

Heart Failure Promotes Cancer Progression in an Integrin β1-Dependent Manner

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
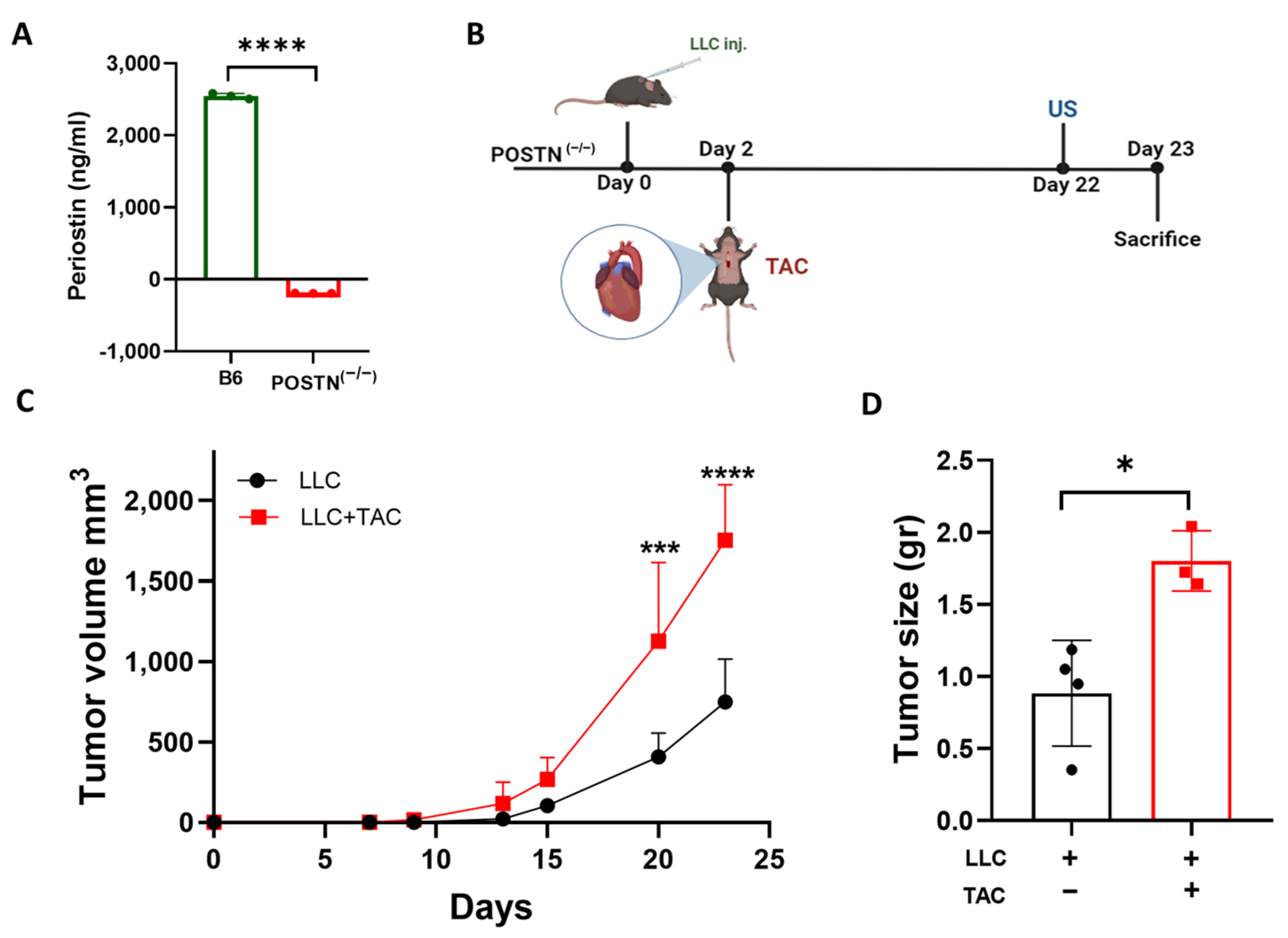
2.1. Cardiac Remodeling following TAC Promotes Tumor Growth in the Periostin Knockout Mice Strain
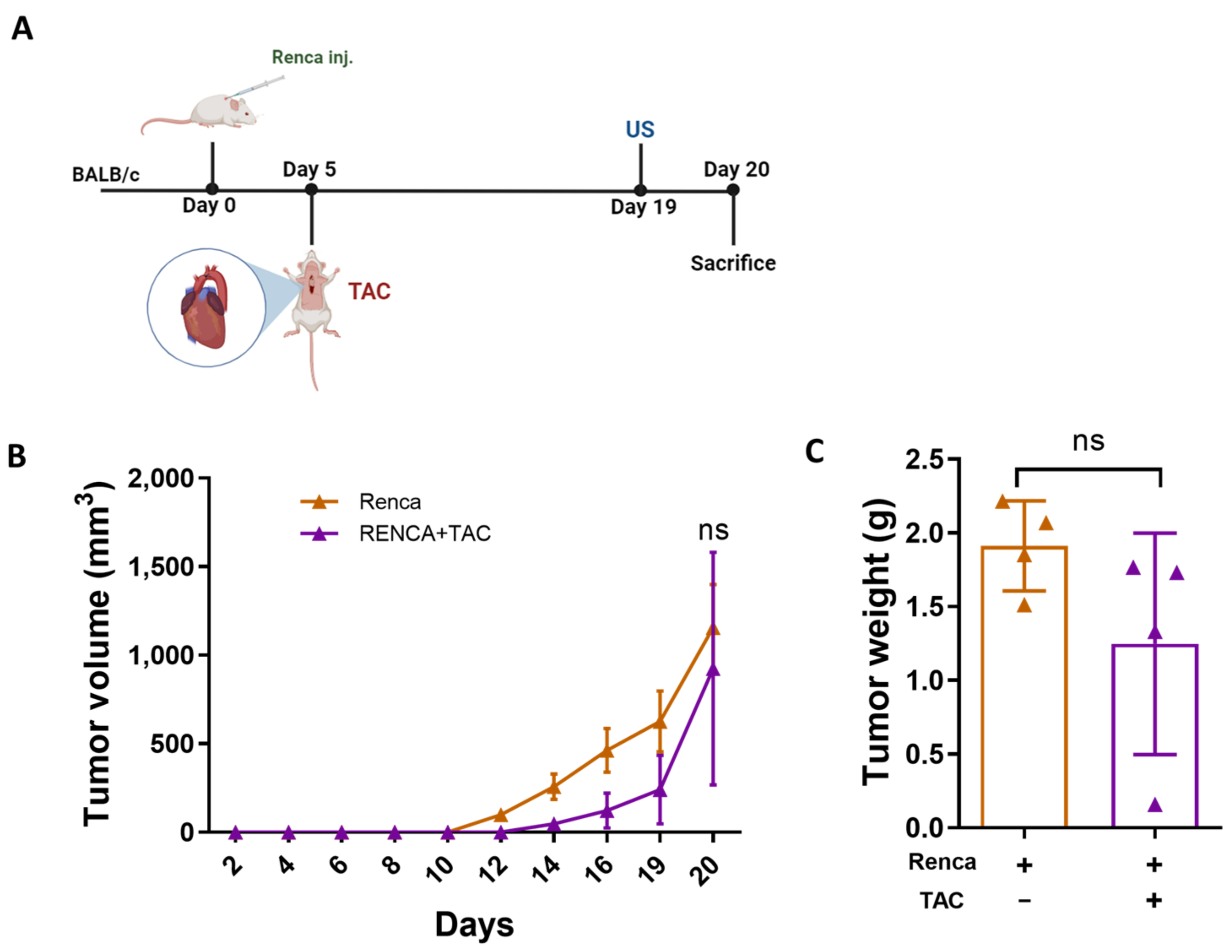
2.2. Tumor Promotion Following Cardiac Remodeling in the POSTN(−/−) Mice Is Independent of Periostin Expression
2.3. Integrin β1 Is Required for Mediating the Connection between the Remodeled Heart and Cancer Cell Promotion
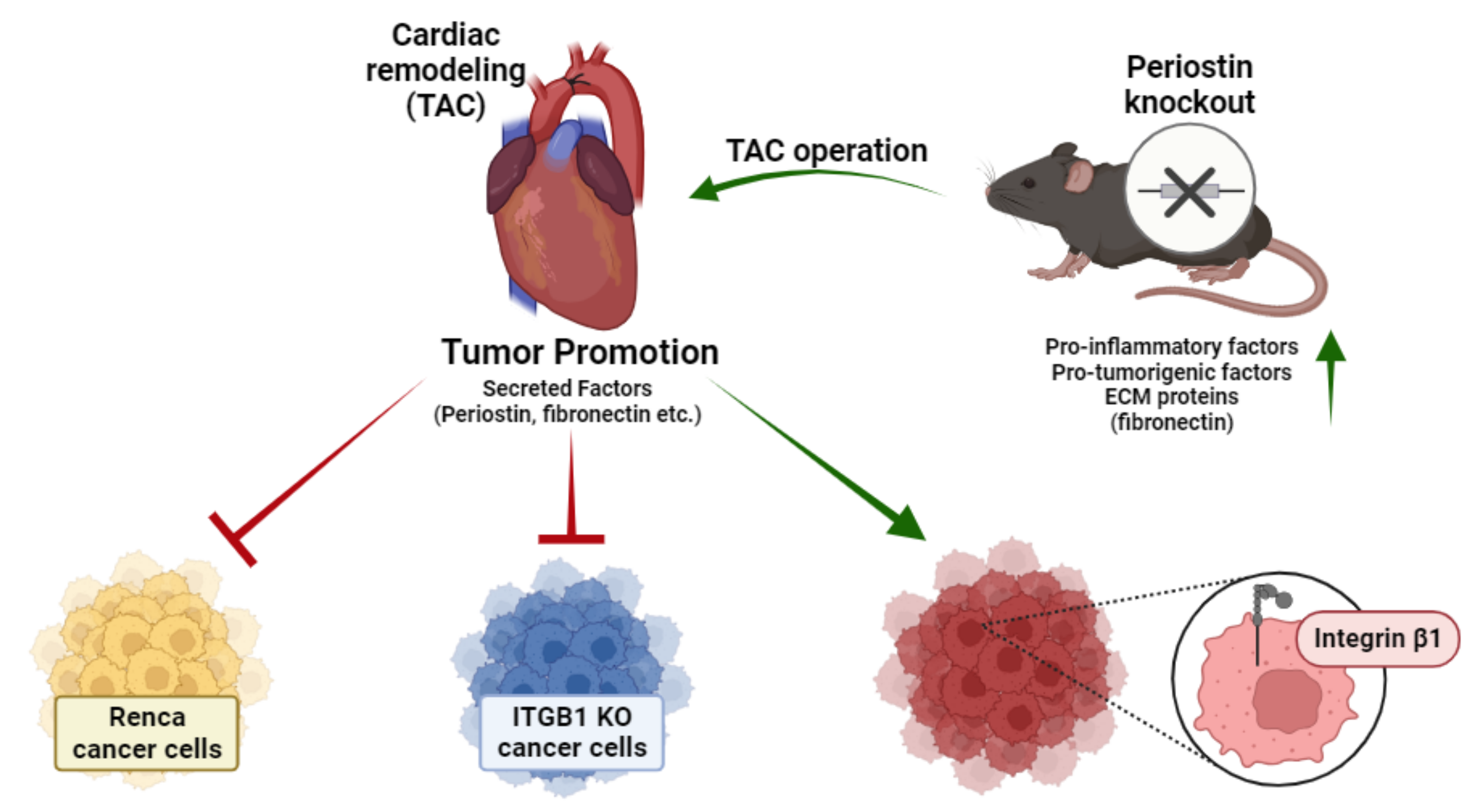
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of ITGB1-KO Cells
4.3. Cell Culture and Transfections
4.4. Antibodies
4.5. Cell Proliferation Assay
4.6. Animals
4.7. Cancer Cells Implantation
4.8. Transverse Aortic Constriction
4.9. Echocardiography
4.10. RNA Extraction
4.11. Quantitative Real-Time PCR
4.12. Blood Serum
4.13. ELISA
4.14. Cytokine Array
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ganau, A.; Devereux, R.B.; Roman, M.J.; de Simone, G.; Pickering, T.G.; Saba, P.S.; Vargiu, P.; Simongini, I.; Laragh, J.H. Patterns of left ventricular hypertrophy and geometric remodeling in essential hypertension. J. Am. Coll. Cardiol. 1992, 19, 1550–1558. [Google Scholar] [CrossRef]
- Maulik, S.K.; Kumar, S. Oxidative stress and cardiac hypertrophy: A review. Toxicol. Mech. Methods 2012, 22, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.F.; Dwyer, T.M.; Grady, A.W.; Reinhart, G.A.; Montani, J.P.; Cockrell, K.; Meydrech, E.F.; Mizelle, H.L. Hypertension, cardiac hypertrophy, and neurohumoral activity in a new animal model of obesity. Am. J. Physiol. Heart Circ. Physiol. 1996, 271, H373–H378. [Google Scholar] [CrossRef]
- Bisping, E.; Wakula, P.; Poteser, M.; Heinzel, F.R. Targeting cardiac hypertrophy: Toward a causal heart failure therapy. J. Cardiovasc. Pharmacol. 2014, 64, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared Risk Factors in Cardiovascular Disease and Cancer. Circulation 2016, 133, 1104–1114. [Google Scholar] [CrossRef]
- Hoshijima, M.; Chien, K.R. Mixed signals in heart failure: Cancer rules. J. Clin. Investig. 2002, 109, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Tocchetti, C.G.; Ameri, P.; A De Boer, R.; D’alessandra, Y.; Russo, M.; Sorriento, D.; Ciccarelli, M.; Kiss, B.; Bertrand, L.; Dawson, D.; et al. Cardiac dysfunction in cancer patients: Beyond direct cardiomyocyte damage of anticancer drugs: Novel cardio-oncology insights from the joint 2019 meeting of the ESC Working Groups of Myocardial Function and Cellular Biology of the Heart. Cardiovasc. Res. 2020, 116, 1820–1834. [Google Scholar] [CrossRef]
- de Boer, R.A.; Hulot, J.-S.; Tocchetti, C.G.; Aboumsallem, J.P.; Ameri, P.; Anker, S.D.; Bauersachs, J.; Bertero, E.; Coats, A.J.S.; Čelutkienė, J.; et al. Common mechanistic pathways in cancer and heart failure. A scientific roadmap on behalf of the Translational Research Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2020, 22, 2272–2289. [Google Scholar] [CrossRef]
- Bertero, E.; Canepa, M.; Maack, C.; Ameri, P. Linking heart failure to cancer: Background evidence and research perspectives. Circulation 2018, 138, 735–742. [Google Scholar] [CrossRef]
- Cuomo, A.; Rodolico, A.; Galdieri, A.; Russo, M.; Campi, G.; Franco, R.; Bruno, D.; Aran, L.; Carannante, A.; Attanasio, U.; et al. Heart Failure and Cancer: Mechanisms of Old and New Cardiotoxic Drugs in Cancer Patients. Card. Fail. Rev. 2019, 5, 112–118. [Google Scholar] [CrossRef]
- Meijers, W.C.; Maglione, M.; Bakker, S.J.L.; Oberhuber, R.; Kieneker, L.M.; de Jong, S.; Haubner, B.J.; Nagengast, W.B.; Lyon, A.R.; van der Vegt, B.; et al. Heart Failure Stimulates Tumor Growth by Circulating Factors. Circulation 2018, 138, 678–691. [Google Scholar] [CrossRef]
- Koelwyn, G.J.; Newman, A.A.C.; Afonso, M.S.; van Solingen, C.; Corr, E.M.; Brown, E.J.; Albers, K.B.; Yamaguchi, N.; Narke, D.; Schlegel, M.; et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat. Med. 2020, 26, 1452–1458. [Google Scholar] [CrossRef]
- Avraham, S.; Abu-Sharki, S.; Shofti, R.; Haas, T.; Korin, B.; Kalfon, R.; Friedman, T.; Shiran, A.; Saliba, W.; Shaked, Y.; et al. Early Cardiac Remodeling Promotes Tumor Growth and Metastasis. Circulation 2020, 142, 670–683. [Google Scholar] [CrossRef]
- Hasin, T.; Gerber, Y.; Weston, S.A.; Jiang, R.; Killian, J.M.; Manemann, S.M.; Cerhan, J.R.; Roger, V.L. Heart Failure After Myocardial Infarction Is Associated With Increased Risk of Cancer. J. Am. Coll. Cardiol. 2016, 68, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Koelwyn, G.J.; Aboumsallem, J.P.; Moore, K.J.; de Boer, R.A. Reverse cardio-oncology: Exploring the effects of cardiovascular disease on cancer pathogenesis. J. Mol. Cell. Cardiol. 2021, 163, 1–8. [Google Scholar] [CrossRef]
- Fountoulaki, K.; Dagres, N.; Iliodromitis, E.K. Cellular communications in the heart. Card. Fail. Rev. 2015, 1, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Tai, I.T.; Dai, M.; Chen, L.B. Periostin induction in tumor cell line explants and inhibition of in vitro cell growth by anti-periostin antibodies. Carcinogen 2005, 26, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Tamai, K.; Shibuya, R.; Nakamura, M.; Mochizuki, M.; Yamaguchi, K.; Abe, J.; Takahashi, S.; Sato, I.; Kudo, A.; et al. Periostin is a negative prognostic factor and promotes cancer cell proliferation in non-small cell lung cancer. Oncotarget 2018, 9, 31187–31199. [Google Scholar] [CrossRef] [PubMed]
- Awwad, L.; Aronheim, A. Cardiac Dysfunction Promotes Cancer Progression via Multiple Secreted Factors. Cancer Res. 2022, 82, 1753–1761. [Google Scholar] [CrossRef]
- Awwad, L.; Goldenberg, T.; Langier-Goncalves, I.; Aronheim, A. Cardiac Remodeling in the Absence of Cardiac Contractile Dysfunction Is Sufficient to Promote Cancer Progression. Cells 2022, 11, 1108. [Google Scholar] [CrossRef]
- Kreidberg, J.A.; Symons, J.M. Integrins in kidney development, function, and disease. Am. J. Physiol. Ren. Physiol. 2000, 279, F233–F242. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1–42. [Google Scholar] [CrossRef]
- Utispan, K.; Sonongbua, J.; Thuwajit, P.; Chau-In, S.; Pairojkul, C.; Wongkham, S.; Thuwajit, C. Periostin activates integrin α5β1 through a PI3K/AKT-dependent pathway in invasion of cholangiocarcinoma. Int. J. Oncol. 2012, 41, 1110–1118. [Google Scholar] [CrossRef]
- Shi, C.; Aboumsallem, J.P.; de Wit, S.; Schouten, E.M.; Bracun, V.; Meijers, W.C.; Silljé, H.H.; de Boer, R.A. Evaluation of renal cancer progression in a mouse model of heart failure. Cancer Commun. 2021, 41, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, M.; Nakamura, K.; Kii, I.; Kashima, T.; Amizuka, N.; Li, M.; Saito, M.; Fukuda, K.; Nishiyama, T.; Kitajima, S.; et al. Periostin is essential for cardiac healing after acute myocardial infarction. J. Cell Biol. 2008, 205, 295–303. [Google Scholar] [CrossRef]
- Oka, T.; Xu, J.; Kaiser, R.A.; Melendez, J.; Hambleton, M.; Sargent, M.A.; Lorts, A.; Brunskill, E.W.; Dorn, G.W.; Conway, S.J.; et al. Genetic Manipulation of Periostin Expression Reveals a Role in Cardiac Hypertrophy and Ventricular Remodeling. Circ. Res. 2007, 101, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sivan, U.; Tan, S.L.; Lippo, L.; De Angelis, J.; Labella, R.; Singh, A.; Chatzis, A.; Cheuk, S.; Medhghalchi, M.; et al. High-resolution 3D imaging uncovers organ-specific vascular control of tissue aging. Sci. Adv. 2021, 7, eabd7819. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Wolford, C.C.; McConoughey, S.J.; Jalgaonkar, S.P.; Leon, M.; Merchant, A.S.; Dominick, J.L.; Yin, X.; Chang, Y.; Zmuda, E.J.; O’Toole, S.A.; et al. Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J. Clin. Investig. 2013, 123, 2893–2906. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Prinz, E.; Aviram, S.; Aronheim, A.; Sakaguchi, M.; Sonegawa, H.; Murata, H.; Kitazoe, M.; Futami, J.-I.; Kataoka, K.; Yamada, H.; et al. WDR62 mediates TNFα-dependent JNK activation via TRAF2-MLK3 axis. Mol. Biol. Cell 2018, 29, 2470–2480. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.C.; van Oort, R.J.; Wehrens, X.H. Transverse aortic constriction in mice. J. Vis. Exp. 2010, 38, e1729. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langier Goncalves, I.; Awwad, L.; Aviram, S.; Izraeli, T.; Achlaug, L.; Aronheim, A. Heart Failure Promotes Cancer Progression in an Integrin β1-Dependent Manner. Int. J. Mol. Sci. 2023, 24, 17367. https://doi.org/10.3390/ijms242417367
Langier Goncalves I, Awwad L, Aviram S, Izraeli T, Achlaug L, Aronheim A. Heart Failure Promotes Cancer Progression in an Integrin β1-Dependent Manner. International Journal of Molecular Sciences. 2023; 24(24):17367. https://doi.org/10.3390/ijms242417367
Chicago/Turabian StyleLangier Goncalves, Irina, Lama Awwad, Sharon Aviram, Talel Izraeli, Laris Achlaug, and Ami Aronheim. 2023. "Heart Failure Promotes Cancer Progression in an Integrin β1-Dependent Manner" International Journal of Molecular Sciences 24, no. 24: 17367. https://doi.org/10.3390/ijms242417367