR-384-5p mimics or inhibitors (50 nM) for 24 h prior to ROT (20 μM) exposure. For gene silencing, cells were transfected with GRP78 siRNA (50 nM) or control siRNA was transfected into SH-SY5Y cells with or without miR-384-5p inhibitors. | GRP78 | miR-384-5p inhibitors, inducing GRP78 overexpression, reduced α-syn-induced neurotoxicity by downregulating ER stress. | [115] |
| MANF | SH-SY5Y cells were cultured under different conditions: control, 6-ODHA (150 μM), 6-OHDA (150 μM) + MANF (4 μg/mL). Gene silencing was achieved using shRNA designed for the HSP70 target sequence (from 3476 to 3494 cDNA). | GRP78 and HSP70 | MANF treatment led to ER stress gene overexpression, such as HSP70 and GRP78. HSP70 silencing suppressed MANF’s protective effect against 6-OHDA-induced cell death. | [118] |
| MANF | The experiment was performed on SH-SY5Y cells treated with different concentrations of 6-OHDA (0–125 μM) and MANF (4 µg/mL or 8 µg/mL) for 48 h. The overexpression of α-syn was obtained through plasmid transfection into cells. Additionally, GRP78 knockdown was carried out via transfecting a vector containing a shRNA specific for a sequence of GRP78. | GRP78 | MANF suppressed apoptosis via GRP78 upregulation. The expression of GRP78 was related to cell survival, as demonstrated by using knockdown cells for GPR78. | [119] |
| CDNF and GDNF | Male Wistar rats received a unilateral stereotaxic injection of 6-OHDA (20 µg) in the left striatum. After 4 weeks, animals received unilateral intrastriatal injections of CDNF (1, 2.5, 5 μg) and GDNF (1, 2.5, 5 μg) alone or in combination. Primary cultured neurons from mouse embryos were exposed to thapsigargin (200 nM) and then to CDNF (100 ng/mL) and GDNF (50 ng/mL). | GRP78 and HSP70 | CDNF + GDNF activated the PI3K-Akt pathway, promoting cell survival. However, only CDNF reduced the expression of ER stress markers, including ATF6, GRP78, and phosphorylation of eIF2α. | [121] |
| HSP90 Inhibitors | ||||
| SNX-0723 and SNX-9114 (Small molecule Hsp90 inhibitors) | Rats were injected unilaterally in the substantia nigra with AAV8 expressing human α-synuclein in order to induce a model of OD. Then, they were treated with SNX-0723 (10 mg/kg) and SNX-9114 (1.5 and 3 mg/kg) for 8 weeks by oral gavage. | HSP90 and HSP70 | SNX-0723 (PF-04924868) and SNX-9114 (PF-04944733) protected against α-syn-dependent nigrostriatal toxicity through inhibition of Hsp90 and via upregulation of HSP70. | [125] |
| HSP90 inhibition (HSP90 siRNA) | Exposure of PC12 cells to 6-OHDA (75, 100, 125, 150, 175, and 200 μM) for 24 h and subsequent transfection with HSP90 siRNA (30 nM). | HSP90, HSF-1 and HSP70 | Inhibition of HSP90 protects cells from death, reducing the expression of pro-apoptotic factors and conversely increasing the expression of antiapoptotic factors. Furthermore, suppression of HSP90 mediated the regulation of other proteins, including the transcription factor HSF-1, which is involved in the activation of protective genes against stress, such as HSP70. | [126] |
| Trichostatin A (a potent inhibitory of HDAC activity) | The UPS impairment model of PD, established by stereotaxic injection of lactacystin (1.25 μg/2 μL) or its vehicle into the right medial forebrain bundle of the mouse (C57BL/6). For treatment, mice received an i.p. injection of trichostatin A (2 mg/kg) or its vehicle every other day. For the in vitro study, SK-N-SH cells and primary ventral midbrain neuron cultures were treated with lactacystin (10 μM) or tubacin, niltubacin (specific HDAC6 inhibitors; 10 μM), and/or HSP90 inhibitors (5 or 10 μM). | HDAC6, HSP90, HSF1, HSP70, and HSP27 | HDAC6 reduced α-syn oligomer levels and ameliorated the dopaminergic neuron survival in the UPS-impairment-induced PD model. This effect was due to HDAC6 binding to α-syn, which led to the triggering of chaperone expression, including HSP70 and HSP27, through dissociation of the HSP90–HSF1 complex. | [130] |
| Tubastatin A (selective inhibitor of HDAC6 activity) | Primary cultures of rat brain oligodendrocytes were incubated with MG-132 (1 μM) alone for 16 h or preincubated with tubastatin A (10 μM) for 3 h followed by MG-132 or for a further 16 h. The plasmid with shRNA for HDAC6 was transiently transfected into OLN-t40 cells (oligodendroglial cell line OLN-93 stably transfected with the longest human tau isoform) and after 24 h the cells were treated with MG-132 for 24 h. | HDAC6 and HSP70 | Tubastatin-A-induced HDAC6 inhibition does not prevent the accumulation of protein aggregates induced by UPS inhibition by MG-132. It caused an altered stress response by repressing the activity of HSP70 and induced the accumulation of autophagosomal vacuoles. | [131] |
| Tubastatin A (selective inhibitor of HDAC6 activity) | A rat model was used in which PD-like neurodegeneration was induced via overexpression of human α-syn in the substantia nigra pars compacta. The HADC6 inhibition was induced by treatment of rats with 15 mg/kg tubastatin A daily by intraperitoneal injection for 12 days, two days after the PD model induction. | HDAC6 and the chaperone proteins HSC70 and Lamp2 | HDAC6 could be potential as a therapeutic target, since inhibiting its activity via tubastatin A protected dopaminergic neurons against α-syn toxicity within the substantia nigra of rats. This effect was associated with an increase in chaperone-mediated autophagy via HSC70 and LAMP2. | [132] |
| Gene therapy | ||||
| Overexpression of HSP70 | A SH-SY5Y cell line stably expressing human Hsp70 was used. For treatment, these cells were treated with proteasome inhibitor PSI (30 nM), 17-AAG (5 μM), and HSP inhibitor I (KNK-473, 20 μM). To assess the PD model, parkin-null flies 24B -Gal4 (muscle-specific) and UAS-Hsp70 (UASHsap/HSPA1L).W Bonini) were used. After eclosion, flies were treated with 17-AAG (5 μM) for 25 days. | HSP70 | HSP70 induction protected cells against proteasome dysfunction. Treatment with 17-AAG, an HSP70 inducer, ameliorated the pathological phenotypes of Drosophila Parkin-null mutants | [134] |
| Overexpression of CHIP | In the animal model of Drosophila with genetic mutations in the PINK1 and PARK genes and wild-type (w1118), the overexpression of the CHIP protein was obtained by genetic transformation via a procedure called “p-element-mediated transformation”. To create flies without the CHIP protein (CHIP null mutant flies), a genome editing technique called CRISPR-Cas9 was employed. | CHIP | Overexpression of Drosophila CHIP suppressed the abnormal phenotypes and mitochondrial dysfunction in PINK1- or PARK-deficient flies thanks to its E3 ubiquitin ligase activity. | [136] |
| Overexpression of CHIP | SH-SY5Y cells were treated with 0.5, 1, 2, and 3 mM MPP+ for 24 h and then transfected with a CHIP-WT plasmid to overexpress human CHIP. Alternatively, cells were transfected with CHIP shRNA and a control shRNA to obtain CHIP knockdown and control cells, respectively. C57BL/6 mice were treated with MPTP to serve as a PD model. The overexpression of the CHIP protein was obtained via intravenous injection with AAV/BBB, whereas CRISPR/CAS9 was used to generate CHIP with a floxed STOP codon in the neural tissue of mice. | CHIP | Overexpression of CHIP reduced MPTP-induced toxicity, improving motor deficits and dopaminergic neuron survival. Moreover, CHIP overexpression suppressed the PD pathological upregulation of Drp1, proving the role of CHIP in mitochondrial maintenance. | [139] |
| AVV-CHIP | The delivery of AAVs-α-syn was employed to induce α-syn aggregates into the substantia nigra pars compacta of rats. Additionally, a separate vector containing CHIP cDNA was used to overexpress. | CHIP | CHIP overexpression reduced α-syn aggregates but may also affect tyrosine hydroxylase. | [140] |
| AVV-HSP70 and AVV-HSP27 | Male Sprague Dawley rats were injected intranigrally with pathogenic AAV-α-syn, to induce the PD model and simultaneously with AAV-HSP27 or AAV-HSP70 viral vectors. | HSP70 and HSP27 | AVV-HSP70 significantly reduced the dystrophy of nigrostriatal dopaminergic neurons and reduced the accumulation of α-syn in the substantia nigra. AVV-HSP27 did not protect animals from α-syn-induced pathology. | [145] |
| Recombinant HSP70 | Male Wistar rats received an intrastriatal injection of 6-OHDA (20 μg/rat) or vehicle. After 5 days, each rat was treated with a daily intranasal dose of recombinant HSP70 (2 μg/rat) or saline for 15 days. | HSP70 | Recombinant HSP70 counteracted the 6-OHDA-induced neurotoxicity, exerting anti-inflammatory effects, protecting dopaminergic neurons from death, and improving locomotor activity in animals. | [146] |
| GRP78 siRNA recombinant AAV | Male Sprague Dawley rats were injected into the substantia nigra pars compacta with pathogenic recombinant AAV-α-syn (1.5 μL) to induce the PD model and simultaneously with GRP78 siRNA recombinant AAV (1.5 μL). | GRP78 | Reducing GRP78 levels in mouse brain cells worsened α-syn-induced neurotoxicity, and the severity increased with reduced protein expression. | [147] |
| AAV6 DNAJB6 | In vitro, the HEK293 cell line was transfected with α-syn fibrils to induce α-syn aggregation. In vivo, stereotaxic injections of AAV6 vector cocktails (AAV-human wild type α-syn and AAV6-GFP or AAV6-human wild type α-syn and AAV6 DNAJB6-GFP) were performed into the substantia nigra of Sprague Dawley rats. | DNAJB6 | DNAJB6 counteracted α-syn aggregation in vitro. In vivo, DNAJB6 prevented α-syn aggregation and this resulted in a decrease in dopaminergic cell death and PD-related motor deficits in an animal model of PD. | [148] |
| AAV6CSPα | PC12 cells stably expressing 1-120hαSyn were transduced with AAV6CSPα (AAV6 vector encoding human CSPα) or with an empty AAV6EV (AAV6 control vector). In vivo, 10-month-old 1-120hαSyn transgenic mice showing a progressive decrease in striatal dopamine release were injected bilaterally with 2 μL of AAVCSPα and AAVEV into the substantia nigra pars compacta. | CSPα | CSPα rescues an α-synuclein-aggregation-related phenotype in 1-120hαSyn mice. Viral CSPα administration ameliorated impaired synaptic function, reduced synaptic α-syn aggregations, and restored normal dopamine release in 1-120hαSyn mice. This also led to the restoration of normal dopamine release in the mice. | [152] |
| Hsp104 mutants | The dopamine transporter gene (dat-1) promoter was used to target the expression of Hsp104 mutants (Hsp104A437W or Hsp104A503V or Hsp104Y507C) and α-syn in C. elegans dopaminergic neurons. | HSP104 | Enhanced HSP104 mutants improve aggregate dissolution, restore proper protein localization, suppress proteo-toxicity, and attenuate dopaminergic neurodegeneration. | [154] |
| Stem cell treatments | ||||
| hBM-MSC secretome | Five weeks after 6-OHDA induction, animals received the hBM-MSC secretome and levodopa. The animals received the hBM-MSC secretome by intracranial injection directly into the substantia nigra and striatum, whereas levodopa (12 mg/kg) was administered via oral gavage. | HSP27 | The multifactorial protein composition of the secretome includes several factors, such as HSP27, that could be important in the neuroprotective and functional recovery effects. | [160] |
| hBM-MSCs secretome and hBM-MSCs transplants | Five weeks after 6-OHDA induction, animals received the hBM-MSC secretome and hBM-MSC transplants (200,000 cells in the substantia nigra pars compacta and in the striatum). In vitro, neural progenitor cells were exposed for 5 days to the hBM-MSC secretome. | UPS | The secretome showed a better effect in restoring dopaminergic neurons and improving motor functions compared with hBM-MSC transplantation. | [162] |
| BDNF-modified hUC-MSCs derived Dopaminergic-like neurons | hUC-MSCs were cultured and differentiated into dopaminergic-like neurons with a neural induction solution for 24 h. After 9 days, cells were supplemented with 50 ng/mL BDNF for 3 days. Lentiviral vectors carrying GFP-BDNF, GFP-null, or BDNF-siRNA were transfected into dopaminergic-like neurons. For cell transplantation, rats were treated with hUC-MSC dopaminergic-like neurons transfected with null vector, with BDNF vector, or with BDNF siRNA. | HSP60 | The BDNF-modified hUC-MSC-derived dopaminergic-like neuron transplantation improved motor deficits, promoted neuroprotection and anti-inflammatory activity, and increased neuronal markers, intracerebral dopamine levels, and expression of proteins such as HSP60. | [165] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silvestro, S.; Raffaele, I.; Mazzon, E. Modulating Stress Proteins in Response to Therapeutic Interventions for Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 16233. https://doi.org/10.3390/ijms242216233
Silvestro S, Raffaele I, Mazzon E. Modulating Stress Proteins in Response to Therapeutic Interventions for Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(22):16233. https://doi.org/10.3390/ijms242216233
Chicago/Turabian StyleSilvestro, Serena, Ivana Raffaele, and Emanuela Mazzon. 2023. "Modulating Stress Proteins in Response to Therapeutic Interventions for Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 22: 16233. https://doi.org/10.3390/ijms242216233
APA StyleSilvestro, S., Raffaele, I., & Mazzon, E. (2023). Modulating Stress Proteins in Response to Therapeutic Interventions for Parkinson’s Disease. International Journal of Molecular Sciences, 24(22), 16233. https://doi.org/10.3390/ijms242216233