
Combined Fluorescence Fluctuation and Spectrofluorometric Measurements Reveal a Red-Shifted, Near-IR Emissive Photo-Isomerized Form of Cyanine 5



Abstract
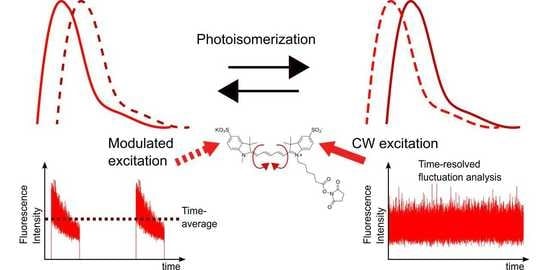
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
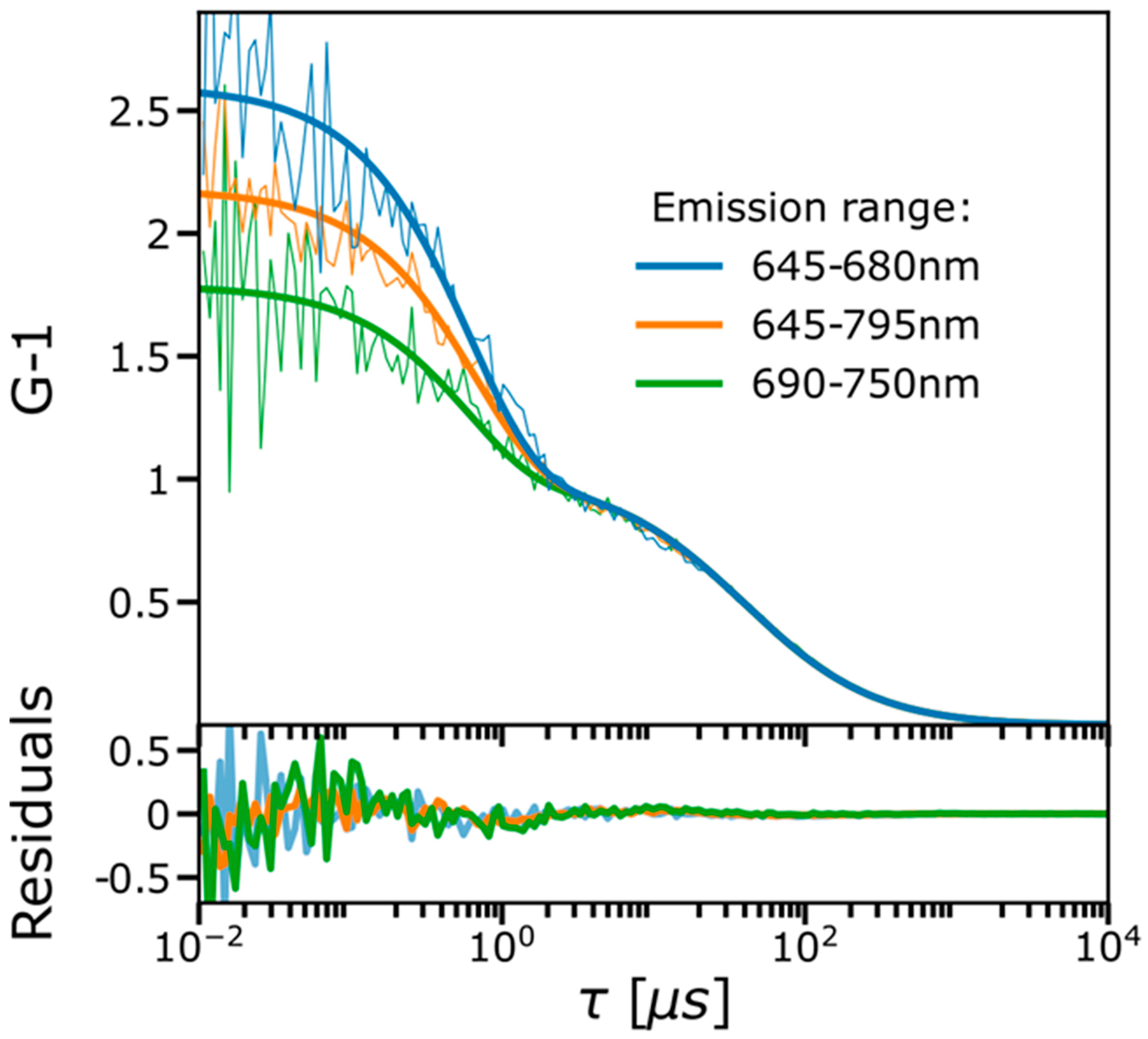
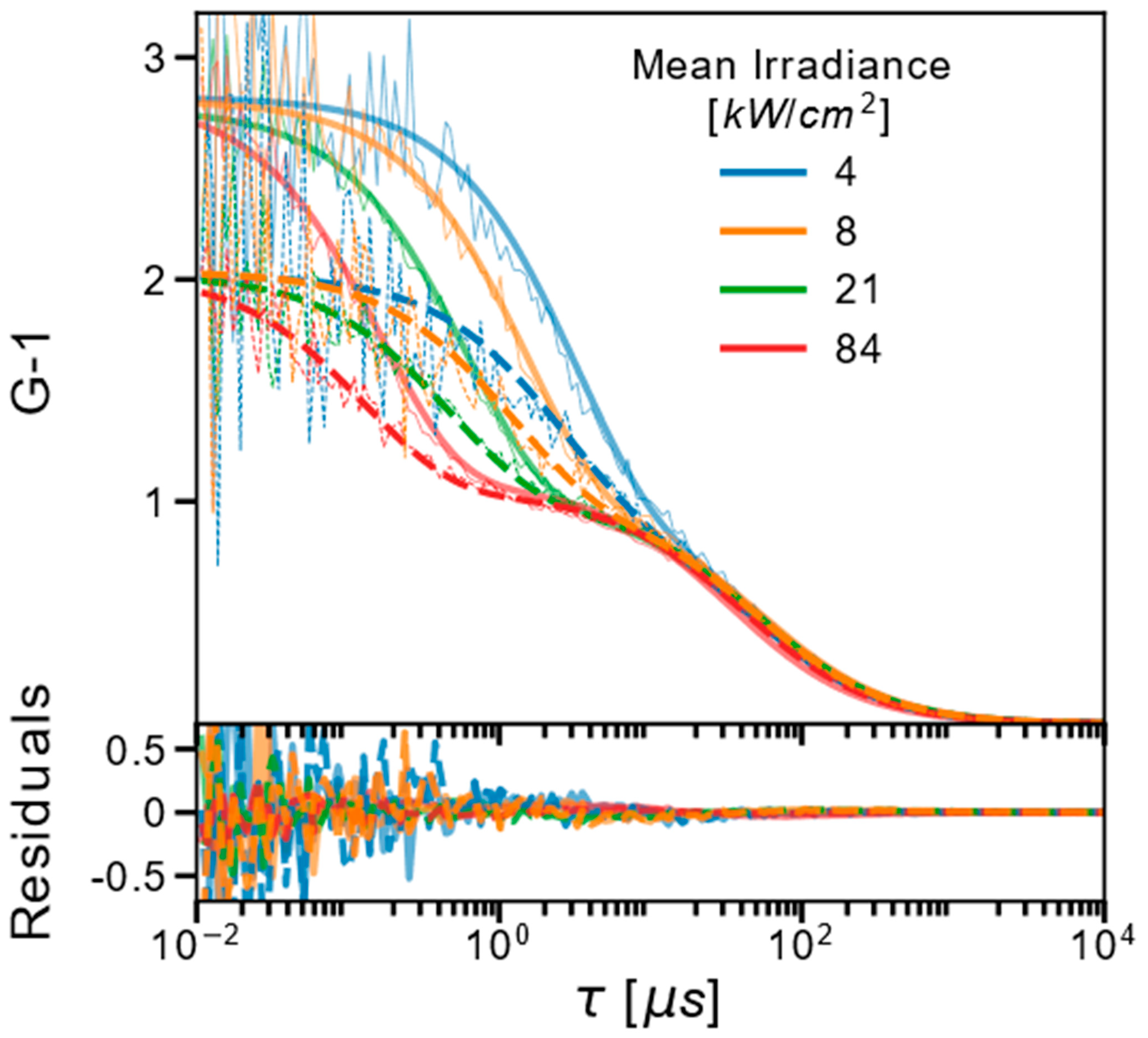
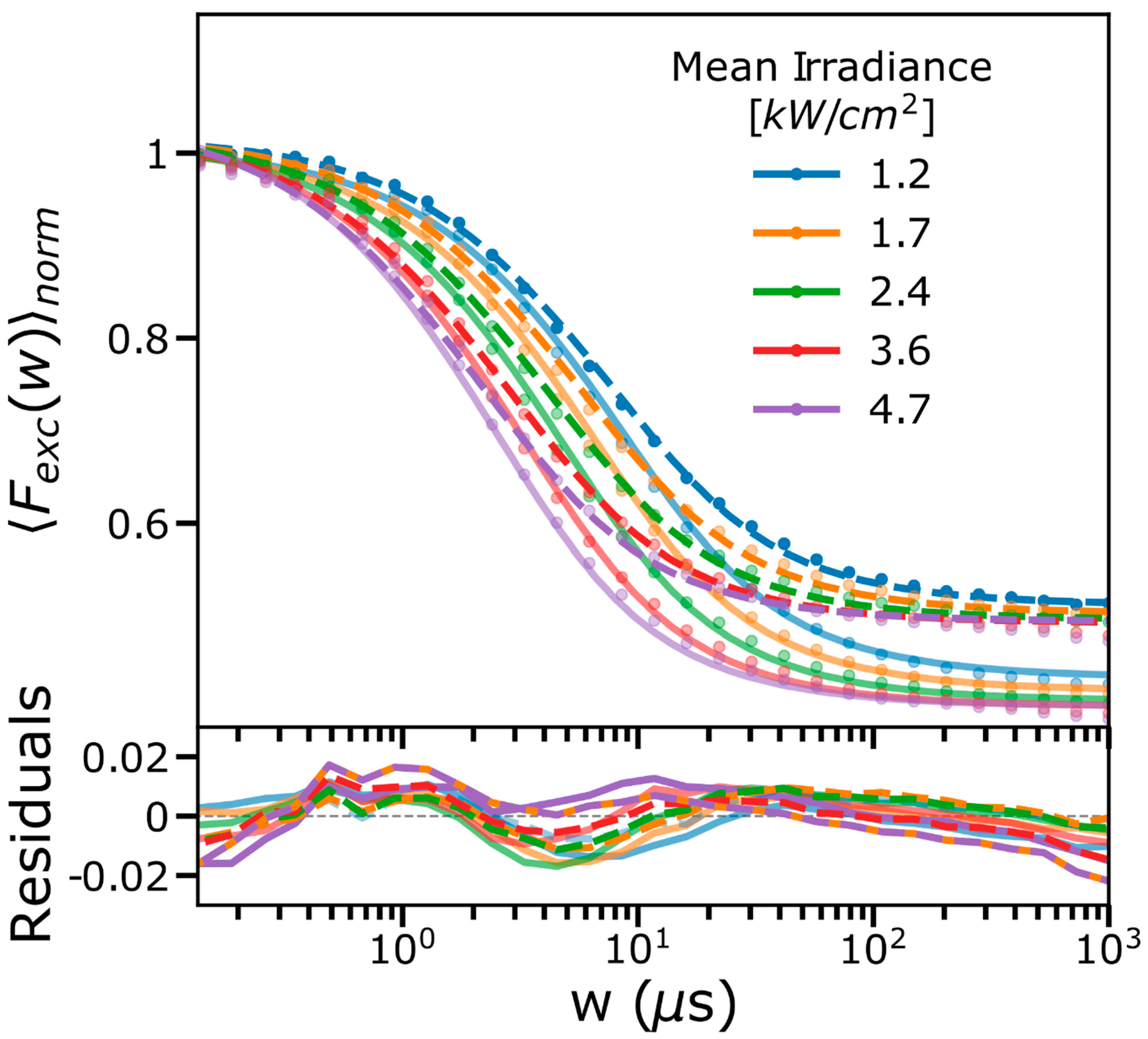
2.1. FCS and TRAST Experiments
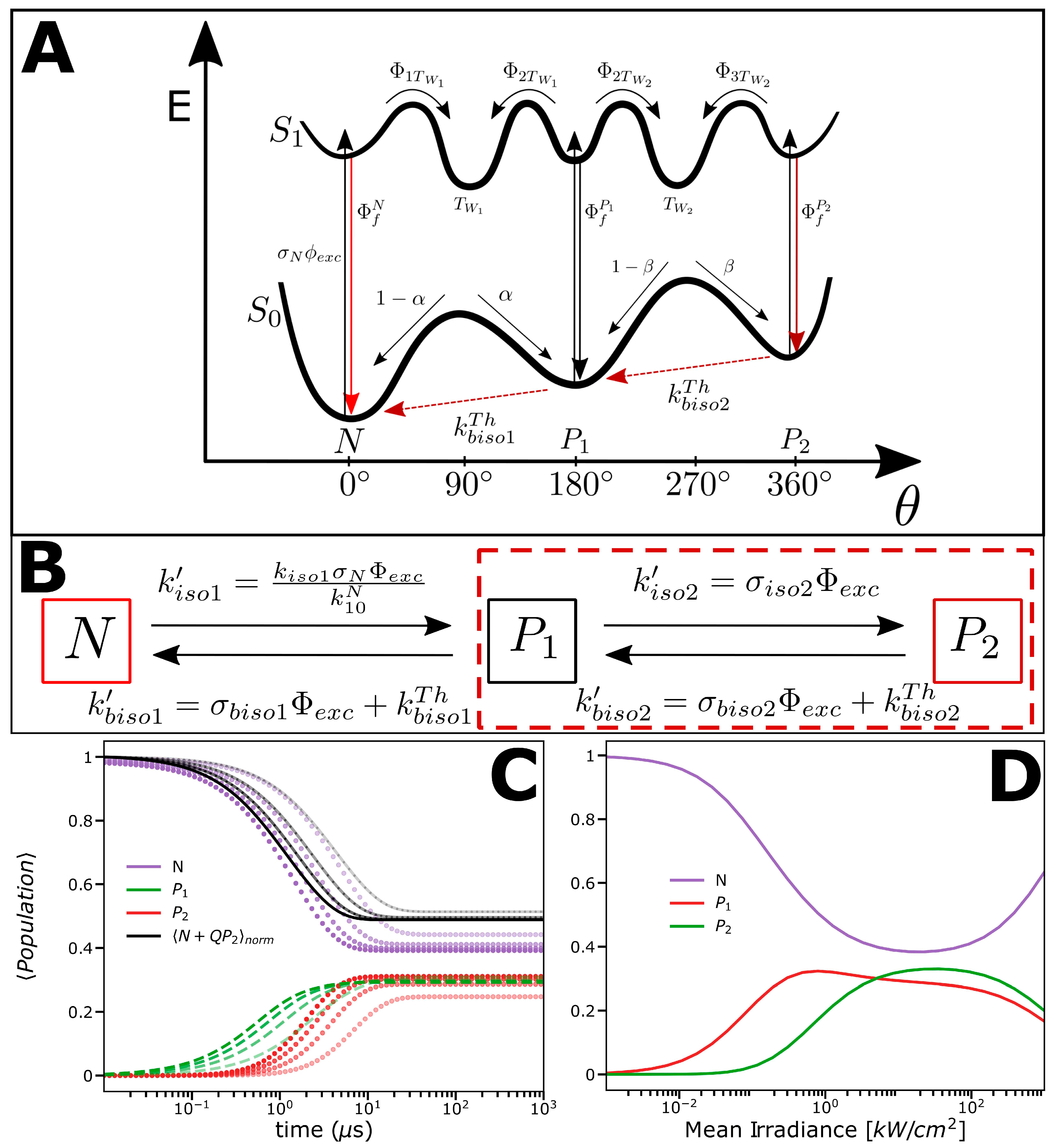
2.2. Photophysical Model Based on the FCS and TRAST Experiments
2.3. Experimental FCS and TRAST Data Fitted to the Photophysical Model
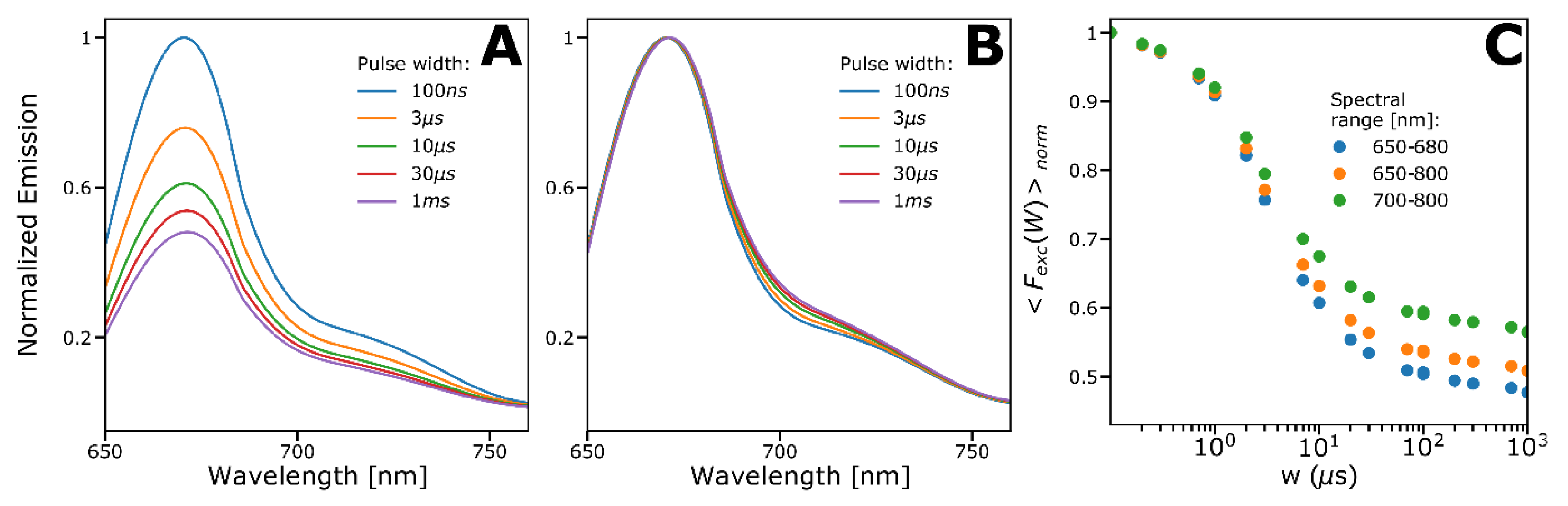
2.4. Spectral-TRAST Experiments
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. TRAST Experiments
4.3. TRAST Analysis
4.4. FCS Experiments
4.5. FCS Analysis
4.6. Fluorescence Lifetime Measurements
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mishra, A.; Behera, R.K.; Behera, P.K.; Mishra, B.K.; Behera, G.B. Cyanines during the 1990s: A review. Chem. Rev. 2000, 100, 1973–2011. [Google Scholar] [CrossRef] [PubMed]
- Kumpulainen, T.; Lang, B.; Rosspeintner, A.; Vauthey, E. Ultrafast Elementary Photochemical Processes of Organic Molecules in Liquid Solution. Chem. Rev. 2017, 117, 10826–10939. [Google Scholar] [CrossRef] [PubMed]
- Aramendia, P.F.; Negri, R.M.; Sanroman, E. Temperature-Dependence of Fluorescence and Photoisomerization in Symmetrical Carbocyanines—Influence of Medium Viscosity and Molecular-Structure. J. Phys. Chem. 1994, 98, 3165–3173. [Google Scholar] [CrossRef]
- Baraldi, I.; Carnevali, A.; Momicchioli, F.; Ponterini, G. Electronic-spectra and trans cis photoisomerism of carbocyanines—A theoretical (CS Indo CI) and experimental study. Spectrochim. Acta Part A-Mol. Biomol. Spectrosc. 1993, 49, 471–495. [Google Scholar] [CrossRef]
- Chibisov, A.K. Triplet-states of cyanine dyes and reactions of electron-transfer with their participation. J. Photochem. 1977, 6, 199–214. [Google Scholar] [CrossRef]
- Chibisov, A.K.; Shvedov, S.V.; Gorner, H. Photosensitized processes in dicarbocyanine dyes induced by energy transfer: Delayed fluorescence, trans -> cis isomerization and electron transfer. J. Photochem. Photobiol. A-Chem. 2001, 141, 39–45. [Google Scholar] [CrossRef]
- Chibisov, A.K.; Zakharova, G.V.; Gorner, H. Effects of substituents in the polymethine chain on the photoprocesses in indodicarbocyanine dyes. J. Chem. Soc. -Faraday Trans. 1996, 92, 4917–4925. [Google Scholar] [CrossRef]
- Chibisov, A.K.; Zakharova, G.V.; Gorner, H.; Sogulyaev, Y.A.; Mushkalo, I.L.; Tolmachev, A.I. Photorelaxation processes in covalently-linked indocarbocyanine and thiacarbocyanine dyes. J. Phys. Chem. 1995, 99, 886–893. [Google Scholar] [CrossRef]
- Huang, Z.X.; Ji, D.M.; Wang, S.F.; Xia, A.D.; Koberling, F.; Patting, M.; Erdmann, R. Spectral identification of specific photophysics of Cy5 by means of ensemble and single molecule measurements. J. Phys. Chem. A 2006, 110, 45–50. [Google Scholar] [CrossRef]
- Krieg, M.; Redmond, R.W. Photophysical properties of 3,3’-dialkylthiacarbocyanine dyes in homogenous solution. Photochem. Photobiol. 1993, 57, 472–479. [Google Scholar] [CrossRef]
- Ponterini, G.; Momicchioli, F. Trans cis photoisomerization mechanism of carbocyanines—Experimental check of theoretical models. Chem. Phys. 1991, 151, 111–126. [Google Scholar] [CrossRef]
- Bilmes, G.M.; Tocho, J.O.; Braslavsky, S.E. Laser-induced optoacoustic studies of the photoisomerization of the laser dye 3,3’-diethyloxadicarbocyanine iodide (DODCI). Chem. Phys. Lett. 1987, 134, 335–340. [Google Scholar] [CrossRef]
- Bilmes, G.M.; Tocho, J.O.; Braslavsky, S.E. Spectrum, energy content, and relaxation mechanism of the photoisomer of the laser-dye 3,3’-diethyloxadicarbocyanine iodide—Laser-induced optoacoustic studies. J. Phys. Chem. 1988, 92, 5958–5962. [Google Scholar] [CrossRef]
- Dipaolo, R.E.; Scaffardi, L.B.; Duchowicz, R.; Bilmes, G.M. Photoisomerization dynamics and spectroscopy of the polymethine dye DTCI. J. Phys. Chem. 1995, 99, 13796–13799. [Google Scholar] [CrossRef]
- Fouassier, J.P.; Lougnot, D.J.; Faure, J. Excited-states and laser properties of tricarbocyanine dyes. Opt. Commun. 1976, 18, 263–267. [Google Scholar] [CrossRef]
- Rentsch, S.; Dopel, E.; Petrov, V. Steady-state photoisomer accumulation in the absorber jet of the passively mode-locked femtosecond dye-laser. Appl. Phys. B-Photophysics Laser Chem. 1988, 46, 357–361. [Google Scholar] [CrossRef]
- Scaffardi, L.; Bilmes, G.M.; Schinca, D.; Tocho, J.O. Back-isomerization from the excited-state photoisomer of the laser-dye 3,3’diethyloxadicarbocyanine iodide (DODCI). Chem. Phys. Lett. 1987, 140, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Vaveliuk, P.; Scaffardi, L.B.; Duchowicz, R. Kinetic analysis of a double photoisomerization of the DTDCI cyanine dye. J. Phys. Chem. 1996, 100, 11630–11635. [Google Scholar] [CrossRef]
- Rulliere, C. Laser action and photoisomerization of 3,3’-diethyl oxadicarbocyanine iodide (DODCI) —Influence of temperature and concentration. Chem. Phys. Lett. 1976, 43, 303–308. [Google Scholar] [CrossRef]
- Momicchioli, F.; Baraldi, I.; Berthier, G. Theoretical study of trans cis photoisomerism in polymethine cyanines. Chem. Phys. 1988, 123, 103–112. [Google Scholar] [CrossRef]
- Momicchioli, F.; Baraldi, I.; Ponterini, G.; Berthier, G. Electronic spectra and trans-cis isomerism of streptopolymethine cyanines—A CS Indo CI study. Spectrochim. Acta Part A-Mol. Biomol. Spectrosc. 1990, 46, 775–791. [Google Scholar] [CrossRef]
- Murphy, S.; Schuster, G.B. Electronic relaxation in a series of cyanine dyes—Evidence for electronic and steric control of the rotational rate. J. Phys. Chem. 1995, 99, 8516–8518. [Google Scholar] [CrossRef]
- Stennett, E.M.S.; Ciuba, M.A.; Lin, S.; Levitus, M. Demystifying PIFE: The Photophysics Behind the Protein-Induced Fluorescence Enhancement Phenomenon in Cy3. J. Phys. Chem. Lett. 2015, 6, 1819–1823. [Google Scholar] [CrossRef] [PubMed]
- Levitus, M.; Ranjit, S. Cyanine dyes in biophysical research: The photophysics of polymethine fluorescent dyes in biomolecular environments. Q. Rev. Biophys. 2011, 44, 123–151. [Google Scholar] [CrossRef]
- Widengren, J.; Chmyrov, A.; Eggeling, C.; Lofdahl, P.A.; Seidel, C.A.M. Strategies to improve photostabilities in ultrasensitive fluorescence spectroscopy. J. Phys. Chem. A 2007, 111, 429–440. [Google Scholar] [CrossRef]
- Blom, H.; Widengren, J. Stimulated Emission Depletion Microscopy. Chem. Rev. 2017, 117, 7377–7427. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X.W. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.; Kwon, J.; Park, J.; Heo, J.; Kim, S.K. Selective thiolation and photoswitching mechanism of Cy5 studied by time-dependent density functional theory. Phys. Chem. Chem. Phys. 2020, 22, 14125–14129. [Google Scholar] [CrossRef]
- Lisovskaya, A.; Carmichael, I.; Harriman, A. Pulse Radiolysis Investigation of Radicals Derived from Water-Soluble Cyanine Dyes: Implications for Super-resolution Microscopy. J. Phys. Chem. A 2021, 125, 5779–5793. [Google Scholar] [CrossRef]
- Li, H.L.; Vaughan, J.C. Switchable Fluorophores for Single-Molecule Localization Microscopy. Chem. Rev. 2018, 118, 9412–9454. [Google Scholar] [CrossRef]
- Widengren, J.; Mets, U.; Rigler, R. Fluorescence correlation spectroscopy of triplet-states in solution—A theoretical and experimental-study. J. Phys. Chem. 1995, 99, 13368–13379. [Google Scholar] [CrossRef]
- Widengren, J.; Schwille, P. Characterization of photoinduced isomerization and back-isomerization of the cyanine dye Cy5 by fluorescence correlation spectroscopy. J. Phys. Chem. A 2000, 104, 6416–6428. [Google Scholar] [CrossRef]
- Du, Z.X.; Piguet, J.; Baryshnikov, G.; Tornmalm, J.; Demirbay, B.; Agren, H.; Widengren, J. Imaging Fluorescence Blinking of a Mitochondrial Localization Probe: Cellular Localization Probes Turned into Multifunctional Sensors br. J. Phys. Chem. B 2022, 126, 3048–3058. [Google Scholar] [CrossRef] [PubMed]
- Sanden, T.; Persson, G.; Thyberg, P.; Blom, H.; Widengren, J. Monitoring kinetics of highly environment sensitive states of fluorescent molecules by modulated excitation and time-averaged fluorescence intensity recording. Anal. Chem. 2007, 79, 3330–3341. [Google Scholar] [CrossRef]
- Sanden, T.; Persson, G.; Widengren, J. Transient State Imaging for Microenvironmental Monitoring by Laser Scanning Microscopy. Anal. Chem. 2008, 80, 9589–9596. [Google Scholar] [CrossRef]
- Tornmalm, J.; Piguet, J.; Chmyrov, V.; Widengren, J. Imaging of intermittent lipid-receptor interactions reflects changes in live cell membranes upon agonist-receptor binding. Sci. Rep. 2019, 9, 18133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornmalm, J.; Widengren, J. Label-free monitoring of ambient oxygenation and redox conditions using the photodynamics of flavin compounds and transient state (TRAST) spectroscopy. Methods 2018, 140, 178–187. [Google Scholar] [CrossRef]
- Widengren, J. Fluorescence-based transient state monitoring for biomolecular spectroscopy and imaging. J. R. Soc. Interface 2010, 7, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.X.; Ji, D.M.; Xia, A.D.; Koberling, F.; Patting, M.; Erdmann, R. Direct observation of delayed fluorescence from a remarkable back-isomerization in Cy5. J. Am. Chem. Soc. 2005, 127, 8064–8066. [Google Scholar] [CrossRef]
- Mets, U.; Widengren, J.; Rigler, R. Application of the antibunching in dye fluorescence: Measuring the excitation rates in solution. Chem. Phys. 1997, 218, 191–198. [Google Scholar] [CrossRef]
- Chmyrov, V.; Spielmann, T.; Hevekerl, H.; Widengren, J. Trans-Cis Isomerization of Lipophilic Dyes Probing Membrane Microviscosity in Biological Membranes and in Live Cells. Anal. Chem. 2015, 87, 5690–5697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spielmann, T.; Xu, L.; Gad, A.K.B.; Johansson, S.; Widengren, J. Transient state microscopy probes patterns of altered oxygen consumption in cancer cells. Febs J. 2014, 281, 1317–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widengren, J.; Seidel, C.A.M. Manipulation and characterization of photo-induced transient states of Merocyanine 540 by fluorescence correlation spectroscopy. Phys. Chem. Chem. Phys. 2000, 2, 3435–3441. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandberg, E.; Piguet, J.; Liu, H.; Widengren, J. Combined Fluorescence Fluctuation and Spectrofluorometric Measurements Reveal a Red-Shifted, Near-IR Emissive Photo-Isomerized Form of Cyanine 5. Int. J. Mol. Sci. 2023, 24, 1990. https://doi.org/10.3390/ijms24031990
Sandberg E, Piguet J, Liu H, Widengren J. Combined Fluorescence Fluctuation and Spectrofluorometric Measurements Reveal a Red-Shifted, Near-IR Emissive Photo-Isomerized Form of Cyanine 5. International Journal of Molecular Sciences. 2023; 24(3):1990. https://doi.org/10.3390/ijms24031990
Chicago/Turabian StyleSandberg, Elin, Joachim Piguet, Haichun Liu, and Jerker Widengren. 2023. "Combined Fluorescence Fluctuation and Spectrofluorometric Measurements Reveal a Red-Shifted, Near-IR Emissive Photo-Isomerized Form of Cyanine 5" International Journal of Molecular Sciences 24, no. 3: 1990. https://doi.org/10.3390/ijms24031990
APA StyleSandberg, E., Piguet, J., Liu, H., & Widengren, J. (2023). Combined Fluorescence Fluctuation and Spectrofluorometric Measurements Reveal a Red-Shifted, Near-IR Emissive Photo-Isomerized Form of Cyanine 5. International Journal of Molecular Sciences, 24(3), 1990. https://doi.org/10.3390/ijms24031990