
Phytaspase Is Capable of Detaching the Endoplasmic Reticulum Retrieval Signal from Tobacco Calreticulin-3


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
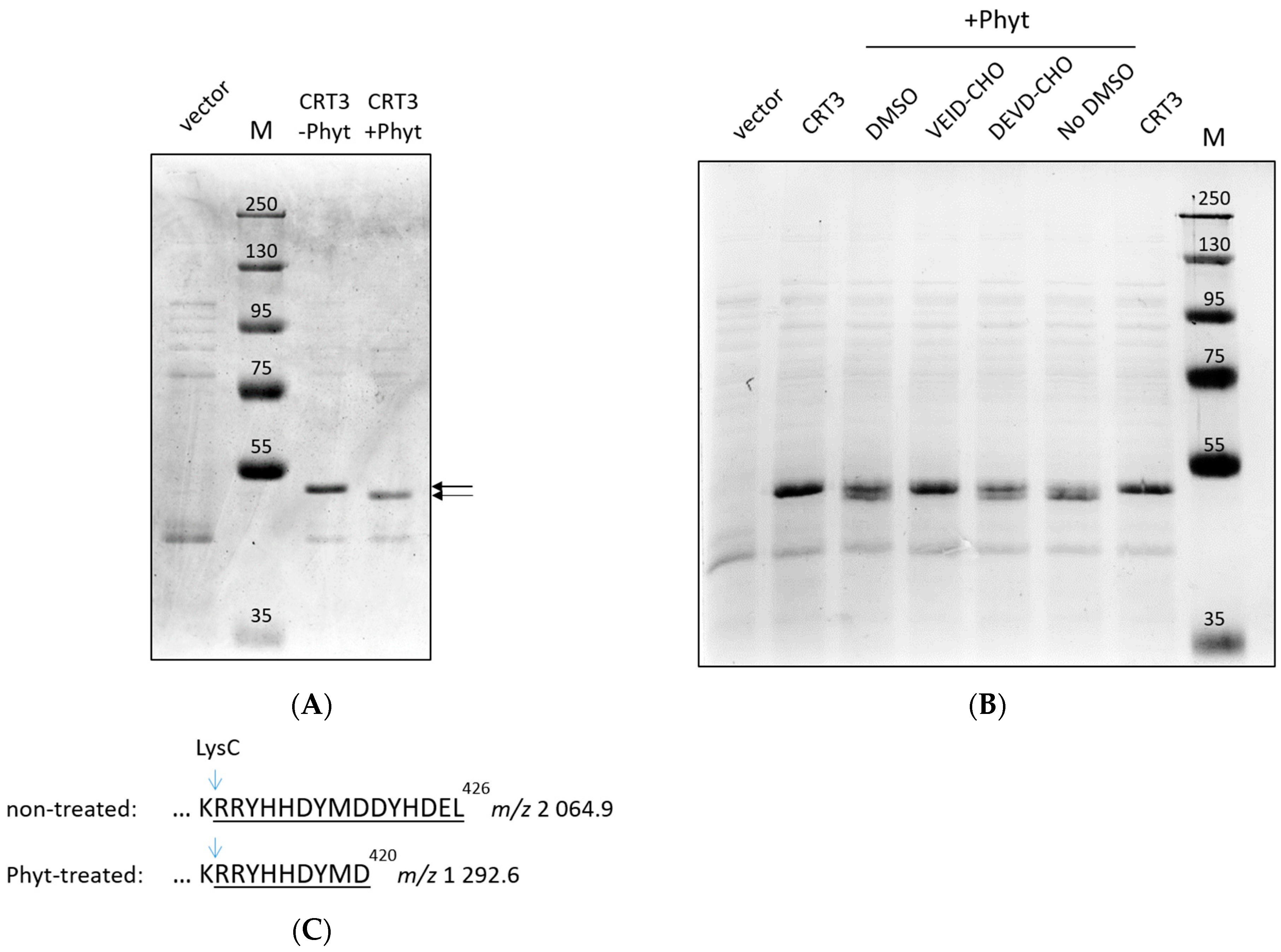
2.1. Phytaspase-Mediated Truncation of N. tabacum CRT3: Evidence with Purified Proteins
2.2. Identification of the NtPhyt Cleavage Site(s) in CRT3
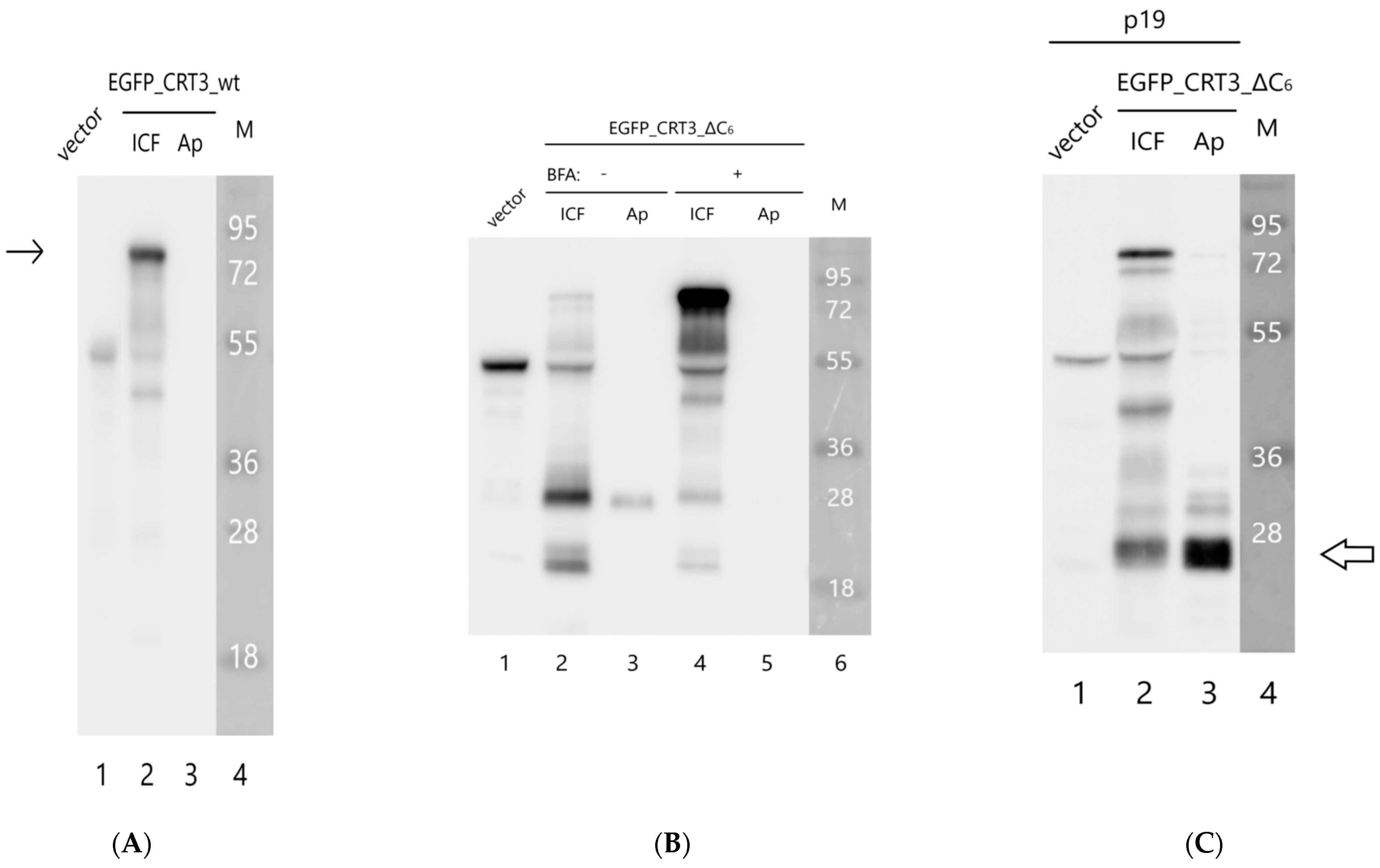
2.3. The Effect of NtCRT3 Truncation on the Protein Localization and Integrity
3. Discussion
4. Materials and Methods
4.1. Plant Growth Conditions
4.2. Plasmid Construction
4.3. Production, Purification, and Treatment of the His-Tagged NtCRT3 with NtPhyt
4.4. MS Analyses
4.5. Transient Expression in N. benthamiana and Protein Fractionation
4.6. Confocal Fluorescence Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blobel, G. Intracellular protein topogenesis. Proc. Natl. Acad. Sci. USA 1980, 77, 1496–1500. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.P.; Zeng, Y. An overview of protein secretion in plant cells. Methods Mol. Biol. 2017, 1662, 19–32. [Google Scholar] [PubMed]
- Liaci, A.M.; Förster, F. Take me home, protein roads: Structural insights into signal peptide interactions during ER translocation. Int. J. Mol. Sci. 2021, 22, 11871. [Google Scholar] [CrossRef]
- Paetzel, M.; Karla, A.; Strynadka, N.C.; Dalbey, R.E. Signal peptidases. Chem. Rev. 2002, 102, 4549–4580. [Google Scholar] [CrossRef]
- Liaci, A.M.; Steigenberger, B.; Telles de Souza, P.C.; Tamara, S.; Gröllers-Mulderij, M.; Ogrissek, P.; Marrink, S.J.; Scheltema, R.A.; Förster, F. Structure of the human signal peptidase complex reveals the determinants for signal peptide cleavage. Mol. Cell 2021, 81, 3934–3948. [Google Scholar] [CrossRef]
- Munro, S.; Pelham, H.R. A C-terminal signal prevents secretion of luminal ER proteins. Cell 1987, 48, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Pelham, H.R. The retention signal for soluble proteins of the endoplasmic reticulum. Trends Biochem. Sci. 1990, 15, 483–486. [Google Scholar] [CrossRef]
- Denecke, J.; De Rycke, R.; Botterman, J. Plant and mammalian sorting signals for protein retention in the endoplasmic reticulum contain a conserved epitope. EMBO J. 1992, 11, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.J.; Pelham, H.R. A human homologue of the yeast HDEL receptor. Nature 1990, 348, 162–163. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.J.; Pelham, H.R. Ligand-induced redistribution of a human KDEL receptor from the Golgi complex to the endoplasmic reticulum. Cell 1992, 68, 353–364. [Google Scholar] [CrossRef]
- Lee, H.I.; Gal, S.; Newman, T.C.; Raikhel, N.V. The Arabidopsis endoplasmic reticulum retention receptor functions in yeast. Proc. Natl. Acad. Sci. USA 1993, 90, 11433–11437. [Google Scholar] [CrossRef]
- Li, J.; Zhao-Hui, C.; Batoux, M.; Nekrasov, V.; Roux, M.; Chinchilla, D.; Zipfel, C.; Jones, J.D. Specific ER quality control components required for biogenesis of the plant innate immune receptor EFR. Proc. Natl. Acad. Sci. USA 2009, 106, 15973–15978. [Google Scholar] [CrossRef]
- Alvim, J.C.; Bolt, R.M.; An, J.; Kamisugi, Y.; Cuming, A.; Silva-Alvim, F.A.L.; Concha, J.O.; daSilva, L.L.P.; Hu, M.; Hirsz, D.; et al. The K/HDEL receptor does not recycle but instead acts as a Golgi-gatekeeper. Nat. Commun. 2023, 14, 1612. [Google Scholar] [CrossRef] [PubMed]
- Borisjuk, N.; Sitailo, L.; Adler, K.; Malysheva, L.; Tewes, A.; Borisjuk, L.; Manteuffel, R. Calreticulin expression in plant cells: Developmental regulation, tissue specificity and intracellular distribution. Planta 1998, 206, 504–514. [Google Scholar] [CrossRef]
- Arosa, F.A.; de Jesus, O.; Porto, G.; Carmo, A.M.; de Sousa, M. Calreticulin is expressed on the cell surface of activated human peripheral blood T lymphocytes in association with major histocompatibility complex class I molecules. J. Biol. Chem. 1999, 274, 16917–16922. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Michalak, M.; Opas, M.; Eggleton, P. The ins and outs of calreticulin: From the ER lumen to the extracellular space. Trends Cell Biol. 2001, 11, 122–129. [Google Scholar] [CrossRef]
- Tarr, J.M.; Young, P.J.; Morse, R.; Shaw, D.J.; Haigh, R.; Petrov, P.G.; Johnson, S.J.; Winyard, P.G.; Eggleton, P. A mechanism of release of calreticulin from cells during apoptosis. J. Mol. Biol. 2010, 401, 799–812. [Google Scholar] [CrossRef]
- Wiersma, V.R.; Michalak, M.; Abdullah, T.M.; Bremer, E.; Eggleton, P. Mechanisms of translocation of ER chaperones to the cell surface and immunomodulatory roles in cancer and autoimmunity. Front. Oncol. 2015, 5, 7. [Google Scholar] [CrossRef]
- Holaska, J.M.; Black, B.E.; Love, D.C.; Hanover, J.A.; Leszyk, J.; Paschal, B.M. Calreticulin is a receptor for nuclear export. J. Cell Biol. 2001, 152, 127–140. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.M.; Michalak, M.; Murphy-Ullrich, J.E. Calreticulin: Non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010, 24, 665–683. [Google Scholar] [CrossRef]
- Raghavan, M.; Wijeyesakere, S.J.; Peters, L.R.; Del Cid, N. Calreticulin in the immune system: Ins and outs. Trends Immunol. 2013, 34, 13–21. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Rano, T.A.; Peterson, E.P.; Rasper, D.M.; Timkey, T.; Garcia-Calvo, M.; Houtzager, V.M.; Nordstrom, P.A.; Roy, S.; Vaillancourt, J.P.; et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997, 272, 17907–17911. [Google Scholar] [CrossRef]
- Talanian, R.V.; Quinlan, C.; Trautz, S.; Hackett, M.C.; Mankovich, J.A.; Banach, D.; Ghayur, T.; Brady, K.D.; Wong, W.W. Substrate specificities of caspase family proteases. J. Biol. Chem. 1997, 272, 9677–9682. [Google Scholar] [CrossRef]
- Timmer, J.C.; Salvesen, G.S. Caspase substrates. Cell Death Differ. 2007, 14, 66–72. [Google Scholar] [CrossRef]
- Chichkova, N.V.; Shaw, J.; Galiullina, R.A.; Drury, G.E.; Tuzhikov, A.I.; Kim, S.H.; Kalkum, M.; Hong, T.B.; Gorshkova, E.N.; Torrance, L.; et al. Phytaspase, a relocalisable cell death promoting plant protease with caspase specificity. EMBO J. 2010, 29, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Galiullina, R.A.; Kasperkiewicz, P.; Chichkova, N.V.; Szalek, A.; Serebryakova, M.V.; Poreba, M.; Drag, M.; Vartapetian, A.B. Substrate specificity and possible heterologous targets of phytaspase, a plant cell death protease. J. Biol. Chem. 2015, 290, 24806–24815. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, S.; Repper, D.; Tuzhikov, A.I.; Galiullina, R.A.; Planas-Marquès, M.; Chichkova, N.V.; Vartapetian, A.B.; Stintzi, A.; Schaller, A. The tomato subtilase family includes several cell death-related proteinases with caspase specificity. Sci. Rep. 2018, 8, 10531. [Google Scholar] [CrossRef]
- Chichkova, N.V.; Kim, S.H.; Titova, E.S.; Kalkum, M.; Morozov, V.S.; Rubtsov, Y.P.; Kalinina, N.O.; Taliansky, M.E.; Vartapetian, A.B. A plant caspase-like protease activated during the hypersensitive response. Plant Cell 2004, 16, 157–171. [Google Scholar] [CrossRef]
- Vartapetian, A.B.; Tuzhikov, A.I.; Chichkova, N.V.; Taliansky, M.; Wolpert, T.J. A plant alternative to animal caspases: Subtilisin-like proteases. Cell Death Differ. 2011, 18, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Chichkova, N.V.; Tuzhikov, A.I.; Taliansky, M.; Vartapetian, A.B. Plant phytaspases and animal caspases: Structurally unrelated death proteases with a common role and specificity. Physiol. Plant. 2012, 145, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Beloshistov, R.E.; Dreizler, K.; Galiullina, R.A.; Tuzhikov, A.I.; Serebryakova, M.V.; Reichardt, S.; Shaw, J.; Taliansky, M.E.; Pfannstiel, J.; Chichkova, N.V.; et al. Phytaspase-mediated precursor processing and maturation of the wound hormone systemin. New Phytol. 2018, 218, 1167–1178. [Google Scholar] [CrossRef]
- Reichardt, S.; Piepho, H.P.; Stintzi, A.; Schaller, A. Peptide signaling for drought-induced tomato flower drop. Science 2020, 367, 1482–1485. [Google Scholar] [CrossRef]
- Trusova, S.V.; Golyshev, S.A.; Chichkova, N.V.; Vartapetian, A.B. Sometimes they come back: Endocytosis provides localization dynamics of a subtilase in cells committed to cell death. J. Exp. Bot. 2019, 70, 2003–2007. [Google Scholar] [CrossRef]
- Trusova, S.V.; Teplova, A.D.; Golyshev, S.A.; Galiullina, R.A.; Morozova, E.A.; Chichkova, N.V.; Vartapetian, A.B. Clathrin-mediated endocytosis delivers proteolytically active phytaspases into plant cells. Front. Plant Sci. 2019, 10, 873. [Google Scholar] [CrossRef]
- Teplova, A.D.; Serebryakova, M.V.; Galiullina, R.A.; Chichkova, N.V.; Vartapetian, A.B. Identification of phytaspase interactors via the proximity-dependent biotin-based identification approach. Int. J. Mol. Sci. 2021, 22, 13123. [Google Scholar] [CrossRef] [PubMed]
- Klausner, R.D.; Donaldson, J.G.; Lippincott-Schwartz, J. Brefeldin A: Insights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992, 116, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Nebenführ, A.; Ritzenthaler, C.; Robinson, D.G. Brefeldin A: Deciphering an enigmatic inhibitor of secretion. Plant Physiol. 2002, 130, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Hong, Z.; Su, W.; Li, J. A plant-specific calreticulin is a key retention factor for a defective brassinosteroid receptor in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2009, 106, 13612–13617. [Google Scholar] [CrossRef]
- Christensen, A.; Svensson, K.; Thelin, L.; Zhang, W.; Tintor, N.; Prins, D.; Funke, N.; Michalak, M.; Schulze-Lefert, P.; Saijo, Y.; et al. Higher plant calreticulins have acquired specialized functions in Arabidopsis. PLoS ONE 2010, 5, e11342. [Google Scholar] [CrossRef]
- Liebrand, T.W.; Smit, P.; Abd-El-Haliem, A.; de Jonge, R.; Cordewener, J.H.; America, A.H.; Sklenar, J.; Jones, A.M.; Robatzek, S.; Thomma, B.P.; et al. Endoplasmic reticulum-quality control chaperones facilitate the biogenesis of Cf receptor-like proteins involved in pathogen resistance of tomato. Plant Physiol. 2012, 159, 1819–1833. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, M.; Shibata, Y.; Ohtsu, M.; Mizutani, A.; Mori, H.; Wang, P.; Ojika, M.; Kawakita, K.; Takemoto, D. Nicotiana benthamiana calreticulin 3a is required for the ethylene-mediated production of phytoalexins and disease resistance against oomycete pathogen Phytophthora infestans. Mol. Plant Microbe Interact. 2013, 26, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.; Paul, M.; Kumar, A.; Pandey, D. Role of calreticulin in biotic and abiotic stress signalling and tolerance mechanisms in plants. Gene 2019, 714, 144004. [Google Scholar] [CrossRef]
- Cedzich, A.; Huttenlocher, F.; Kuhn, B.M.; Pfannstiel, J.; Gabler, L.; Stintzi, A.; Schaller, A. The protease-associated domain and C-terminal extension are required for zymogen processing, sorting within the secretory pathway, and activity of tomato subtilase 3 (SlSBT3). J. Biol. Chem. 2009, 284, 14068–14078. [Google Scholar] [CrossRef]
- Meyer, M.; Leptihn, S.; Welz, M.; Schaller, A. Functional characterization of propeptides in plant subtilases as intramolecular chaperones and inhibitors of the mature protease. J. Biol. Chem. 2016, 291, 19449–19461. [Google Scholar] [CrossRef] [PubMed]
- Stührwohldt, N.; Scholl, S.; Lang, L.; Katzenberger, J.; Schumacher, K.; Schaller, A. The biogenesis of CLEL peptides involves several processing events in consecutive compartments of the secretory pathway. eLife 2020, 9, e55580. [Google Scholar] [CrossRef]
- Sandvig, K.; Grimmer, S.; Lauvrak, S.U.; Torgersen, M.L.; Skretting, G.; van Deurs, B.; Iversen, T.G. Pathways followed by ricin and Shiga toxin into cells. Histochem. Cell Biol. 2002, 117, 131–141. [Google Scholar] [CrossRef]
- Lord, J.M.; Spooner, R.A. Ricin trafficking in plant and mammalian cells. Toxins 2011, 3, 787–801. [Google Scholar] [CrossRef]
- Sowa-Rogozińska, N.; Sominka, H.; Nowakowska-Gołacka, J.; Sandvig, K.; Słomińska-Wojewódzka, M. Intracellular transport and cytotoxicity of the protein toxin ricin. Toxins 2019, 11, 350. [Google Scholar] [CrossRef]
- Sandvig, K.; Kavaliauskiene, S.; Skotland, T. The protein toxins ricin and Shiga toxin as tools to explore cellular mechanisms of internalization and intracellular transport. Toxins 2021, 13, 377. [Google Scholar] [CrossRef]
- Pike, S.E.; Yao, L.; Jones, K.D.; Cherney, B.; Appella, E.; Sakaguchi, K.; Nakhasi, H.; Teruya-Feldstein, J.; Wirth, P.; Gupta, G.; et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J. Exp. Med. 1998, 188, 2349–2356. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teplova, A.D.; Pigidanov, A.A.; Serebryakova, M.V.; Golyshev, S.A.; Galiullina, R.A.; Chichkova, N.V.; Vartapetian, A.B. Phytaspase Is Capable of Detaching the Endoplasmic Reticulum Retrieval Signal from Tobacco Calreticulin-3. Int. J. Mol. Sci. 2023, 24, 16527. https://doi.org/10.3390/ijms242216527
Teplova AD, Pigidanov AA, Serebryakova MV, Golyshev SA, Galiullina RA, Chichkova NV, Vartapetian AB. Phytaspase Is Capable of Detaching the Endoplasmic Reticulum Retrieval Signal from Tobacco Calreticulin-3. International Journal of Molecular Sciences. 2023; 24(22):16527. https://doi.org/10.3390/ijms242216527
Chicago/Turabian StyleTeplova, Anastasia D., Artemii A. Pigidanov, Marina V. Serebryakova, Sergei A. Golyshev, Raisa A. Galiullina, Nina V. Chichkova, and Andrey B. Vartapetian. 2023. "Phytaspase Is Capable of Detaching the Endoplasmic Reticulum Retrieval Signal from Tobacco Calreticulin-3" International Journal of Molecular Sciences 24, no. 22: 16527. https://doi.org/10.3390/ijms242216527