

Tip60/KAT5 Histone Acetyltransferase Is Required for Maintenance and Neurogenesis of Embryonic Neural Stem Cells
 ,
,
Abstract
:1. Introduction
2. Results
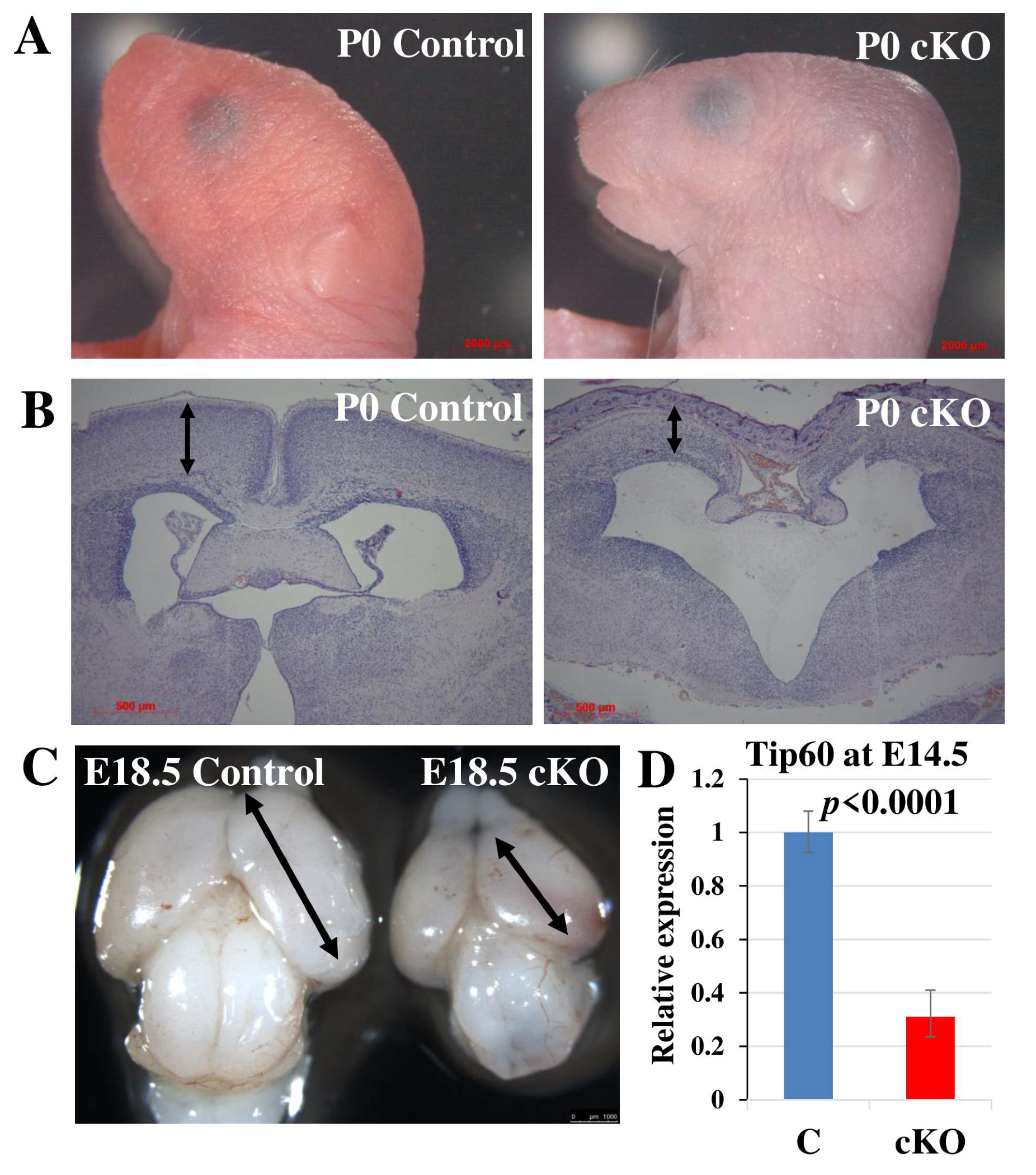
2.1. Tip60 Deletion in Neural Stem Cells Causes Perinatal Lethality
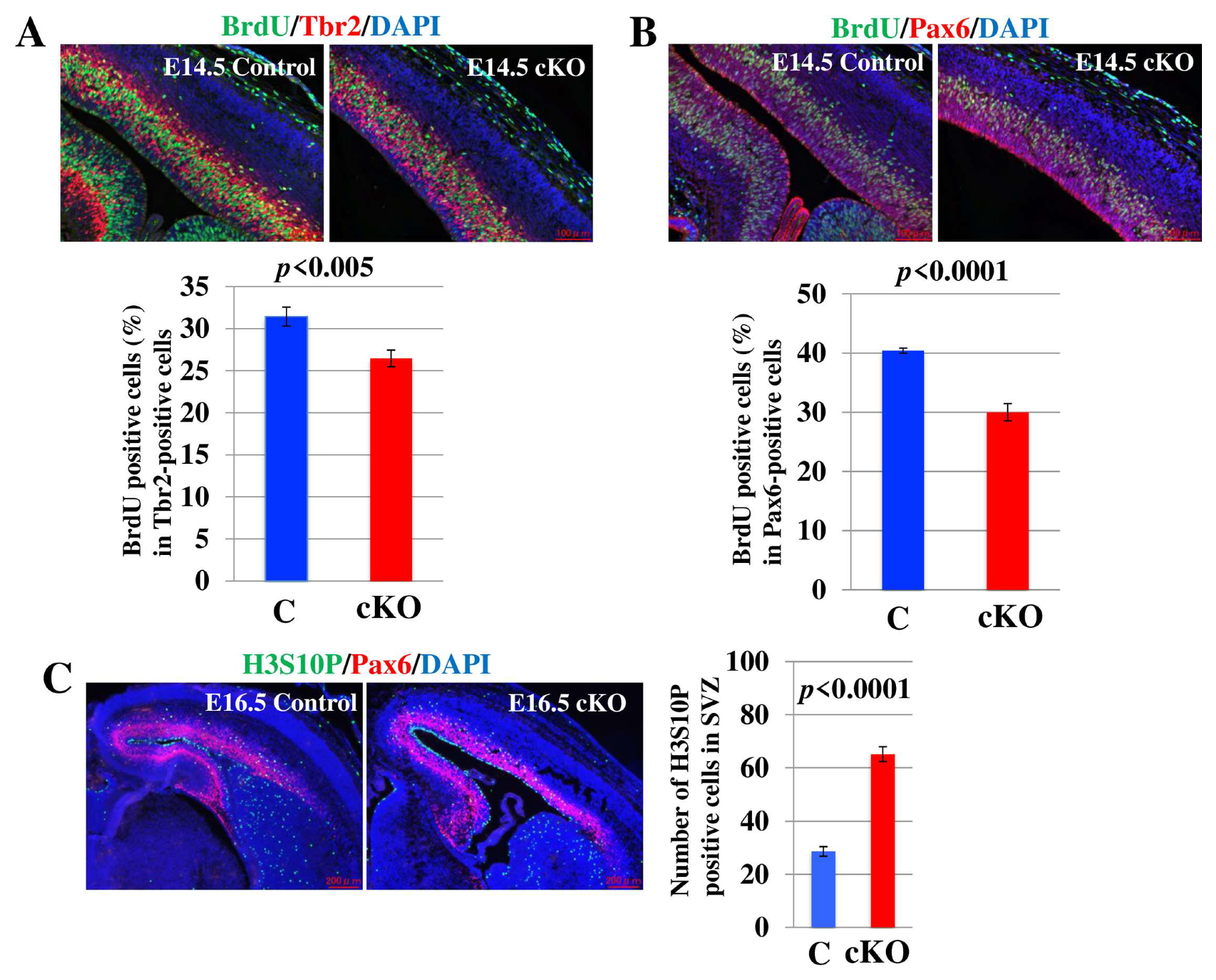
2.2. Tip60 Deletion in NSCs Causes Abnormalities in Neurogenesis and Neuronal Migration
2.3. Loss of Tip60 in NSCs Shows Cell Proliferation Defect and Accumulation of M Phase Cells in the Brain
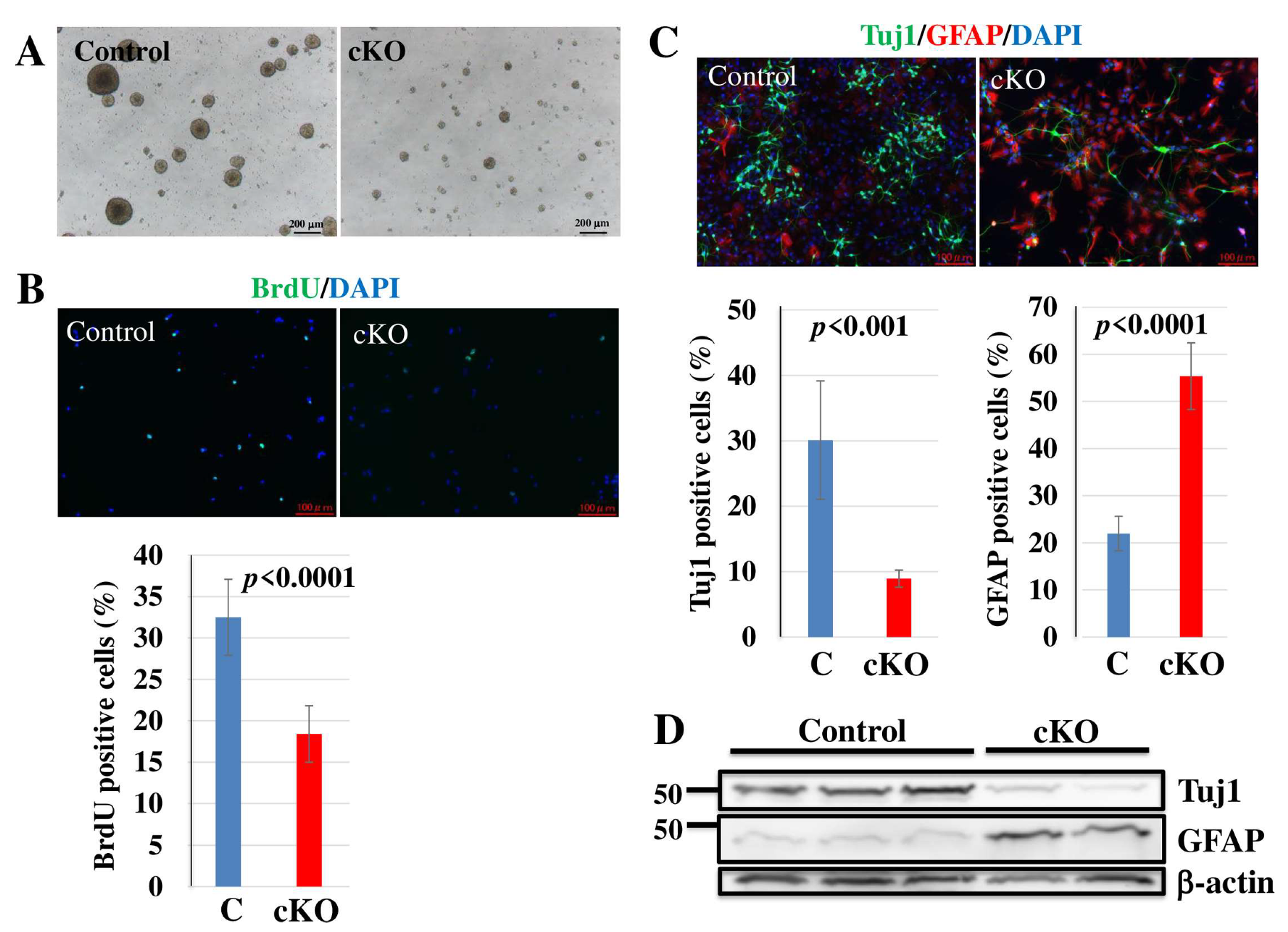
2.4. Loss of Tip60 in NSCs Affects Cell Growth and Differentiation In Vitro
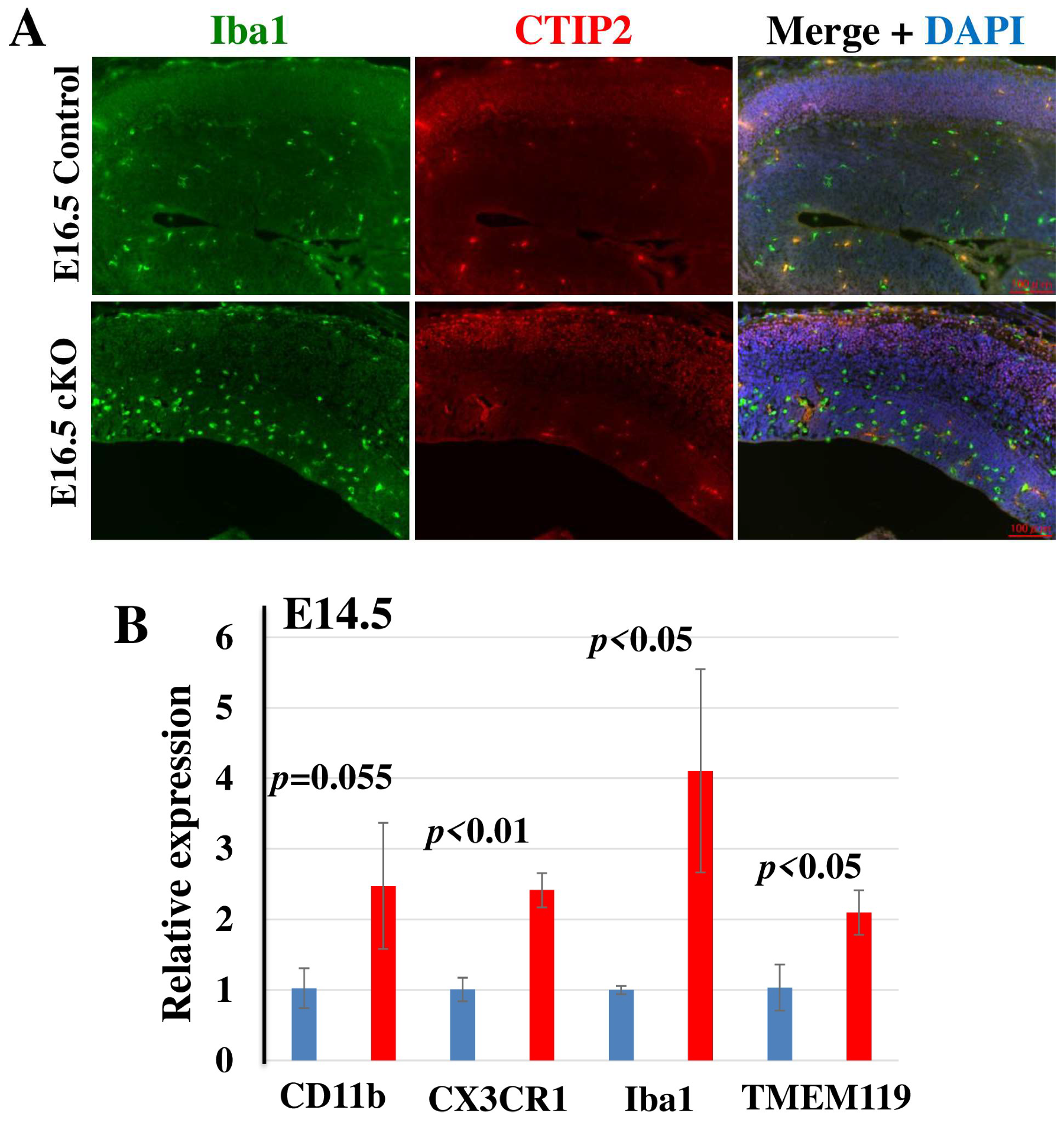
2.5. Microglia Are Activated in Tip60 cKO Brain
2.6. Tip60 Regulates Neurogenesis in Developing Brain
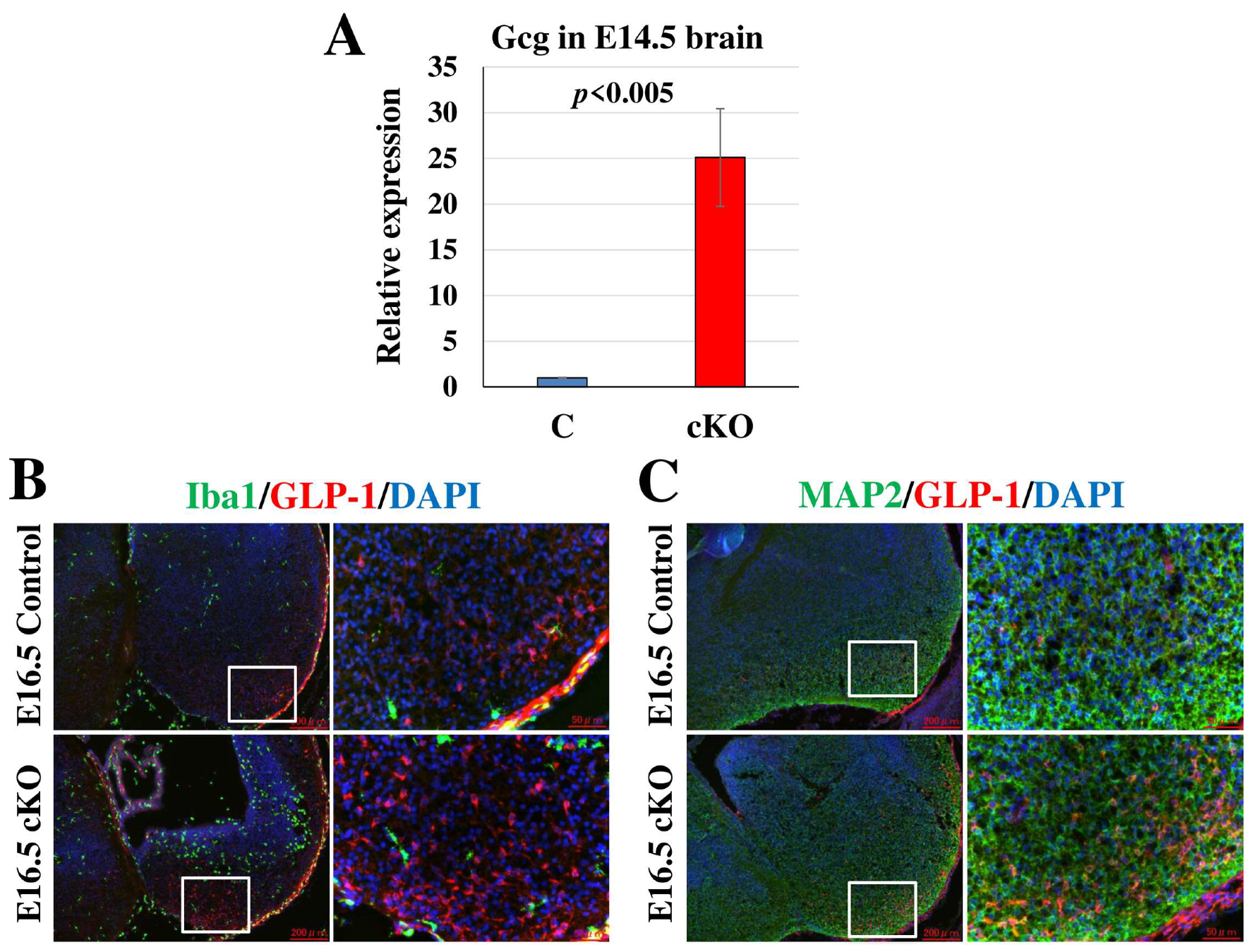
2.7. Glucagon-like Peptide-1 (GLP-1) Positive Cell Population Is Increased in Tip60 cKO Brains
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Histology and IHC
4.3. NSC Culture
4.4. BrdU Incorporation Assay
4.5. Differentiation Assay
4.6. Quantitative RT-PCR (qRT-PCR)
4.7. Western Blots
4.8. RNA Sequencing (RNA-Seq) and Data Processing
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, J.C.; Maze, I. Nothing Is Yet Set in (Hi)stone: Novel Post-Translational Modifications Regulating Chromatin Function. Trends Biochem. Sci. 2020, 45, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Macrae, T.A.; Fothergill-Robinson, J.; Ramalho-Santos, M. Regulation, functions and transmission of bivalent chromatin during mammalian development. Nat. Rev. Mol. Cell Biol. 2022, 24, 6–26. [Google Scholar] [CrossRef] [PubMed]
- Balsalobre, A.; Drouin, J. Pioneer factors as master regulators of the epigenome and cell fate. Nat. Rev. Mol. Cell Biol. 2022, 23, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Tapias, A.; Zhou, Z.; Shi, Y.; Chong, Z.; Wang, P.; Groth, M.; Platzer, M.; Huttner, W.; Herceg, Z.; Yang, Y.-G.; et al. Trrap-Dependent Histone Acetylation Specifically Regulates Cell-Cycle Gene Transcription to Control Neural Progenitor Fate Decisions. Cell Stem Cell 2014, 14, 632–643. [Google Scholar] [CrossRef] [Green Version]
- Tapias, A.; Wang, Z.-Q. Lysine Acetylation and Deacetylation in Brain Development and Neuropathies. Genom. Proteom. Bioinform. 2017, 15, 19–36. [Google Scholar] [CrossRef]
- Tang, T.; Zhang, Y.; Wang, Y.; Cai, Z.; Lu, Z.; Li, L.; Huang, R.; Hagelkruys, A.; Matthias, P.; Zhang, H.; et al. HDAC1 and HDAC2 Regulate Intermediate Progenitor Positioning to Safeguard Neocortical Development. Neuron 2019, 101, 1117–1133.e5. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Rousseau, J.; Machol, K.; Cross, L.A.; Agre, K.E.; Gibson, C.F.; Goverde, A.; Engleman, K.L.; Verdin, H.; De Baere, E.; et al. Deficient histone H3 propionylation by BRPF1-KAT6 complexes in neurodevelopmental disorders and cancer. Sci. Adv. 2020, 6, eaax0021. [Google Scholar] [CrossRef] [Green Version]
- Katada, S.; Takouda, J.; Nakagawa, T.; Honda, M.; Igarashi, K.; Imamura, T.; Ohkawa, Y.; Sato, S.; Kurumizaka, H.; Nakashima, K. Neural stem/precursor cells dynamically change their epigenetic landscape to differentially respond to BMP signaling for fate switching during brain development. Genes Dev. 2021, 35, 1431–1444. [Google Scholar] [CrossRef]
- Tapias, A.; Lázaro, D.; Yin, B.-K.; Rasa, S.M.M.; Krepelova, A.; Sacramento, E.K.; Grigaravicius, P.; Koch, P.; Kirkpatrick, J.; Ori, A.; et al. HAT cofactor TRRAP modulates microtubule dynamics via SP1 signaling to prevent neurodegeneration. Elife 2021, 10, e61531. [Google Scholar] [CrossRef]
- Lee, M.Y.; Lee, J.; Hyeon, S.J.; Cho, H.; Hwang, Y.J.; Shin, J.-Y.; McKee, A.C.; Kowall, N.W.; Kim, J.-I.; Stein, T.D.; et al. Epigenome signatures landscaped by histone H3K9me3 are associated with the synaptic dysfunction in Alzheimer’s disease. Aging Cell 2020, 19, e13153. [Google Scholar] [CrossRef]
- Nativio, R.; Lan, Y.; Donahue, G.; Sidoli, S.; Berson, A.; Srinivasan, A.R.; Shcherbakova, O.; Amlie-Wolf, A.; Nie, J.; Cui, X.; et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat. Genet. 2020, 52, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, L.; Channakkar, A.S.; Muralidharan, B. Chromatin remodelling complexes in cerebral cortex development and neurodevelopmental disorders. Neurochem. Int. 2021, 147, 105055. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Jefri, M. Epigenetic priming in neurodevelopmental disorders. Trends Mol. Med. 2021, 27, 1106–1114. [Google Scholar] [CrossRef]
- Valencia, A.M.; Pașca, S.P. Chromatin dynamics in human brain development and disease. Trends Cell Biol. 2022, 32, 98–101. [Google Scholar] [CrossRef]
- Voss, A.K.; Thomas, T. Histone Lysine and Genomic Targets of Histone Acetyltransferases in Mammals. Bioessays 2018, 40, e1800078. [Google Scholar] [CrossRef]
- D’Mello, S.R. Regulation of Central Nervous System Development by Class I Histone Deacetylases. Dev. Neurosci. 2019, 41, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Nitsch, S.; Shahidian, L.Z.; Schneider, R. Histone acylations and chromatin dynamics: Concepts, challenges, and links to metabolism. EMBO Rep. 2021, 22, e52774. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.C.; Koutelou, E.; Dent, S.Y.R. Now open: Evolving insights to the roles of lysine acetylation in chromatin organization and function. Mol. Cell 2022, 82, 716–727. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Voss, A.K.; Thomas, T. MYST family histone acetyltransferases take center stage in stem cells and development. Bioessays 2009, 31, 1050–1061. [Google Scholar] [CrossRef]
- Sapountzi, V.; Côté, J. MYST-family histone acetyltransferases: Beyond chromatin. Cell. Mol. Life Sci. 2010, 68, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, H.; Zhu, M.; Bai, H.; Huang, X. Roles of the MYST Family in the Pathogenesis of Alzheimer’s Disease via Histone or Non-histone Acetylation. Aging Dis. 2021, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Sapountzi, V.; Logan, I.R.; Robson, C.N. Cellular functions of TIP60. Int. J. Biochem. Cell Biol. 2006, 38, 1496–1509. [Google Scholar] [CrossRef]
- Li, Z.; Rasmussen, L.J. TIP60 in aging and neurodegeneration. Ageing Res. Rev. 2020, 64, 101195. [Google Scholar] [CrossRef] [PubMed]
- Mir, U.S.; Bhat, A.; Mushtaq, A.; Pandita, S.; Altaf, M.; Pandita, T.K. Role of histone acetyltransferases MOF and Tip60 in genome stability. DNA Repair 2021, 107, 103205. [Google Scholar] [CrossRef]
- Lashgari, A.; Tchara, P.-E.K.; Lambert, J.-P.; Côté, J. New insights into the DNA repair pathway choice with NuA4/TIP60. DNA Repair 2022, 113, 103315. [Google Scholar] [CrossRef]
- Xu, Y.; Wan, W. Acetylation in the regulation of autophagy. Autophagy 2022, 19, 379–387. [Google Scholar] [CrossRef]
- Stilling, R.M.; Rönicke, R.; Benito, E.; Urbanke, H.; Capece, V.; Burkhardt, S.; Bahari-Javan, S.; Barth, J.; Sananbenesi, F.; Schütz, A.L.; et al. K-Lysine acetyltransferase 2a regulates a hippocampal gene expression network linked to memory formation. EMBO J. 2014, 33, 1912–1927. [Google Scholar] [CrossRef] [Green Version]
- Urban, I.; Kerimoglu, C.; Sakib, M.S.; Wang, H.; Benito, E.; Thaller, C.; Zhou, X.; Yan, J.; Fischer, A.; Eichele, G. TIP60/KAT5 is required for neuronal viability in hippocampal CA1. Sci. Rep. 2019, 9, 16173. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Teubner, B.J.W.; Hevi, C.; Hara, K.; Kobayashi, A.; Dave, R.M.; Shintaku, T.; Jaikhan, P.; Yamagata, H.; Suzuki, T.; et al. CRTC1 Nuclear Translocation Following Learning Modulates Memory Strength via Exchange of Chromatin Remodeling Complexes on the Fgf1 Gene. Cell Rep. 2017, 18, 352–366. [Google Scholar] [CrossRef]
- Humbert, J.; Salian, S.; Makrythanasis, P.; Lemire, G.; Rousseau, J.; Ehresmann, S.; Garcia, T.; Alasiri, R.; Bottani, A.; Hanquinet, S.; et al. De Novo KAT5 Variants Cause a Syndrome with Recognizable Facial Dysmorphisms, Cerebellar Atrophy, Sleep Disturbance, and Epilepsy. Am. J. Hum. Genet. 2020, 107, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Panikker, P.; Xu, S.-J.; Zhang, H.; Sarthi, J.; Beaver, M.; Sheth, A.; Akhter, S.; Elefant, F. Restoring Tip60 HAT/HDAC2 Balance in the Neurodegenerative Brain Relieves Epigenetic Transcriptional Repression and Reinstates Cognition. J. Neurosci. 2018, 38, 4569–4583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaver, M.; Bhatnagar, A.; Panikker, P.; Zhang, H.; Snook, R.; Parmar, V.; Vijayakumar, G.; Betini, N.; Akhter, S.; Elefant, F. Disruption of Tip60 HAT mediated neural histone acetylation homeostasis is an early common event in neurodegenerative diseases. Sci. Rep. 2020, 10, 18265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Karisetty, B.C.; Bhatnagar, A.; Armour, E.M.; Beaver, M.; Roach, T.V.; Mortazavi, S.; Mandloi, S.; Elefant, F. Tip60 protects against amyloid-β-induced transcriptomic alterations via different modes of action in early versus late stages of neurodegeneration. Mol. Cell. Neurosci. 2020, 109, 103570. [Google Scholar] [CrossRef]
- Beaver, M.; Karisetty, B.C.; Zhang, H.; Bhatnagar, A.; Armour, E.; Parmar, V.; Brown, R.; Xiang, M.; Elefant, F. Chromatin and transcriptomic profiling uncover dysregulation of the Tip60 HAT/HDAC2 epigenomic landscape in the neurodegenerative brain. Epigenetics 2022, 17, 786–807. [Google Scholar] [CrossRef]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schütz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef]
- Kwan, K.; Šestan, N.; Anton, E.S. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 2012, 139, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Mukhtar, T.; Taylor, V. Untangling Cortical Complexity During Development. J. Exp. Neurosci. 2018, 12, 1179069518759332. [Google Scholar] [CrossRef] [Green Version]
- Englund, C.; Fink, A.; Lau, C.; Pham, D.; Daza, R.A.M.; Bulfone, A.; Kowalczyk, T.; Hevner, R.F. Pax6, Tbr2, and Tbr1 Are Expressed Sequentially by Radial Glia, Intermediate Progenitor Cells, and Postmitotic Neurons in Developing Neocortex. J. Neurosci. 2005, 25, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia Regulate the Number of Neural Precursor Cells in the Developing Cerebral Cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef]
- Thion, M.S.; Garel, S. On place and time: Microglia in embryonic and perinatal brain development. Curr. Opin. Neurobiol. 2017, 47, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Naito, Y.; Tsugawa, Y.; Nonaka, S.; Wake, H.; Nagasawa, T.; Kawaguchi, A.; Miyata, T. Transient microglial absence assists postmigratory cortical neurons in proper differentiation. Nat. Commun. 2020, 11, 1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Moriyama, Y.; Hasegawa, T.; Endo, T.A.; Toyoda, T.; Gotoh, Y. Scratch regulates neuronal migration onset via an epithelial-mesenchymal transition–like mechanism. Nat. Neurosci. 2013, 16, 416–425. [Google Scholar] [CrossRef]
- Sokpor, G.; Brand-Saberi, B.; Nguyen, H.P.; Tuoc, T. Regulation of Cell Delamination During Cortical Neurodevelopment and Implication for Brain Disorders. Front. Neurosci. 2022, 16, 824802. [Google Scholar] [CrossRef]
- Sandoval, D.A.; D’Alessio, D.A. Physiology of Proglucagon Peptides: Role of Glucagon and GLP-1 in Health and Disease. Physiol. Rev. 2015, 95, 513–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, M.A.; Wong, C.K.; Campbell, J.E.; Hodson, D.J.; Trapp, S.; Drucker, D.J. Revisiting the Complexity of GLP-1 Action from Sites of Synthesis to Receptor Activation. Endocr. Rev. 2021, 42, 101–132. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T.; Ito, S.; Tanimitsu, K.; Udagawa, S.; Oka, J.-I. Glucagon-like peptide-1 inhibits LPS-induced IL-1β production in cultured rat astrocytes. Neurosci. Res. 2006, 55, 352–360. [Google Scholar] [CrossRef]
- Kappe, C.; Tracy, L.M.; Patrone, C.; Iverfeldt, K.; Sjöholm, Å. GLP-1 secretion by microglial cells and decreased CNS expression in obesity. J. Neuroinflamm. 2012, 9, 276. [Google Scholar] [CrossRef] [Green Version]
- Rust, K.; Tiwari, M.D.; Mishra, V.K.; Grawe, F.; Wodarz, A. Myc and the Tip60 chromatin remodeling complex control neuroblast maintenance and polarity in Drosophila. EMBO J. 2018, 37, e98659. [Google Scholar] [CrossRef]
- Messina, G.; Prozzillo, Y.; Monache, F.D.; Santopietro, M.V.; Dimitri, P. Evolutionary conserved relocation of chromatin remodeling complexes to the mitotic apparatus. BMC Biol. 2022, 20, 172. [Google Scholar] [CrossRef]
- Mo, F.; Zhuang, X.; Liu, X.; Yao, P.Y.; Qin, B.; Su, Z.; Zang, J.; Wang, Z.; Zhang, J.; Dou, Z.; et al. Acetylation of Aurora B by TIP60 ensures accurate chromosomal segregation. Nat. Chem. Biol. 2016, 12, 226–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, X.; Liu, H.; Liu, X.; Ruan, K.; Zhang, Y.; Zhang, Z.; Yonghui, Z.; Liu, Y.; Akram, S.; Zhang, J.; et al. Mitosis-specific acetylation tunes Ran effector binding for chromosome segregation. J. Mol. Cell Biol. 2018, 10, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Sudevan, S.; Rao, V.J.; Shima, H.; Trivedi, A.K.; Igarashi, K.; Kundu, T.K. Haploinsufficient tumor suppressor Tip60 negatively regulates oncogenic Aurora B kinase. J. Biosci. 2019, 44, 147. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Cheng, Y.; Gui, P.; Cui, M.; Liu, W.; Wang, W.; Wang, X.; Ali, M.; Dou, Z.; Niu, L.; et al. Dynamic acetylation of the kinetochore-associated protein HEC1 ensures accurate microtubule–kinetochore attachment. J. Biol. Chem. 2019, 294, 576–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Yang, F.; Liu, X.; Xia, P.; Yin, W.; Wang, Z.; Wang, Y.; Yuan, X.; Dou, Z.; Jiang, K.; et al. Dynamic crotonylation of EB1 by TIP60 ensures accurate spindle positioning in mitosis. Nat. Chem. Biol. 2021, 17, 1314–1323. [Google Scholar] [CrossRef]
- Akbar, H.; Cao, J.; Wang, D.; Yuan, X.; Zhang, M.; Muthusamy, S.; Song, X.; Liu, X.; Aikhionbare, F.; Yao, X.; et al. Acetylation of Nup62 by TIP60 ensures accurate chromosome segregation in mitosis. J. Mol. Cell Biol. 2022. published online ahead of print. [Google Scholar] [CrossRef]
- Frank, S.R.; Parisi, T.; Taubert, S.; Fernandez, P.C.; Fuchs, M.; Chan, H.M.; Livingston, D.M.; Amati, B. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep. 2003, 4, 575–580. [Google Scholar] [CrossRef]
- Taubert, S.; Gorrini, C.; Frank, S.R.; Parisi, T.; Fuchs, M.; Chan, H.-M.; Livingston, D.M.; Amati, B. E2F-Dependent Histone Acetylation and Recruitment of the Tip60 Acetyltransferase Complex to Chromatin in Late G1. Mol. Cell. Biol. 2004, 24, 4546–4556. [Google Scholar] [CrossRef] [Green Version]
- Wichmann, J.; Pitt, C.; Eccles, S.; Garnham, A.L.; Li-Wai-Suen, C.S.N.; May, R.; Allan, E.; Wilcox, S.; Herold, M.J.; Smyth, G.K.; et al. Loss of TIP60 (KAT5) abolishes H2AZ lysine 7 acetylation and causes p53, INK4A, and ARF-independent cell cycle arrest. Cell Death Dis. 2022, 13, 627. [Google Scholar] [CrossRef]
- Bayraktar, O.A.; Fuentealba, L.C.; Alvarez-Buylla, A.; Rowitch, D.H. Astrocyte Development and Heterogeneity. Cold Spring Harb. Perspect. Biol. 2014, 7, a020362. [Google Scholar] [CrossRef]
- Takouda, J.; Katada, S.; Nakashima, K. Emerging mechanisms underlying astrogenesis in the developing mammalian brain. Proc. Jpn. Acad. Ser. B 2017, 93, 386–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, A.L.; Wicki-Stordeur, L.E.; Murai, K.K.; Swayne, L.A. Foundations and implications of astrocyte heterogeneity during brain development and disease. Trends Neurosci. 2022, 45, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Namihira, M.; Kohyama, J.; Semi, K.; Sanosaka, T.; Deneen, B.; Taga, T.; Nakashima, K. Committed Neuronal Precursors Confer Astrocytic Potential on Residual Neural Precursor Cells. Dev. Cell 2009, 16, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Nagao, M.; Ogata, T.; Sawada, Y.; Gotoh, Y. Zbtb20 promotes astrocytogenesis during neocortical development. Nat. Commun. 2016, 7, 11102. [Google Scholar] [CrossRef]
- Pereira, J.D.; Sansom, S.N.; Smith, J.; Dobenecker, M.-W.; Tarakhovsky, A.; Livesey, F.J. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 15957–15962. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, K.; Yanagisawa, M.; Arakawa, H.; Kimura, N.; Hisatsune, T.; Kawabata, M.; Miyazono, K.; Taga, T. Synergistic Signaling in Fetal Brain by STAT3-Smad1 Complex Bridged by p300. Science 1999, 284, 479–482. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Ge, W.; Martinowich, K.; Becker-Catania, S.; Coskun, V.; Zhu, W.; Wu, H.; Castro, D.; Guillemot, F.; Fan, G.; et al. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat. Neurosci. 2005, 8, 616–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Chung, J.; Kao, H.-Y.; Yang, Y.-C. Tip60 Is a Co-repressor for STAT3. J. Biol. Chem. 2003, 278, 11197–11204. [Google Scholar] [CrossRef] [Green Version]
- Merchenthaler, I.; Lane, M.; Shughrue, P. Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J. Comp. Neurol. 1998, 403, 261–280. [Google Scholar] [CrossRef]
- Holt, M.K. Mind affects matter: Hindbrain GLP1 neurons link stress, physiology and behaviour. Exp. Physiol. 2021, 106, 1853–1862. [Google Scholar] [CrossRef]
- Kabahizi, A.; Wallace, B.; Lieu, L.; Chau, D.; Dong, Y.; Hwang, E.; Williams, K.W. Glucagon-like peptide-1 (GLP-1) signalling in the brain: From neural circuits and metabolism to therapeutics. Br. J. Pharmacol. 2022, 179, 600–624. [Google Scholar] [CrossRef] [PubMed]
- Trapp, S.; Brierley, D.I. Brain GLP-1 and the regulation of food intake: GLP-1 action in the brain and its implications for GLP-1 receptor agonists in obesity treatment. Br. J. Pharmacol. 2022, 179, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Fazzio, T.G.; Huff, J.T.; Panning, B. An RNAi Screen of Chromatin Proteins Identifies Tip60-p400 as a Regulator of Embryonic Stem Cell Identity. Cell 2008, 134, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, D.; Hainer, S.J.; Yoon, Y.; Wang, F.; Bach, I.; Rivera-Pérez, J.A.; Fazzio, T.G. KAT-Independent Gene Regulation by Tip60 Promotes ESC Self-Renewal but Not Pluripotency. Cell Rep. 2017, 19, 671–679. [Google Scholar] [CrossRef]
- Barry, R.M.; Sacco, O.; Mameri, A.; Stojaspal, M.; Kartsonis, W.; Shah, P.; De Ioannes, P.; Hofr, C.; Côté, J.; Sfeir, A. Rap1 regulates TIP60 function during fate transition between two-cell-like and pluripotent states. Genes Dev. 2022, 36, 313–330. [Google Scholar] [CrossRef]
- Chen, P.B.; Hung, J.-H.; Hickman, T.L.; Coles, A.H.; Carey, J.F.; Weng, Z.; Chu, F.; Fazzio, T.G. Hdac6 regulates Tip60-p400 function in stem cells. Elife 2013, 2, e01557. [Google Scholar] [CrossRef]
- Petrif, F.; Giles, R.H.; Dauwerse, H.G.; Saris, J.J.; Hennekam, R.C.M.; Masuno, M.; Tommerup, N.; Van Ommen, G.-J.B.; Goodman, R.H.; Peters, D.J.M.; et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 1995, 376, 348–351. [Google Scholar] [CrossRef]
- Roelfsema, J.H.; White, S.J.; Ariyürek, Y.; Bartholdi, D.; Niedrist, D.; Papadia, F.; Bacino, C.A.; Dunnen, J.T.D.; van Ommen, G.-J.B.; Breuning, M.H.; et al. Genetic Heterogeneity in Rubinstein-Taybi Syndrome: Mutations in Both the CBP and EP300 Genes Cause Disease. Am. J. Hum. Genet. 2005, 76, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ghorbani, M.; Weisz-Hubshman, M.; Rousseau, J.; Thiffault, I.; Schnur, R.E.; Breen, C.; Oegema, R.; Weiss, M.M.; Waisfisz, Q.; et al. Lysine acetyltransferase 8 is involved in cerebral development and syndromic intellectual disability. J. Clin. Investig. 2020, 130, 1431–1445. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Guhathakurta, S.; Tsang, T.H.; Schwabenland, M.; Renschler, G.; Herquel, B.; Bhardwaj, V.; Holz, H.; Stehle, T.; Bondareva, O.; et al. Neural metabolic imbalance induced by MOF dysfunction triggers pericyte activation and breakdown of vasculature. Nature 2020, 22, 828–841. [Google Scholar] [CrossRef]
- Janas, J.A.; Zhang, L.; Luu, J.H.; Demeter, J.; Meng, L.; Marro, S.G.; Mall, M.; Mooney, N.A.; Schaukowitch, K.; Ng, Y.H.; et al. Tip60-mediated H2A.Z acetylation promotes neuronal fate specification and bivalent gene activation. Mol. Cell 2022, 82, 4627–4646.e14. [Google Scholar] [CrossRef] [PubMed]
- Champagne, N.; Bertos, N.R.; Pelletier, N.; Wang, A.H.; Vezmar, M.; Yang, Y.; Heng, H.H.; Yang, X.-J. Identification of a Human Histone Acetyltransferase Related to Monocytic Leukemia Zinc Finger Protein. J. Biol. Chem. 1999, 274, 28528–28536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, T.; Voss, A.K.; Chowdhury, K.; Gruss, P. Querkopf, a MYST family histone acetyltransferase, is required for normal cerebral cortex development. Development 2000, 127, 2537–2548. [Google Scholar] [CrossRef] [PubMed]
- Merson, T.D.; Dixon, M.P.; Collin, C.; Rietze, R.L.; Bartlett, P.F.; Thomas, T.; Voss, A.K. The Transcriptional Coactivator Querkopf Controls Adult Neurogenesis. J. Neurosci. 2006, 26, 11359–11370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, B.N.; Dixon, M.P.; Thomas, T.; Voss, A.K. Querkopf is a key marker of self-renewal and multipotency of adult neural stem cells. J. Cell Sci. 2012, 125, 295–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campeau, P.M.; Kim, J.C.; Lu, J.T.; Schwartzentruber, J.A.; Abdul-Rahman, O.A.; Schlaubitz, S.; Murdock, D.M.; Jiang, M.-M.; Lammer, E.J.; Enns, G.M.; et al. Mutations in KAT6B, Encoding a Histone Acetyltransferase, Cause Genitopatellar Syndrome. Am. J. Hum. Genet. 2012, 90, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Simpson, M.A.; Deshpande, C.; Dafou, D.; Vissers, L.E.; Woollard, W.J.; Holder, S.E.; Gillessen-Kaesbach, G.; Derks, R.; White, S.M.; Cohen-Snuijf, R.; et al. De Novo Mutations of the Gene Encoding the Histone Acetyltransferase KAT6B Cause Genitopatellar Syndrome. Am. J. Hum. Genet. 2012, 90, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.; Corcoran, L.M.; Gugasyan, R.; Dixon, M.P.; Brodnicki, T.; Nutt, S.L.; Metcalf, D.; Voss, A.K. Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev. 2006, 20, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Katsumoto, T.; Aikawa, Y.; Iwama, A.; Ueda, S.; Ichikawa, H.; Ochiya, T.; Kitabayashi, I. MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 2006, 20, 1321–1330. [Google Scholar] [CrossRef] [Green Version]
- Perez-Campo, F.M.; Costa, G.; Lie-A-Ling, M.; Stifani, S.; Kouskoff, V.; Lacaud, G. MOZ-Mediated Repression of p16INK4a Is Critical for the Self-Renewal of Neural and Hematopoietic Stem Cells. STEM CELLS 2013, 32, 1591–1601. [Google Scholar] [CrossRef]
- Wiesel-Motiuk, N.; Assaraf, Y.G. The key roles of the lysine acetyltransferases KAT6A and KAT6B in physiology and pathology. Drug Resist. Updat. 2020, 53, 100729. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J. MOZ and MORF acetyltransferases: Molecular interaction, animal development and human disease. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 1818–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, L.; Zou, J.; Zhao, H.; Bertos, N.R.; Park, M.; Wang, E.; Yang, X.-J. Deficiency of the Chromatin Regulator Brpf1 Causes Abnormal Brain Development. J. Biol. Chem. 2015, 290, 7114–7129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, L.; Yan, K.; Zhou, J.; Zhao, H.; Bertos, N.R.; Park, M.; Wang, E.; Yang, X.-J. The Lysine Acetyltransferase Activator Brpf1 Governs Dentate Gyrus Development through Neural Stem Cells and Progenitors. PLoS Genet. 2015, 11, e1005034. [Google Scholar] [CrossRef] [Green Version]
- Mattioli, F.; Schaefer, E.; Magee, A.; Mark, P.; Mancini, G.M.; Dieterich, K.; Von Allmen, G.; Alders, M.; Coutton, C.; van Slegtenhorst, M.; et al. Mutations in Histone Acetylase Modifier BRPF1 Cause an Autosomal-Dominant Form of Intellectual Disability with Associated Ptosis. Am. J. Hum. Genet. 2016, 100, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Kueh, A.J.; Dixon, M.P.; Voss, A.K.; Thomas, T. HBO1 Is Required for H3K14 Acetylation and Normal Transcriptional Activity during Embryonic Development. Mol. Cell. Biol. 2011, 31, 845–860. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kueh, A.J.; Grant, Z.L.; Abeysekera, W.; Garnham, A.L.; Wilcox, S.; Hyland, C.D.; Di Rago, L.; Metcalf, D.; Alexander, W.S.; et al. The histone lysine acetyltransferase HBO1 (KAT7) regulates hematopoietic stem cell quiescence and self-renewal. Blood 2022, 139, 845–858. [Google Scholar] [CrossRef]
- Bardeesy, N.; Sinha, M.; Hezel, A.F.; Signoretti, S.; Hathaway, N.A.; Sharpless, N.E.; Loda, M.; Carrasco, D.R.; DePinho, R.A. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature 2002, 419, 162–167. [Google Scholar] [CrossRef]
- Iwamori, N.; Tominaga, K.; Sato, T.; Riehle, K.; Iwamori, T.; Ohkawa, Y.; Coarfa, C.; Ono, E.; Matzuk, M.M. MRG15 is required for pre-mRNA splicing and spermatogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E5408–E5415. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Takano-Maruyama, M.; Pereira-Smith, O.M.; Gaufo, G.O.; Tominaga, K. MRG15, a component of HAT and HDAC complexes, is essential for proliferation and differentiation of neural precursor cells. J. Neurosci. Res. 2008, 87, 1522–1531. [Google Scholar] [CrossRef]
- Kuroda, R.; Tominaga, K.; Kasashima, K.; Kuroiwa, K.; Sakashita, E.; Hayakawa, H.; Kouki, T.; Ohno, N.; Kawai, K.; Endo, H. Loss of mitochondrial transcription factor A in neural stem cells leads to immature brain development and triggers the activation of the integral stress response in vivo. PLoS ONE 2021, 16, e0255355. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Kirtane, B.; Jackson, J.G.; Ikeno, Y.; Ikeda, T.; Hawks, C.; Smith, J.R.; Matzuk, M.M.; Pereira-Smith, O.M. MRG15 Regulates Embryonic Development and Cell Proliferation. Mol. Cell. Biol. 2005, 25, 2924–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Pereira-Smith, O.M.; Tominaga, K. Loss of the chromatin regulator MRG15 limits neural stem/progenitor cell proliferation via increased expression of the p21 Cdk inhibitor. Stem Cell Res. 2011, 7, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emig, D.; Salomonis, N.; Baumbach, J.; Lengauer, T.; Conklin, B.R.; Albrecht, M. AltAnalyze and DomainGraph: Analyzing and visualizing exon expression data. Nucleic Acids Res. 2010, 38, W755–W762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pups | % | % Expected | |
|---|---|---|---|
| Tip60F/F;Nes-Cre+ | 56 | 24.6 | 25 |
| Tip60F/F;Nes-Cre- | 63 | 27.6 | 25 |
| Tip60F/+;Nes-Cre+ | 52 | 22.8 | 25 |
| Tip60F/+;Nes-Cre- | 57 | 25.0 | 25 |
| Total | 228 | 100 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tominaga, K.; Sakashita, E.; Kasashima, K.; Kuroiwa, K.; Nagao, Y.; Iwamori, N.; Endo, H. Tip60/KAT5 Histone Acetyltransferase Is Required for Maintenance and Neurogenesis of Embryonic Neural Stem Cells. Int. J. Mol. Sci. 2023, 24, 2113. https://doi.org/10.3390/ijms24032113
Tominaga K, Sakashita E, Kasashima K, Kuroiwa K, Nagao Y, Iwamori N, Endo H. Tip60/KAT5 Histone Acetyltransferase Is Required for Maintenance and Neurogenesis of Embryonic Neural Stem Cells. International Journal of Molecular Sciences. 2023; 24(3):2113. https://doi.org/10.3390/ijms24032113
Chicago/Turabian StyleTominaga, Kaoru, Eiji Sakashita, Katsumi Kasashima, Kenji Kuroiwa, Yasumitsu Nagao, Naoki Iwamori, and Hitoshi Endo. 2023. "Tip60/KAT5 Histone Acetyltransferase Is Required for Maintenance and Neurogenesis of Embryonic Neural Stem Cells" International Journal of Molecular Sciences 24, no. 3: 2113. https://doi.org/10.3390/ijms24032113
APA StyleTominaga, K., Sakashita, E., Kasashima, K., Kuroiwa, K., Nagao, Y., Iwamori, N., & Endo, H. (2023). Tip60/KAT5 Histone Acetyltransferase Is Required for Maintenance and Neurogenesis of Embryonic Neural Stem Cells. International Journal of Molecular Sciences, 24(3), 2113. https://doi.org/10.3390/ijms24032113