
Ubiquitination of Major Histocompatibility Complex II Changes Its Immunological Recognition Structure



Abstract
:1. Introduction
2. Results
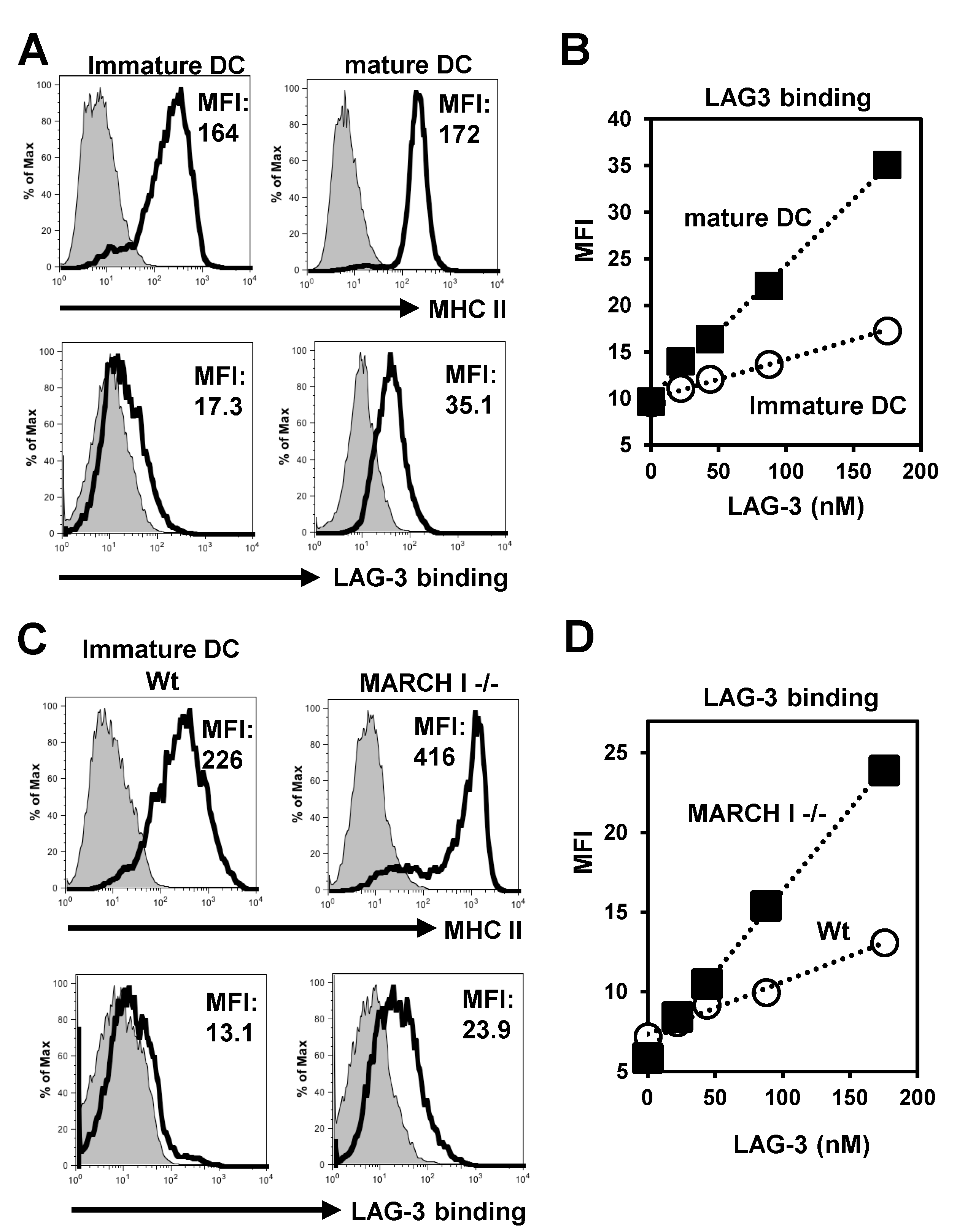
2.1. LAG-3 Preferentially Binds to Non-Ubiquitinated MHC II
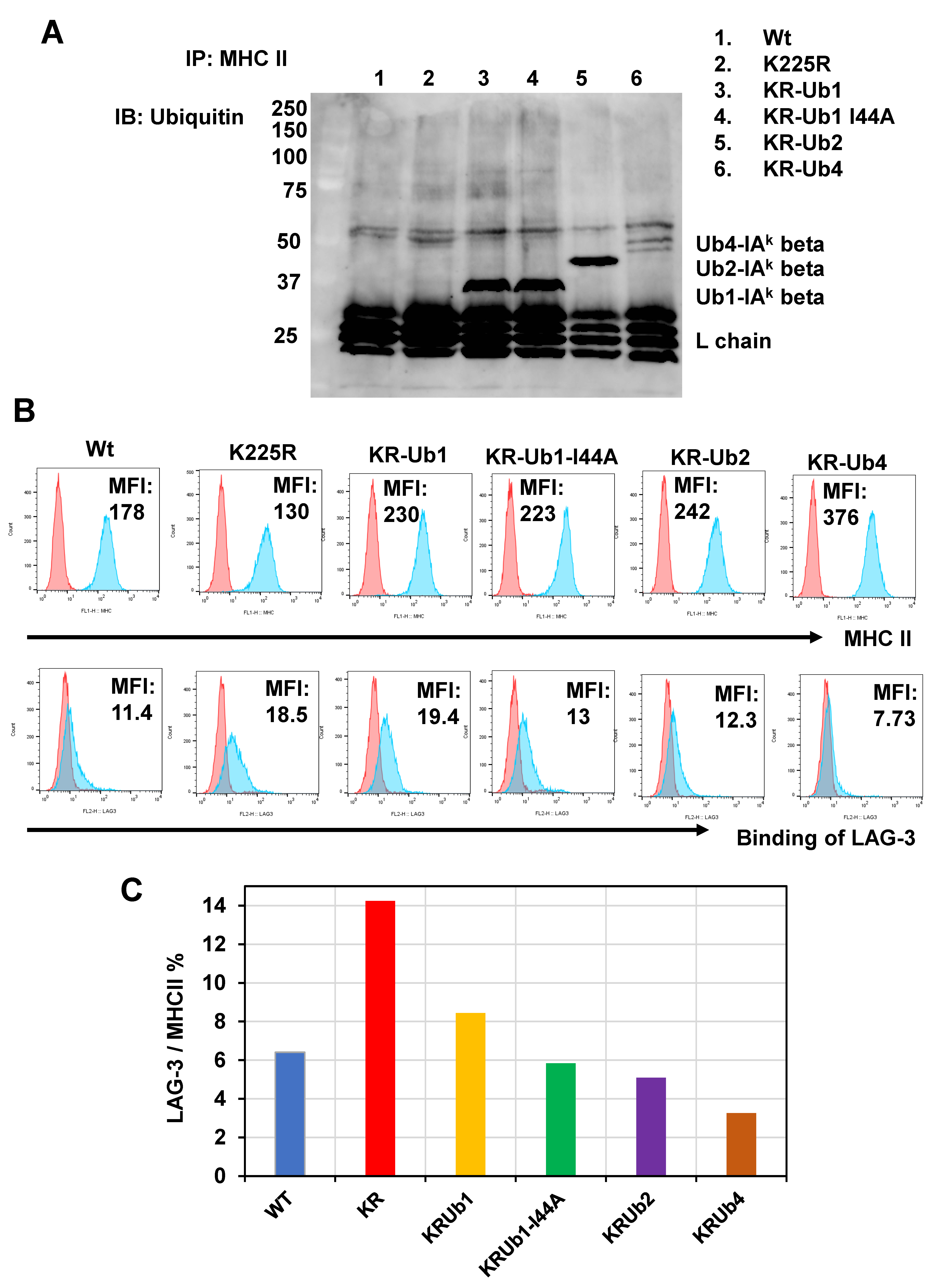
2.2. Ubiquitin Inhibits LAG-3 Binding to MHC II Conjugated Ubiquitin Number-Dependently
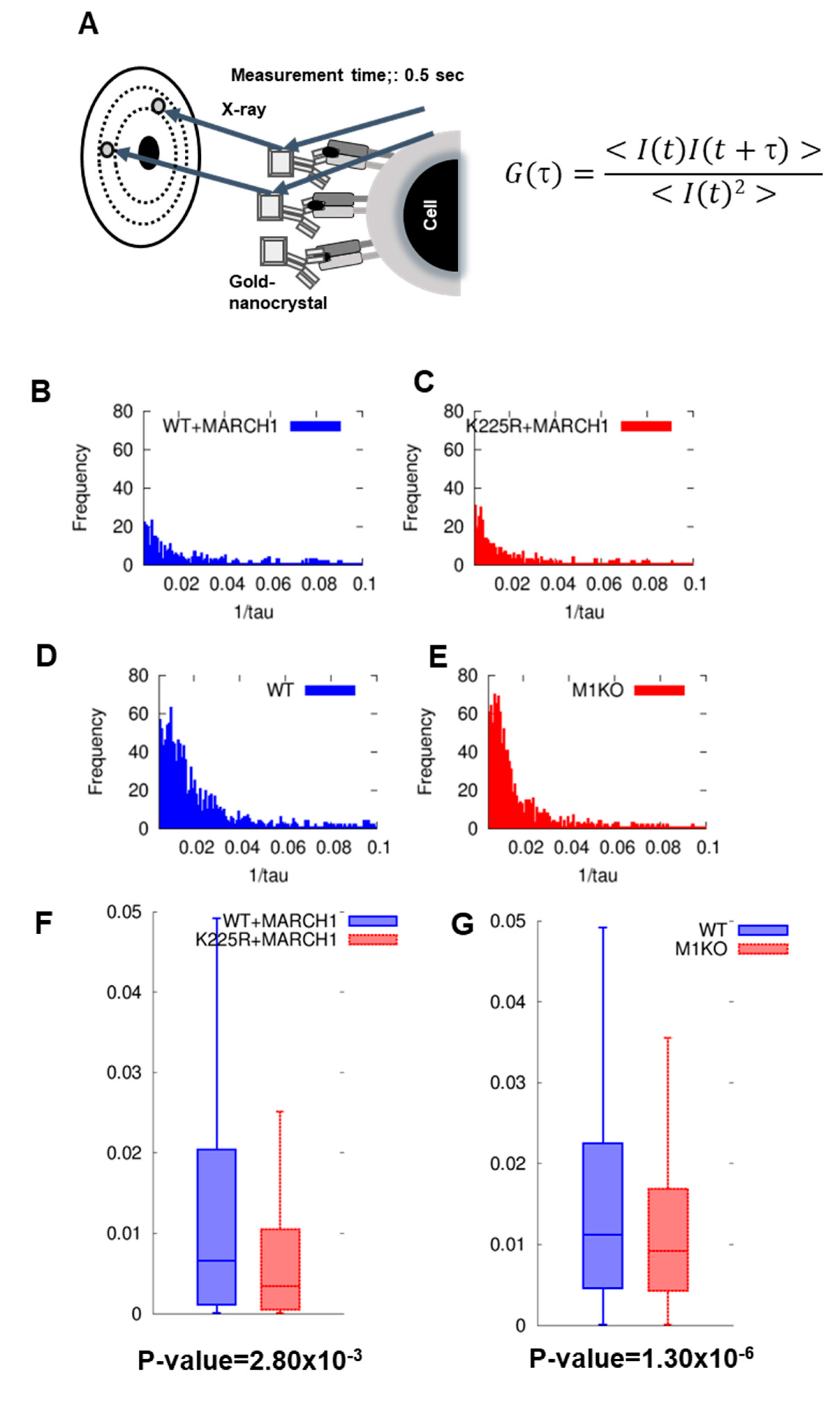
2.3. DXB Reveals Increased Dynamics of Ubiquitinated MHC II
3. Discussion
4. Materials and Methods
4.1. Mice and Cells
4.2. Reagents
4.3. Flow Cytometry
4.4. Generation of MHC II- and MARCHI-Expressing Cells
4.5. LAG-3 Tetramers
4.6. Diffracted X-ray Blinking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [PubMed]
- Kambayashi, T.; Laufer, T.M. Atypical MHC class II-expressing antigen-presenting cells: Can anything replace a dendritic cell? Nat. Rev. Immunol. 2014, 14, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.S.; Lee, H.M.; Lio, C.W. Selection of regulatory T cells in the thymus. Nat. Rev. Immunol. 2012, 12, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Ishido, S.; Goto, E.; Matsuki, Y.; Ohmura-Hoshino, M. E3 ubiquitin ligases for MHC molecules. Curr. Opin. Immunol. 2009, 21, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, Y.; Ohmura-Hoshino, M.; Goto, E.; Aoki, M.; Mito-Yoshida, M.; Uematsu, M.; Hasegawa, T.; Koseki, H.; Ohara, O.; Nakayama, M.; et al. Novel regulation of MHC class II function in B cells. EMBO J. 2007, 26, 846–854. [Google Scholar] [CrossRef] [PubMed]
- De Gassart, A.; Camosseto, V.; Thibodeau, J.; Ceppi, M.; Catalan, N.; Pierre, P.; Gatti, E. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 3491–3496. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.S.; Ebersold, M.; Pypaert, M.; Delamarre, L.; Hartley, A.; Mellman, I. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature 2006, 444, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Wubbolts, R.; Ten Broeke, T.; Buschow, S.I.; Ossendorp, F.A.; Melief, C.J.; Raposo, G.; van Balkom, B.W.; Stoorvogel, W. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity 2006, 25, 885–894. [Google Scholar] [CrossRef]
- Walseng, E.; Furuta, K.; Bosch, B.; Weih, K.A.; Matsuki, Y.; Bakke, O.; Ishido, S.; Roche, P.A. Ubiquitination regulates MHC class II-peptide complex retention and degradation in dendritic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 20465–20470. [Google Scholar] [CrossRef]
- Chow, A.; Toomre, D.; Garrett, W.; Mellman, I. Dendritic cell maturation triggers retrograde MHC class II transport from lysosomes to the plasma membrane. Nature 2002, 418, 988–994. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Workman, C.J.; Dugger, K.J.; Vignali, D.A. Cutting edge: Molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. 2002, 169, 5392–5395. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Workman, C.; Lee, J.; Chew, C.; Dale, B.M.; Colonna, L.; Flores, M.; Li, N.; Schweighoffer, E.; Greenberg, S.; et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class, II. J. Immunol. 2008, 180, 5916–5926. [Google Scholar] [CrossRef] [PubMed]
- Bauche, D.; Joyce-Shaikh, B.; Jain, R.; Grein, J.; Ku, K.S.; Blumenschein, W.M.; Ganal-Vonarburg, S.C.; Wilson, D.C.; McClanahan, T.K.; Malefyt, R.W.; et al. LAG3(+) Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1(+) Gut-Resident Macrophages during Group 3 Innate Lymphoid Cell-Driven Colitis. Immunity 2018, 49, 342–352.e345. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Okazaki, I.M.; Sugiura, D.; Takahashi, S.; Maeda, T.K.; Shimizu, K.; Okazaki, T. LAG-3 inhibits the activation of CD4(+) T cells that recognize stable pMHCII through its conformation-dependent recognition of, pMHCII. Nat. Immunol. 2018, 19, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Kozono, H.; Matsushita, Y.; Ogawa, N.; Kozono, Y.; Miyabe, T.; Sekiguchi, H.; Ichiyanagi, K.; Okimoto, N.; Taiji, M.; Kanagawa, O.; et al. Single-molecule motions of MHC class II rely on bound peptides. Biophys. J. 2015, 108, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, H.; Kuramochi, M.; Ikezaki, K.; Okamura, Y.; Yoshimura, K.; Matsubara, K.; Chang, J.W.; Ohta, N.; Kubo, T.; Mio, K.; et al. Diffracted X-ray Blinking Tracks Single Protein Motions. Sci. Rep. 2018, 8, 17090. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.K.; Platt, M.Y.; Eastham-Anderson, J.; Shin, J.S.; Mellman, I. MHC class II distribution in dendritic cells and B cells is determined by ubiquitin chain length. Proc. Natl. Acad. Sci. USA 2012, 109, 8820–8827. [Google Scholar] [CrossRef]
- Jahnke, M.; Trowsdale, J.; Kelly, A.P. Ubiquitination of human leukocyte antigen (HLA)-DM by different membrane-associated RING-CH (MARCH) protein family E3 ligases targets different endocytic pathways. J. Biol. Chem. 2012, 287, 7256–7264. [Google Scholar] [CrossRef]
- Jahnke, M.; Trowsdale, J.; Kelly, A.P. Ubiquitination of HLA-DO by MARCH family E3 ligases. Eur. J. Immunol. 2013, 43, 1153–1161. [Google Scholar] [CrossRef]
- Marks, M.S.; Roche, P.A.; van Donselaar, E.; Woodruff, L.; Peters, P.J.; Bonifacino, J.S. A lysosomal targeting signal in the cytoplasmic tail of the beta chain directs HLA-DM to MHC class II compartments. J. Cell Biol. 1995, 131, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Liljedahl, M.; Kuwana, T.; Fung-Leung, W.P.; Jackson, M.R.; Peterson, P.A.; Karlsson, L. HLA-DO is a lysosomal resident which requires association with HLA-DM for efficient intracellular transport. EMBO J. 1996, 15, 4817–4824. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Tu, L.; Miller, A.S.; Bock, C.; Fujimoto, M.; Doyle, C.; Steeber, D.A.; Tedder, T.F. CD83 expression influences CD4+ T cell development in the thymus. Cell 2002, 108, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Tze, L.E.; Horikawa, K.; Domaschenz, H.; Howard, D.R.; Roots, C.M.; Rigby, R.J.; Way, D.A.; Ohmura-Hoshino, M.; Ishido, S.; Andoniou, C.E.; et al. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J. Exp. Med. 2011, 208, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Von Rohrscheidt, J.; Petrozziello, E.; Nedjic, J.; Federle, C.; Krzyzak, L.; Ploegh, H.L.; Ishido, S.; Steinkasserer, A.; Klein, L. Thymic CD4 T cell selection requires attenuation of March8-mediated MHCII turnover in cortical epithelial cells through CD83. J. Exp. Med. 2016, 213, 1685–1694. [Google Scholar] [CrossRef]
- Chang, J.; Arai, T.; Kuramochi, M.; Inamasu, R.; Lee, Z.; Ohkubo, T.; Mio, K.; Sasaki, Y.C. Dynamic observations of various oligomers in amyloid β isoforms using laboratory diffracted X-ray blinking. Biochem. Biophys. Rep. 2022, 31, 101298. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Ghosh, M.; Pal, T.K.; Chakrabarti, S.; Roy, S. Cholesterol lowering drug may influence cellular immune response by altering MHC II function. J. Lipid Res. 2013, 54, 3106–3115. [Google Scholar] [CrossRef]
- Roy, K.; Mandloi, S.; Chakrabarti, S.; Roy, S. Cholesterol Corrects Altered Conformation of MHC-II Protein in Leishmania donovani Infected Macrophages: Implication in Therapy. PLoS Neglected Trop. Dis. 2016, 10, e0004710. [Google Scholar] [CrossRef]
- Glimcher, L.H.; McKean, D.J.; Choi, E.; Seidman, J.G. Complex regulation of class II gene expression: Analysis with class II mutant cell lines. J. Immunol. 1985, 135, 3542–3550. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT + MARCH I | K225R + MARCH I | WT DC | M1KO DC | |
|---|---|---|---|---|
| Attenuation constant (s−1) | 6.75 × 10−3 | 3.25 × 10−3 | 12.15 × 10−3 | 9.65 × 10−3 |
| Verocity of dffraction spots (μm s−1) | 0.73 | 0.35 | 1.2 | 1.04 |
| Angular verosity (mrad s−1) | 1.4 | 0.67 | 2.3 | 2 |
| Distance (Å s−1) | 0.38 | 0.182 | 0.62 | 0.54 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozono, Y.; Kuramochi, M.; Sasaki, Y.C.; Kozono, H. Ubiquitination of Major Histocompatibility Complex II Changes Its Immunological Recognition Structure. Int. J. Mol. Sci. 2023, 24, 17083. https://doi.org/10.3390/ijms242317083
Kozono Y, Kuramochi M, Sasaki YC, Kozono H. Ubiquitination of Major Histocompatibility Complex II Changes Its Immunological Recognition Structure. International Journal of Molecular Sciences. 2023; 24(23):17083. https://doi.org/10.3390/ijms242317083
Chicago/Turabian StyleKozono, Yuko, Masahiro Kuramochi, Yuji C. Sasaki, and Haruo Kozono. 2023. "Ubiquitination of Major Histocompatibility Complex II Changes Its Immunological Recognition Structure" International Journal of Molecular Sciences 24, no. 23: 17083. https://doi.org/10.3390/ijms242317083
APA StyleKozono, Y., Kuramochi, M., Sasaki, Y. C., & Kozono, H. (2023). Ubiquitination of Major Histocompatibility Complex II Changes Its Immunological Recognition Structure. International Journal of Molecular Sciences, 24(23), 17083. https://doi.org/10.3390/ijms242317083