
The Role H-Bonding and Supramolecular Structures in Homogeneous and Enzymatic Catalysis
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. The Role of the Secondary Interactions in Homogeneous Mechanisms and Enzymatic Catalysis
2.1.1. H-Bonding and the O2 Activation
2.1.2. Reactions in the Second Coordination Sphere of Metal Complexes including O2 Addition, Proton or Hydrogen Transfer, H-Bonding
2.2. Nanostructure Science and Supramolecular Chemistry
2.2.1. Models Ni(Fe)ARD Dioxygenases
2.2.2. Possible Effect of the Tyr Fragment in the Second Coordination Sphere of Ni(Fe)ARD
2.2.3. Models of Family Fe-Mono Oxygenases P450: The Possible Role of Tyrosine and Histidine Fragments
3. Materials and Methods
4. Conclusions
5. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AFM method | Atomic force microscopy method |
| (acac)− | Acetylacetonate ion |
| AP | Acetophenone |
| ARD | Acireductone ligand |
| 18C6 | 18-crown-6 |
| CO | Carbon monoxide |
| CHCl3 | Chloroform |
| Hem | Hemin |
| His | L-histidine |
| HMPA | Hexamethylphosphorotriamide |
| Hacac | Acetylacetone |
| L1 | acac− |
| L1ox | OAc− |
| L2 | Electron-donating mono- or multidentate activating ligand |
| MPC | Methyl phenyl carbinol |
| MSt | Stearates of alkaline metals (M = Li, Na, K) |
| Ni(Fe)ARD | Ni(Fe) Acireductone Dioxygenase |
| NMP | N-methyl-2-pirrolidone |
| (OAc)− | Acetate ion |
| PEH | α—phenyl ethyl hydroperoxide |
| RH | Ethylbenzene |
| SPEH | Selectivity of RH oxidation |
| Tyr | L-tyrosine |
| UV spectrum | Ultraviolet spectrum |
References
- Jacobsen, E.N.; Taylor, M.S. Asymmetric catalysis by chiral hydrogen bond donors. Angew. Chem. Int. Ed. 2006, 45, 1521–1539. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; Knowles, R.R. Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. Proc. Natl. Acad. Sci. USA 2010, 107, 20678–20685. [Google Scholar] [CrossRef]
- Matienko, L.I.; Mosolova, L.A.; Zaikov, G.E. Selective Catalytic Hydrocarbons Oxidation. In New Perspectives; Nova Science Publ. Inc.: New York, NY, USA, 2010; 150p. [Google Scholar]
- Borovik, A.S. Bioinspired Hydrogen Bond Motifs in Ligand Design: The Role of Noncovalent Interactions in Metal Ion Mediated Activation of Dioxygen. Acc. Chem. Res. 2005, 38, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Holm, R.H.; Solomon, E.I. Biomimetic Inorganic Chemistry. Chem. Rev. 2004, 104, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Reek, J.N.H.; de Bruin, B.; Pullen, S.; Mooibroek, T.J.; Kluwer, A.M.; Caumes, X. Transition Metal Catalysis Controlled by Hydrogen Bonding in the Second Coordination Sphere. Chem Rev. 2022, 122, 12308–12369. [Google Scholar] [CrossRef] [PubMed]
- Zamaraev, K.I. Prospects of catalysis: From studies at the molecular level to new industrial catalysts and processes. Rus. Chem. Rev. 1993, 62, 1051–1063. (In Russian) [Google Scholar] [CrossRef]
- Matienko, L.I.; Mosolova, L.A. Mechanism of selective catalysis with triple system {bis(acetylacetonate)Ni(II)+metalloligand+phenol} in ethylbenzene oxidation with dioxygen. Role of H-bonding interactions. Oxid. Commun. 2014, 37, 20–31. [Google Scholar]
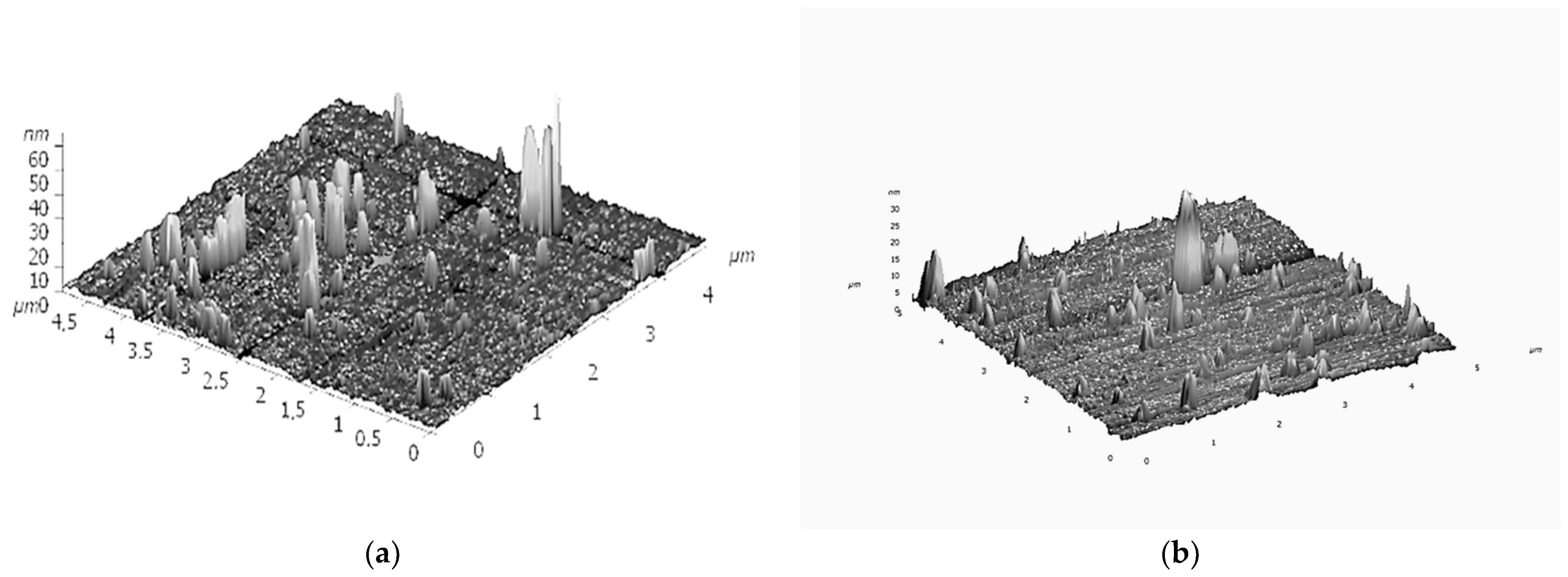
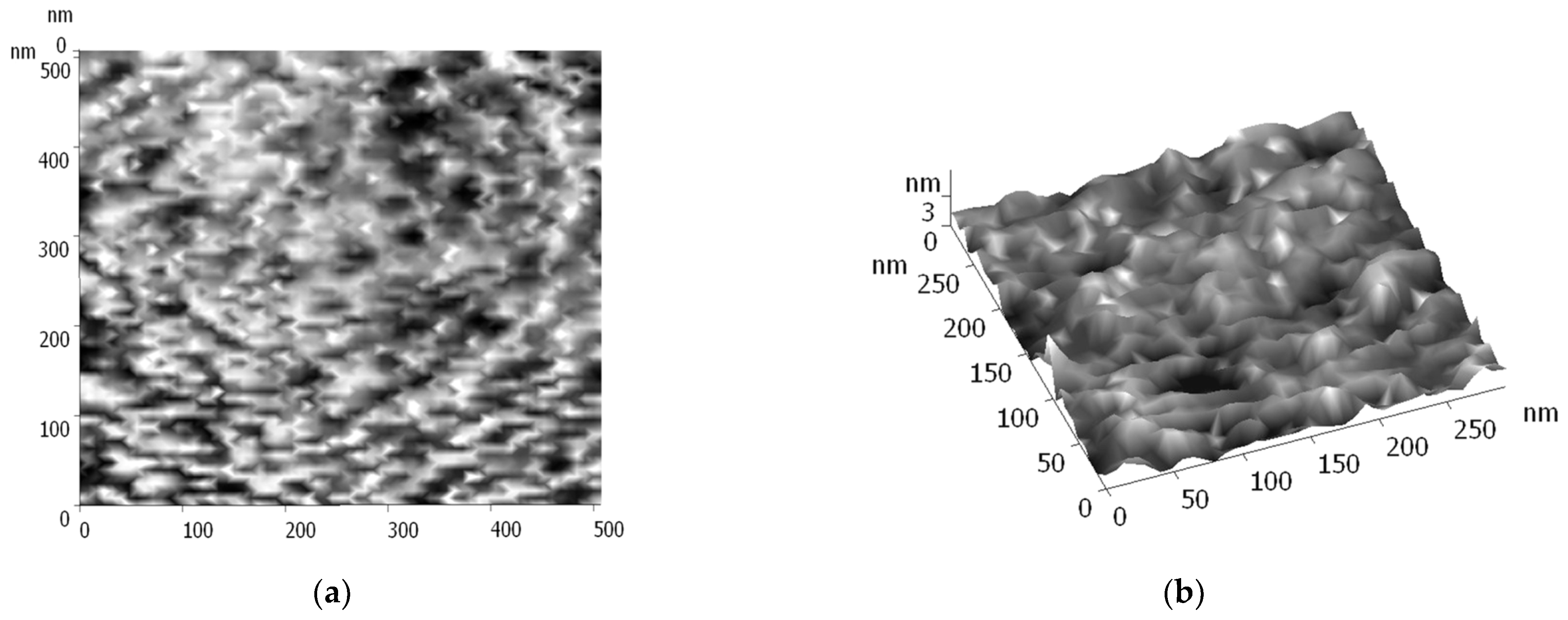
- Matienko, L.I.; Binyukov, V.I.; Mil, E.M. AFM Research of Supramolecular Structures. Russ. J. Phys. Chem. B 2020, 14, 559–563. [Google Scholar] [CrossRef]
- Sobczyk, L.; Grabowski, S.J.; Krygowski, T.M. Interrelation between H-Bond and Pi-Electron Delocalisation. Chem. Rev. 2005, 105, 3513–3560. [Google Scholar] [CrossRef]
- Saggu, M.; Levinson, N.M.; Boxer, S.G. Direct Measurements of Electric Fields in Weak OH···π Hydrogen Bonds. J. Am. Chem. Soc. 2011, 133, 17414–17419. [Google Scholar] [CrossRef]
- Ma, J.C.; Dougherty, D.A. The Cation−π Interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.D.; Buytendyk, A.M.; Wang, D.; Bowen, K.H.; Collins, K.D. Strong, Low-Barrier Hydrogen Bonds May Be Available to Enzymes. Biochemistry 2014, 53, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Golubev, N.S.; Smirnov, S.N.; Denisov, G.S.; Limbach, H.-H. Nuclear Scalar Spin–Spin Coupling Reveals Novel Properties of Low-Barrier Hydrogen Bonds in a Polar Environment. Chem. A Eur. J. 1999, 5, 492–497. [Google Scholar] [CrossRef]
- Shook, R.L.; Borovik, A.S. Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg. Chem. 2010, 49, 3646–3660. [Google Scholar] [CrossRef]
- Lucas, R.L.; Zart, M.K.; Murkerjee, J.; Sorrell, T.N.; Powell, D.R.; Borovik, A.S. A Modular Approach toward Regulating the Secondary Coordination Sphere of Metal Ions: Differential Dioxygen Activation Assisted by Intramolecular Hydrogen Bonds. J. Am. Chem. Soc. 2006, 128, 15476–15489. [Google Scholar] [CrossRef] [PubMed]
- Tomchick, D.R.; Phan, P.; Cymborovski, M.; Minor, W.; Holm, T.R. Structural and Functional Characterization of Second-Coordination Sphere Mutants of Soybean Lipoxygenase-1. Biochemistry 2001, 40, 7509–7517. [Google Scholar] [CrossRef]
- Perutz, M.F.; Fermi, G.; Luisi, B.; Shaanan, B.; Liddington, R.C. Stereochemistry of cooperative mechanisms in hemoglobin. Acc. Chem. Res. 1987, 20, 309–321. [Google Scholar] [CrossRef]
- Schlichting, I.; Berendzen, J.; Chu, K.; Stock, A.M.; Maves, S.A.; Benson, D.E.; Sweet, R.M.; Ringe, D.; Petsko, G.A.; Sligar, S.G. The catalytic Pathway of Cytochrome P450cam at Atomic Resolution. Science 2000, 287, 1615–1622. [Google Scholar] [CrossRef]
- Nekipelov, V.M.; Zamaraev, K.I. Outer-sphere coordination of organic moleculs to electrically neutral metal complexes. Coord. Chem. Rev. 1985, 61, 185–240. [Google Scholar] [CrossRef]
- Uehara, K.; Ohashi, Y.; Tanaka, M. Bis(acetylacetonato) metal(II)–Catalyzed Addition of Acceptor Molecules to Acetylacetone. Bull. Chem. Soc. Jpn. 1976, 49, 1447–1448. [Google Scholar] [CrossRef]
- Nelson, J.H.; Howels, P.N.; Landen, G.L.; De Lullo, G.S.; Henry, R.A. Catalytic addition of electrophiles to β—Dicarbonyles. In Fundamental Research in Homogeneous Catalysis; Plenum Press: New York, NY, USA; London, UK, 1979; Volume 3, pp. 921–939. [Google Scholar]
- Dai, Y.; Pochapsky, T.C.; Abeles, R.H. Mechanistic Studies of Two Dioxygenases in the Methionine Salvage Pathway of Klebsiella pneumonia. Biochemistry 2001, 40, 6379–6387. [Google Scholar] [CrossRef] [PubMed]
- Gopal, B.; Madan, L.L.; Betz, S.F.; Kossiakoff, A.A. The Crystal Structure of a Quercetin 2,3-Dioxygenase from Bacillus subtilis Suggests Modulation of Enzyme Activity by a Change in the Metal Ion at the Active Site(s). Biochemistry 2005, 44, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Balogh-Hergovich, E.; Kaizer, J.; Speier, G. Kinetics and mechanism of the Cu(I) and Cu(II) flavonolate-catalyzed oxygenation of flavonols, Functional quercetin 2,3-dioxygenase models. J. Mol. Catal. A Chem. 2000, 159, 215–224. [Google Scholar] [CrossRef]
- Straganz, G.D.; Nidetzky, B. Reaction Coordinate Analysis for β-Diketone Cleavage by the Non-Heme Fe2+-Dependent Dioxygenase Dke 1. J. Am. Chem. Soc. 2005, 127, 12306–12314. [Google Scholar] [CrossRef] [PubMed]
- Matienko, L.I.; Mosolova, L.A. Modeling of Catalytic of Complexes FeII,III(acac)n with R4NBr or 18-Crown-6 in the Ethyl benzene Oxidation by Dioxygen in the Presence of Small Amounts of H2O. Oxid. Commun. 2008, 31, 285–298. [Google Scholar]
- Matienko, L.I.; Mosolova, L.A. The Effect of Small Amounts of H2O on Mechanism of the Ethyl benzene Oxidation by Dioxygen, Catalyzed with Complexes FeII,III(acac)n with 18-Crown-6. Pet. Chem. 2008, 48, 371–380. [Google Scholar] [CrossRef]
- Allpress, C.J.; Grubel, K.; Szajna-Fuller, E.; Arif, A.M.; Berreau, L.M. Regioselective Aliphatic Carbon-Carbon Bond Cleavage by Model System of Relevance to Iron-Contaning Acireductone Dioxygenase. J. Am. Chem. Soc. 2013, 135, 659–668. [Google Scholar] [CrossRef]
- Ball, P. Water as an active constituent in cell biology. Chem. Rev. 2008, 108, 74–108. [Google Scholar] [CrossRef]
- Derat, E.; Shaik, S.; Rovira, C.; Vidossich, P.; Alfonso-Prieto, M. The Effect of a Water Molecule on the Mechanism of Formation of Compound 0 in Horseradish Peroxidase. J. Am. Chem. Soc. 2007, 129, 6346–6347. [Google Scholar] [CrossRef]
- Ma, L.-H.; Liu, Y.; Zhang, X.; Yoshida, T.; Langry, K.C.; Smith, K.M.; LaMar, G.N. Modulation of the Axial Water Hydrogen-Bonding Properties by Chemical Modification of the Substrate in Resting State, Substrate-Bound Heme Oxygenase from Neisseria meningitides; Coupling to the Distal H-Bond Network via Ordered Water Molecules. J. Am. Chem. Soc. 2006, 128, 6391–6399. [Google Scholar] [CrossRef]
- Funabiki, T.; Yoneda, I.; Ishikawa, M.; Ujiie, M.; Nagai, Y.; Yoshida, S. Extradiol Oxygenation of 3,5-Di-tert-butylcatechol with O2 by Iron Chlorides in Tetrahydrofuran-Water as a Model Reaction for Catechol-2,3-dioxygenases. J. Chem. Soc. Chem. Commun. 1994, 1453–1454. [Google Scholar] [CrossRef]
- Leninger, S.; Olenyuk, B.; Stang, P.J. Self-Assembly of Discrete Cyclic Nanostructures Mediated by Transition Metals. Chem. Rev. 2000, 100, 853–908. [Google Scholar] [CrossRef] [PubMed]
- Stang, P.J.; Olenyuk, B. Self-Assembly, Symmetry, and Molecular Architecture: Coordination as the Motif in the Rational Design of Supramolecular Metallacyclic Polygons and Polyhedra. Acc. Chem. Res. 1997, 30, 502–518. [Google Scholar] [CrossRef]
- Lehn, J.-M. Toward Self-Organization and Complex Matter. Science 2002, 295, 2400–2403. [Google Scholar] [CrossRef] [PubMed]
- Drain, C.M.; Varotto Radivojevic, A.I. Self-Organized Porphyrinic Materials. Chem. Rev. 2009, 109, 1630–1658. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.; Tyurin, V.S.; Tsivadze, A.Y.; Guilard, R.R.; Stem, C. Supramolecular Chemistry of Metalloporphyrins. Chem. Rev. 2009, 109, 1659–1713. [Google Scholar] [CrossRef]
- Liu, X.; Garber, A.; Ryan, J.; Deshpande, A.; Ringe, D.; Pochapsky, T.C. A Model for the Solution Structure of Human Fe(II)-Bound Acireductone Dioxygenase and Interactions with the Regulatory Domain of Matrix Metalloproteinase I (MMP-I). Biochemistry 2020, 59, 4238–4249. [Google Scholar] [CrossRef]
- Leitgeb, S.; Straganz, G.D.; Nidetzky, B. Functional characterization of an orphan cupin protein from Burkholderia xenovorans reveals a mononuclear nonheme Fe2+-dependent oxygenase that cleaves β-diketones. FEBS J. 2009, 276, 5983–5997. [Google Scholar] [CrossRef]
- Deshpande, A.R.; Pochapsky, T.C.; Ringe, D. Metal controls chemistry: Dual functions of acireductone dioxygenase. Chem. Rev. 2017, 117, 10474–10501. [Google Scholar] [CrossRef]
- Matienko, L.I.; Binyukov, V.I.; Mil, E.M.; Goloshchapov, A.A. Role of PhOH and Tyrosine in Selective Oxidation of Hydrocarbons. Catalysts 2021, 11, 1032. [Google Scholar] [CrossRef]
- Smirnov, V.V.; Roth, J.P. Tyrosine oxidation in heme oxygenase: Examination of long-range proton-coupled electron transfer. J. Biol. Inorg. Chem. 2014, 19, 1137–1148. [Google Scholar] [CrossRef]
- Mbughuni, M.M.; Meier, K.K.; Münck, E.; Lipscomb, J.D. Substrate-Mediated Oxygen Activation by Homoprotocatechuate 2,3-Dioxygenase: Intermediates Formed by a Tyrosine 257 Variant. Biochemistry 2012, 51, 8743–8754. [Google Scholar] [CrossRef]
- Zhang, J.; Klinman, J.P. Enzymatic Methyl Transfer: Role of an Active Site Residue in Generating Active Site Compactio That Correlates with Catalytic Efficiency. J. Am. Chem. Soc. 2011, 133, 17134–17137. [Google Scholar] [CrossRef]
- Horowitz, S.; Dirk, L.M.A.; Yesselman, J.D.; Nimtz, J.S.; Adhikari, U.U.; Mehl, R.A.; Scheiner, S.; Houtz, R.L.; Al-Hashimi, H.M.; Trievel, R.C. Conservation and Functional Importance of Carbon–Oxygen Hydrogen Bonding in AdoMet-Dependent Methyltransferases. J. Am. Chem. Soc. 2013, 135, 15536–15548. [Google Scholar] [CrossRef] [PubMed]
- Matienko, L.I.; Zhigacheva, I.V.; Mil, E.M.; Albantova, A.A.; Goloshchapov, A.A. The Dual Function of PhOH Included in the Coordination Sphere of the Nickel Complexes in the Processes of Oxidation with Dioxygen. Molecules 2022, 27, 3502. [Google Scholar] [CrossRef]
- Dubey, M.; Koner, R.R.; Ray, M. Sodium and Potassium Ion Directed Self-assembled Multinuclear Assembly of Divalent Nickel or Copper and L-Leucine Derived Ligand. Inorg. Chem. 2009, 48, 9294–9302. [Google Scholar] [CrossRef] [PubMed]
- Basiuk, E.V.; Basiuk, V.V.; Gomez-Lara, J.; Toscano, R.A. A bridged high-spin complex bis-[Ni(II)(rac-5,5,7,12,12,14-hexamethyl-1,4,8,11-tetraazacyclotetradecane)]-2,5-pyridinedicaboxylate diperchlorate monohydrate. J. Incl. Phenom. Macrocycl. Chem. 2000, 38, 45–56. [Google Scholar] [CrossRef]
- Mukherjee, P.; Drew, M.G.B.; Gómez-Garcia, C.J.; Ghosh, A. (Ni2), (Ni3), and (Ni2 + Ni3): A Unique Example of Isolated and Cocrystallized Ni2 and Ni3 Complexes. Inorg. Chem. 2009, 48, 4817–4825. [Google Scholar] [CrossRef] [PubMed]
- Gentili, D.; Valle, F.; Albonetti, C.; Liscio, F.; Cavallini, M. Self-Organization of Functional Materials in Confinement. Acc. Chem. Res. 2014, 47, 2692–2699. [Google Scholar] [CrossRef]
- Kneipp, J.; Balakrishnan, G.; Chen, R.; Shen, T.J.; Sahu, S.C.; Ho, N.T.; Giovannelli, J.L.; Simplaceanu, V.; Ho, C.; Spiro, T. Dynamics of allostery in hemoglobin: Roles of the penultimate tyrosine H bonds. J. Mol. Biol. 2005, 356, 335–353. [Google Scholar] [CrossRef]
- Cook, S.A.; Hill, E.A.; Borovik, A.S. Lessons from Nature: A Bio-Inspired Approach to Molecular Design. Biochemistry 2015, 54, 4167–4180. [Google Scholar] [CrossRef] [PubMed]
- Murry, D.T.; Tysko, R. Side Chain Hydrogen-Bonding Interactions within Amiloid-like Fibrils Formed by the Low-Complexity Domain of FUS: Evidence from Solid State Nuclear Magnetic Resonance Spectroscopy. Biochemistry 2020, 59, 304–378. [Google Scholar] [CrossRef]
- Biedermann, F.; Schneider, H.-J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef] [PubMed]
- Basom, E.J.; Bryce, A.M.; Thielges, M.C. Conformational Heterogeneity and the Affinity of Substrate Molecular Recognition by Cytochrome P450cam. Biochemistry 2017, 56, 3248–3256. [Google Scholar] [CrossRef]
- Binyukov, V.I.; Mil, E.M.; Matienko, L.I.; Albantova, A.A.; Goloshchapov, A.N. Methodic Approach of Atomic-Force Microscopy (AFM) to study of morphological changes of cells and of model systems. Micro 2023, 3, 382–390. [Google Scholar] [CrossRef]
- Mukhopadhyay, R. Molecular level structural studies of metalloproteins/metalloenzymes by scanning tunneling microscopy; Scopes and promises. Curr. Sci. 2003, 84, 1202–1210. [Google Scholar]
- Sarkar, R.; Xie, T.-Z.; Endres, K.J.; Wang, Z.; Moorefield, C.N.; Saunders, M.J.; Ghorai, S.; Patri, A.K.; Wesdemiotis, C.; Dobrynin, A.V.; et al. Sierpinski Pyramids by Molecular Entanglement. J. Am. Chem. Soc. 2020, 142, 5526–5530. [Google Scholar] [CrossRef]

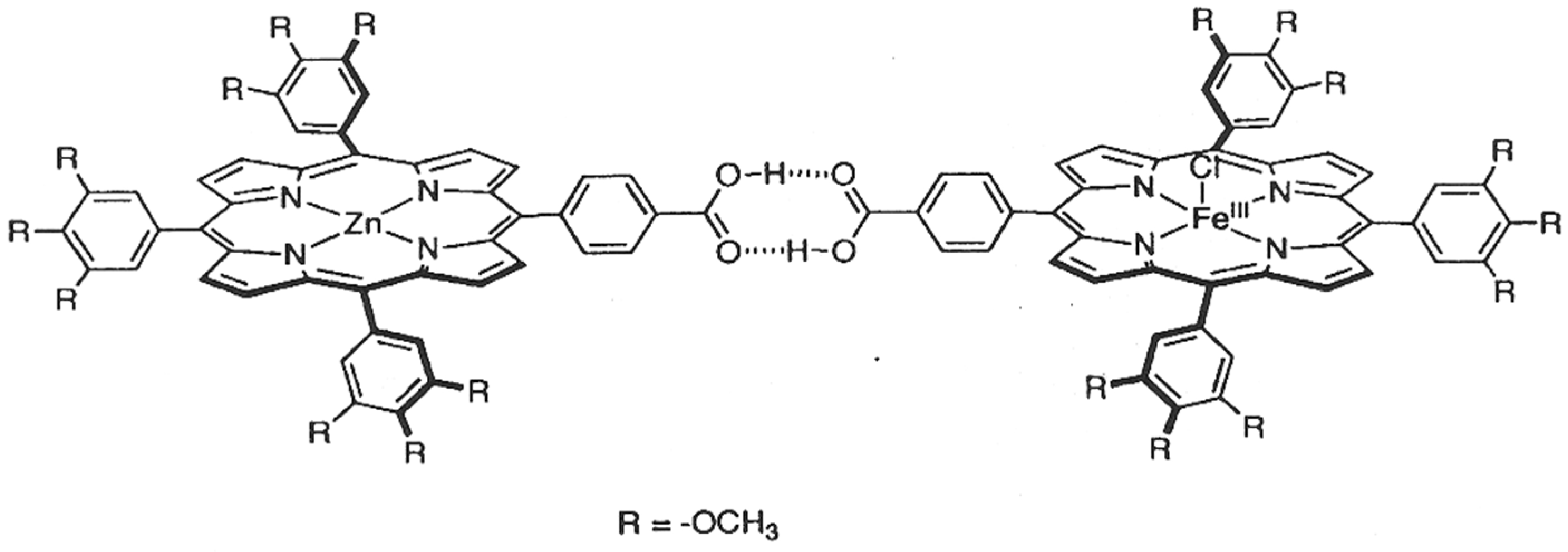



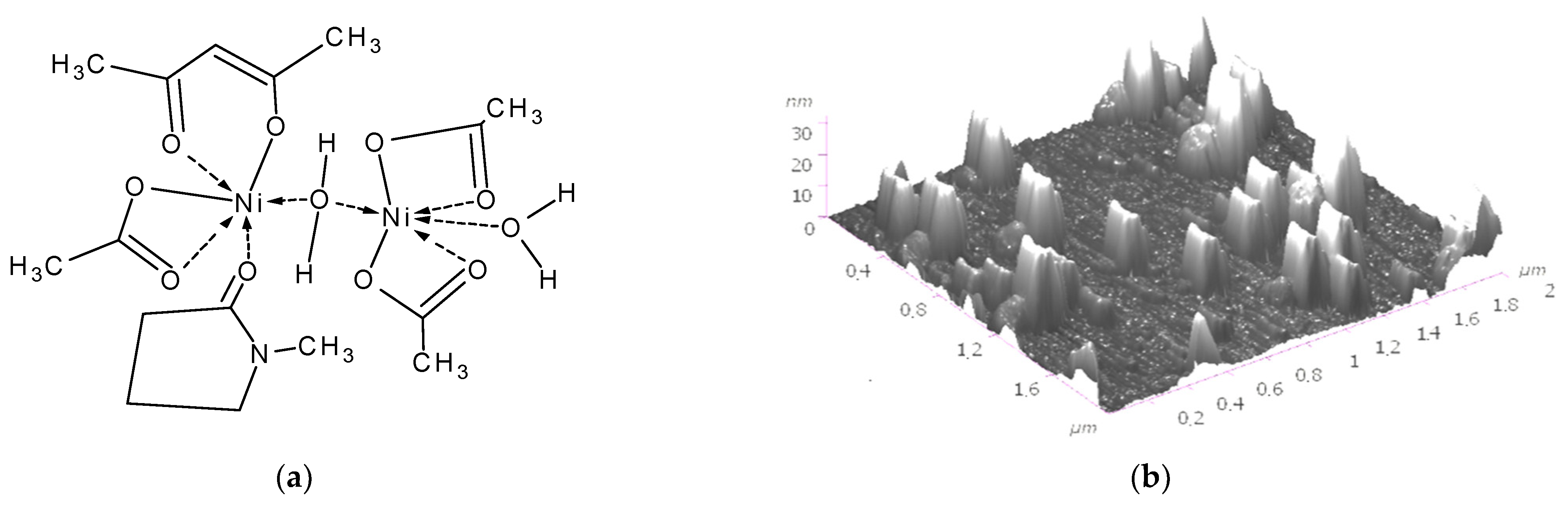
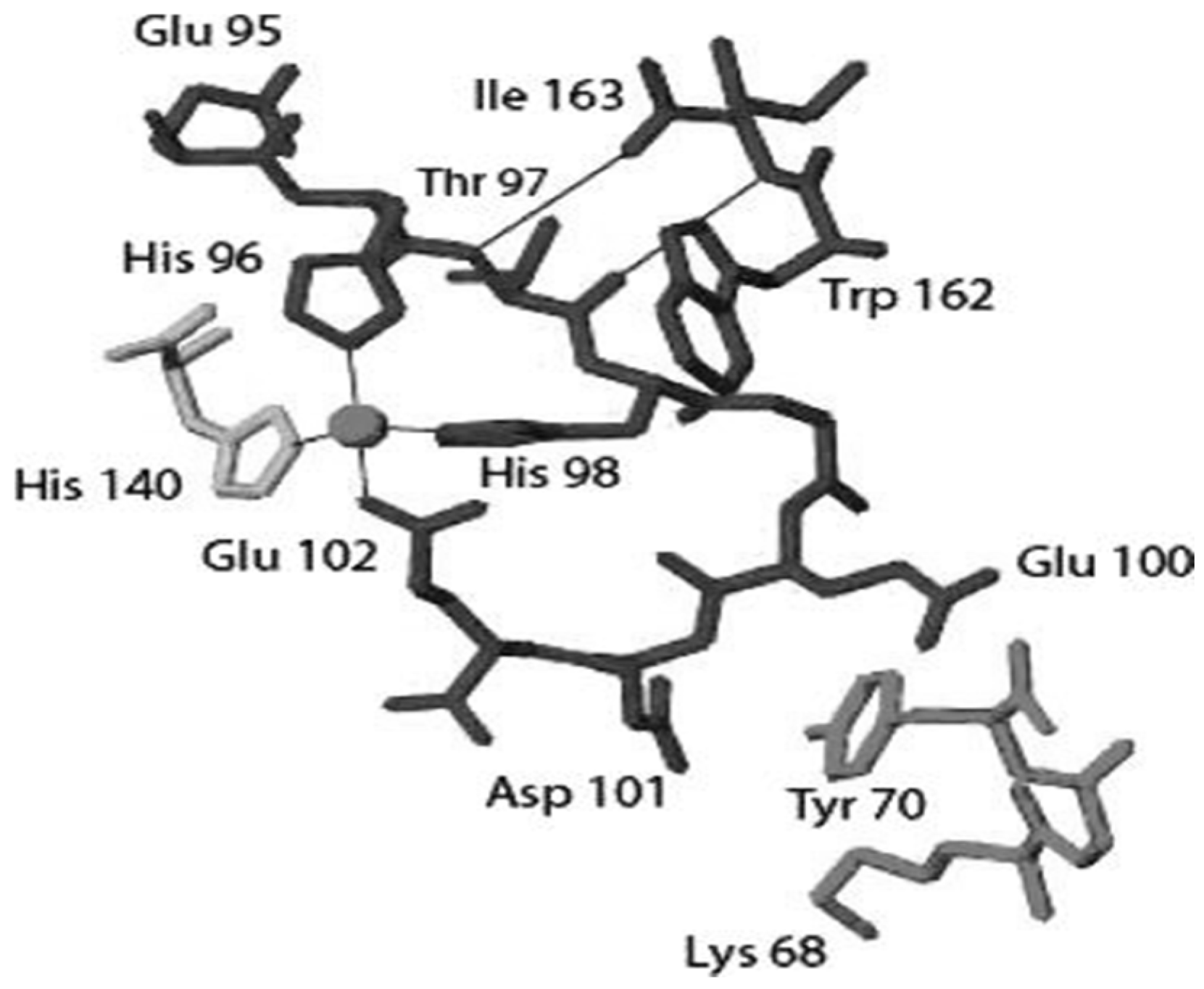
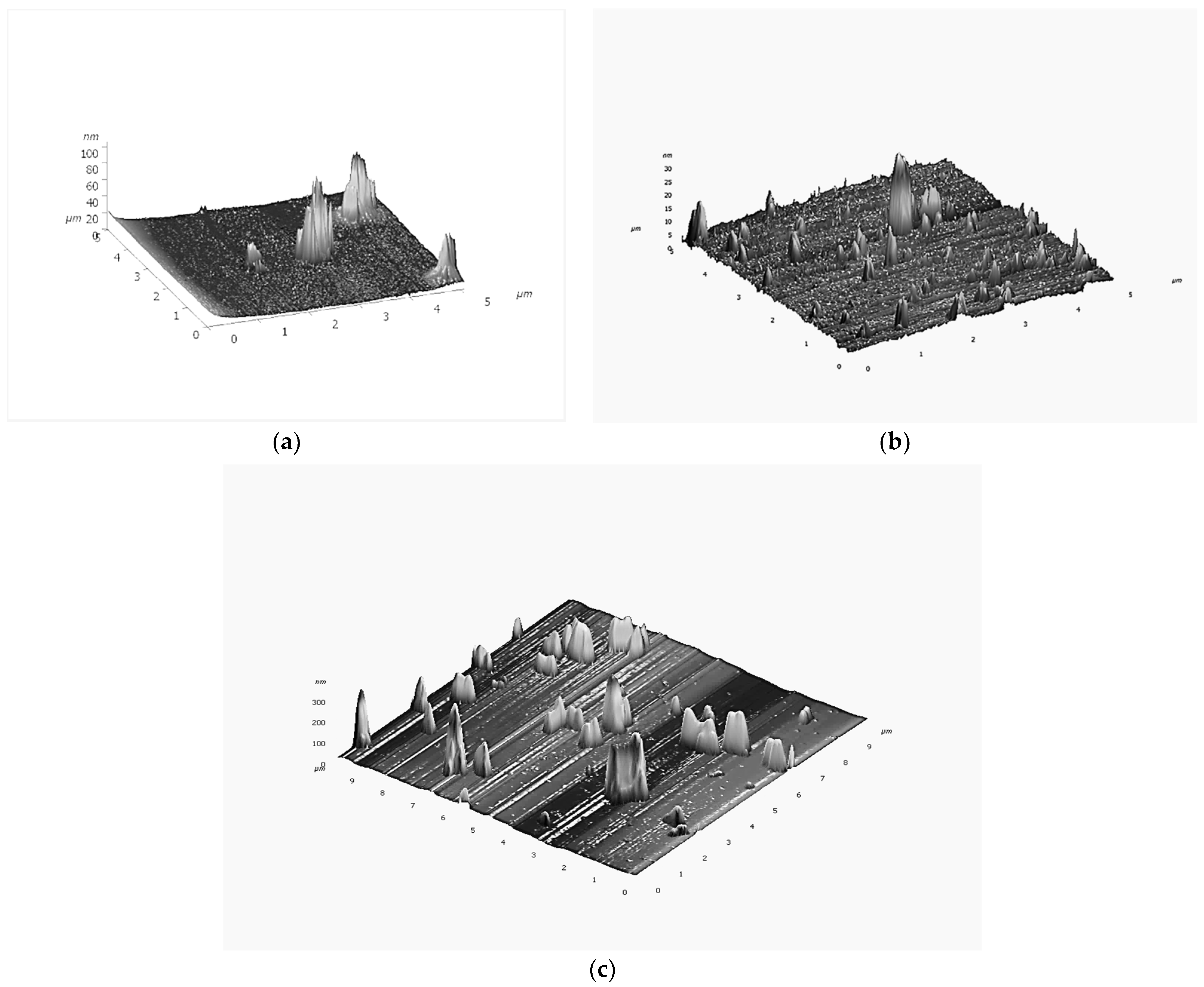


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matienko, L.I.; Mil, E.M.; Albantova, A.A.; Goloshchapov, A.N. The Role H-Bonding and Supramolecular Structures in Homogeneous and Enzymatic Catalysis. Int. J. Mol. Sci. 2023, 24, 16874. https://doi.org/10.3390/ijms242316874
Matienko LI, Mil EM, Albantova AA, Goloshchapov AN. The Role H-Bonding and Supramolecular Structures in Homogeneous and Enzymatic Catalysis. International Journal of Molecular Sciences. 2023; 24(23):16874. https://doi.org/10.3390/ijms242316874
Chicago/Turabian StyleMatienko, Ludmila I., Elena M. Mil, Anastasia A. Albantova, and Alexander N. Goloshchapov. 2023. "The Role H-Bonding and Supramolecular Structures in Homogeneous and Enzymatic Catalysis" International Journal of Molecular Sciences 24, no. 23: 16874. https://doi.org/10.3390/ijms242316874
APA StyleMatienko, L. I., Mil, E. M., Albantova, A. A., & Goloshchapov, A. N. (2023). The Role H-Bonding and Supramolecular Structures in Homogeneous and Enzymatic Catalysis. International Journal of Molecular Sciences, 24(23), 16874. https://doi.org/10.3390/ijms242316874