
Mapping of QTLs and Screening Candidate Genes Associated with the Ability of Sugarcane Tillering and Ratooning




Abstract
:1. Introduction
2. Results
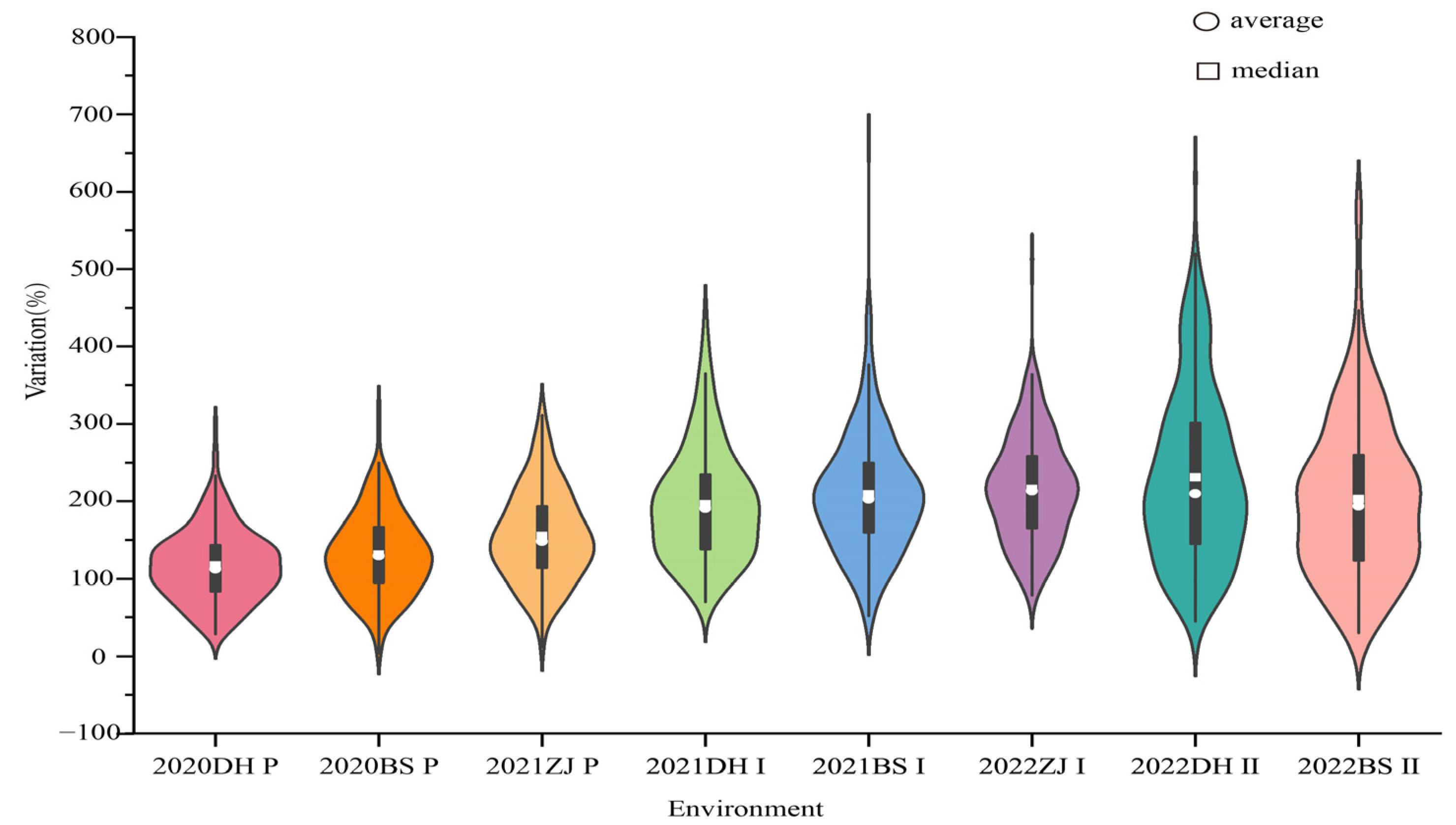
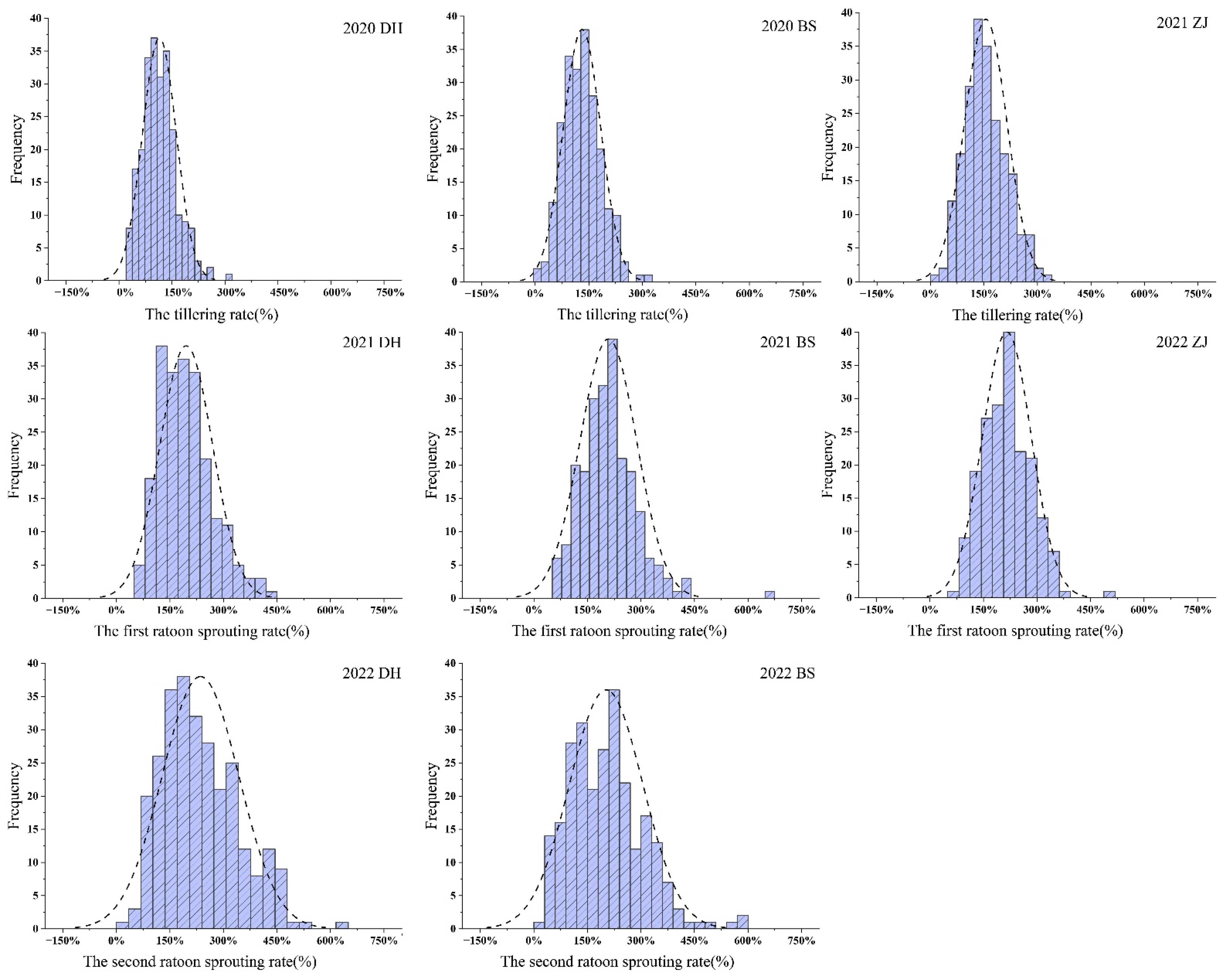
2.1. Phenotypic Heritability and Correlation Evaluation
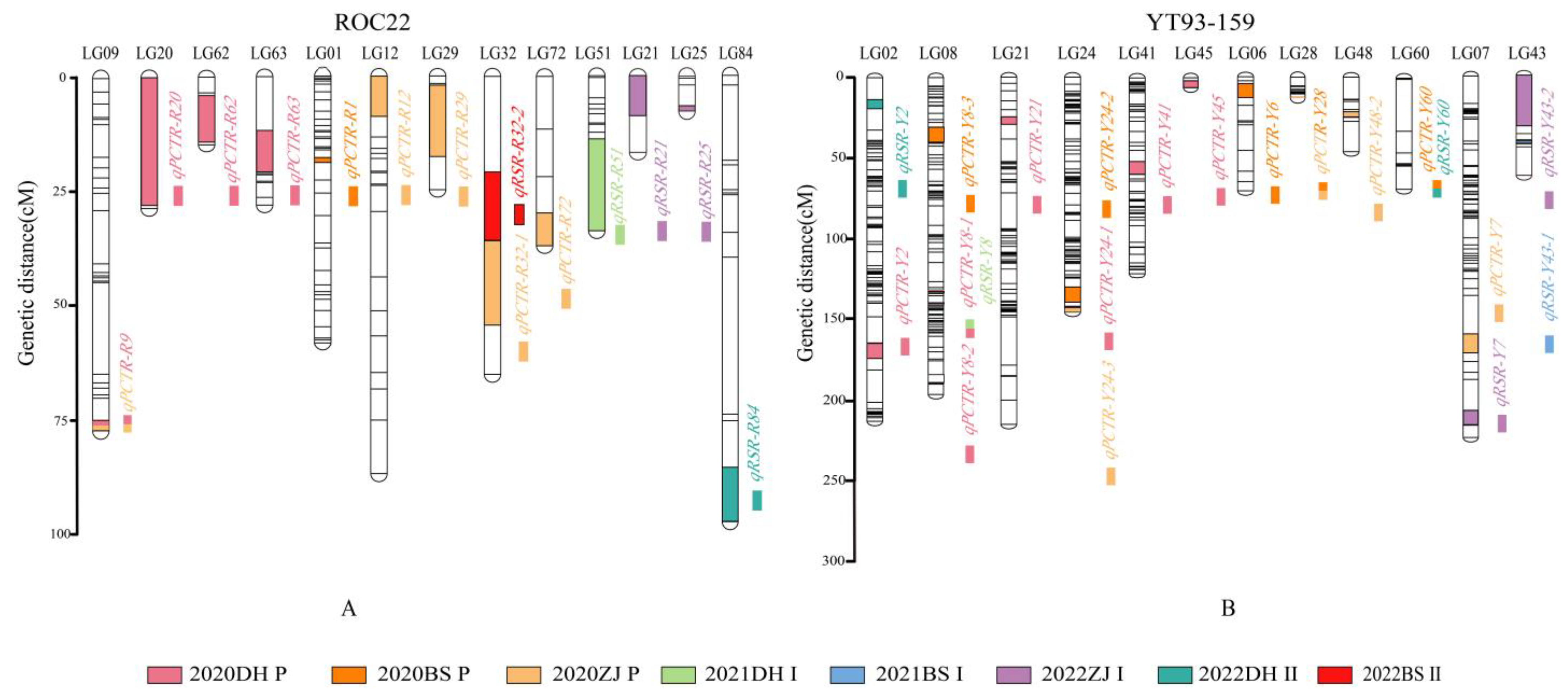
2.2. Mapping QTLs Associated with Sugarcane Tillering and Ratooning
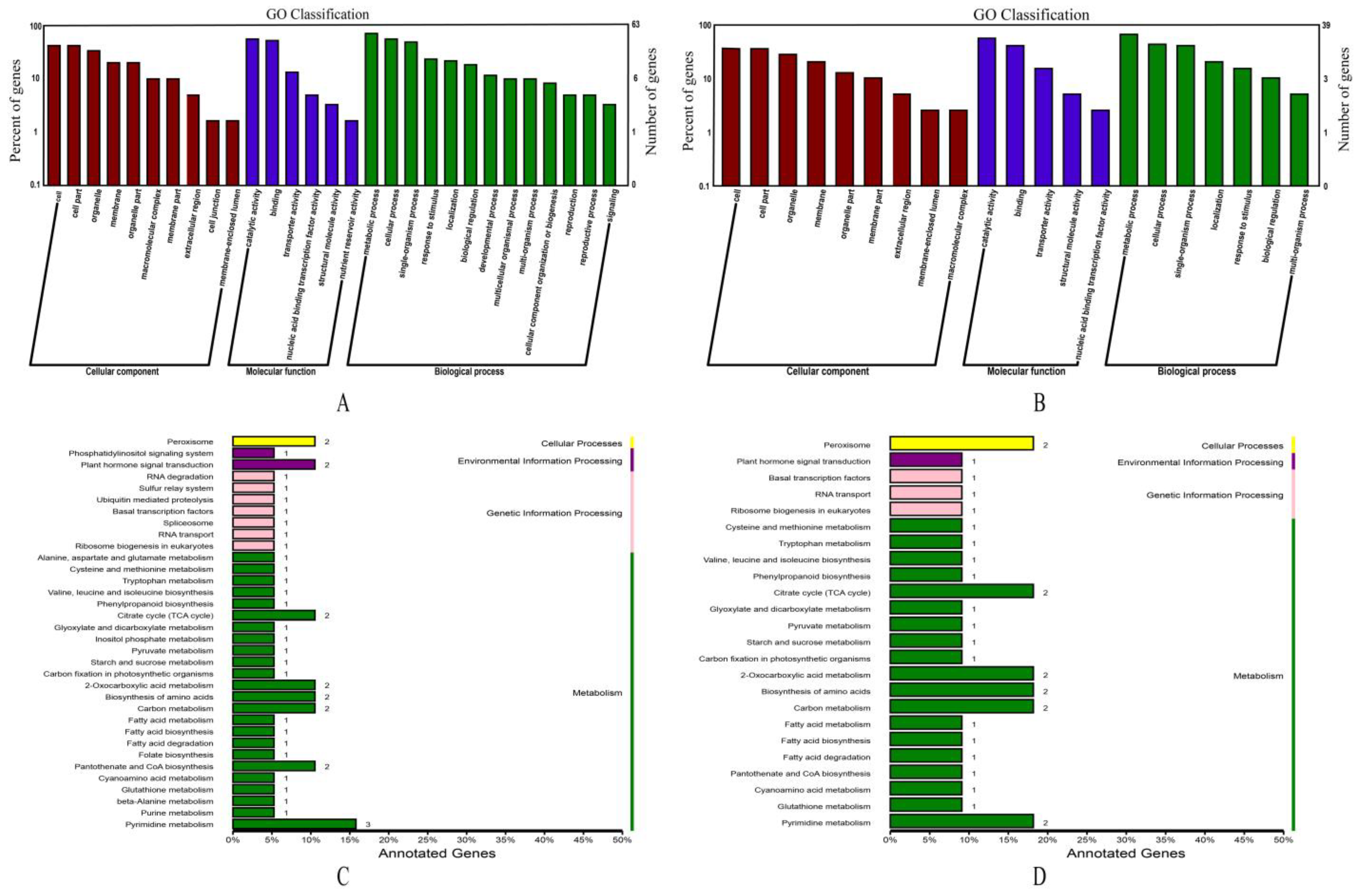
2.3. GO and KEGG Analysis of Genes
2.4. Screening of the Candidate Genes Related to Tillering and Ratooning
3. Discussion
4. Materials and Methods
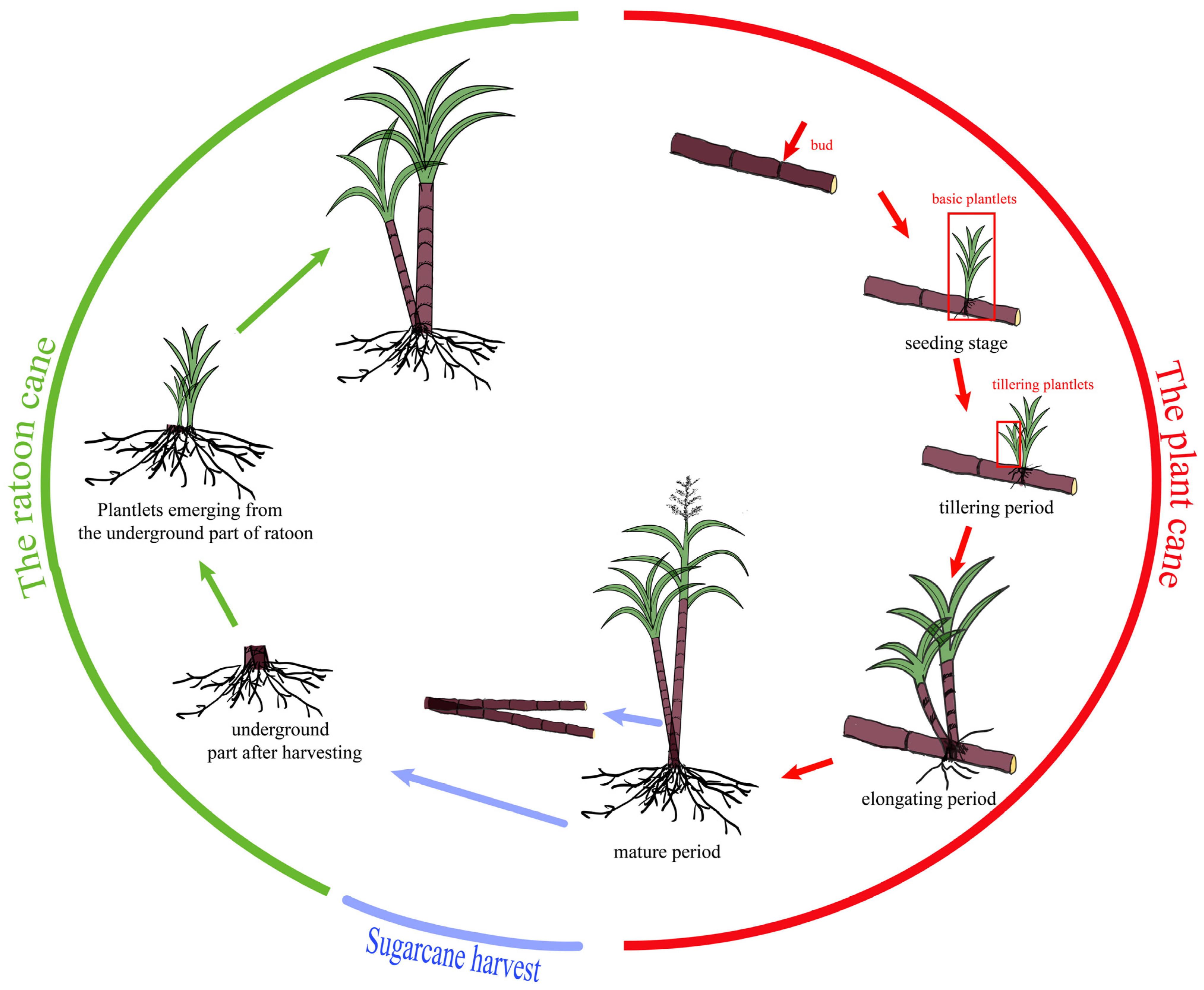
4.1. Plant Material
4.2. Phenotypic Investigation and Data Analysis
4.3. Mapping of QTLs Associated with Tillering and Ratooning
4.4. Gene Extraction and Functional Annotation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xu, F.; Wang, Z.; Lu, G.; Zeng, R.; Que, Y. Sugarcane ratooning ability: Research status, shortcomings, and prospects. Biology 2021, 10, 1052. [Google Scholar] [CrossRef]
- Milligan, S.B.; Gravois, K.A.; Bischoff, K.P.; Martin, F.A. Crop effects on genetic relationships among sugarcane traits. Crop Sci. 1990, 30, 927–931. [Google Scholar] [CrossRef]
- Osman, A.M.; Kumar, G.V.; Natarajan, U.S.; Babu, C. Investigations on sugar cane families for ratooning ability and iron chlorosis tolerance under field conditions. Indian Sugar 2006, 56, 25–30. [Google Scholar]
- Ren, M.; Huang, M.; Qiu, H.; Chun, Y.; Li, L.; Kumar, A.; Fang, J.; Zhao, J.; He, H.; Li, X. Genome-wide association study of the genetic basis of effective tiller number in rice. Rice 2021, 14, 56. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Wang, J.; Liu, X.; Yang, J.; Zhou, M.; Kong, W.; Jiang, Y.; Ke, S.; Sun, T.; Li, Y. QTL analysis and heterosis loci of effective tiller using three genetic populations derived from Indica-Japonica crosses in rice. Agronomy 2022, 12, 2171. [Google Scholar] [CrossRef]
- Govindarajulu, R.; Hostetler, A.N.; Xiao, Y.; Chaluvadi, S.R.; Mauro-Herrera, M.; Siddoway, M.L.; Whipple, C.; Bennetzen, J.L.; Devos, K.M.; Doust, A.N.; et al. Integration of high-density genetic mapping with transcriptome analysis uncovers numerous agronomic QTL and reveals candidate genes for the control of tillering in sorghum. G3 2021, 11, jkab024. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Wang, Y.-H.; Sharma, S.; Singh, S.; Hasenstein, K.H. SSR markers linked to kernel weight and tiller number in sorghum identified by association mapping. Euphytica 2012, 187, 401–410. [Google Scholar] [CrossRef]
- Liu, J.; Tang, H.; Qu, X.; Liu, H.; Li, C.; Tu, Y.; Li, S.; Habib, A.; Mu, Y.; Dai, S.; et al. A novel, major, and validated QTL for the effective tiller number located on chromosome arm 1BL in bread wheat. Plant Mol. Biol. 2020, 104, 173–185. [Google Scholar] [CrossRef]
- Bilal, M.; Saeed, M.; Nasir, I.A.; Tabassum, B.; Zameer, M.; Khan, A.; Tariq, M.; Javed, M.A.; Husnain, T. Association mapping of cane weight and tillers per plant in sugarcane. Biotechnol. Biotech. Equip. 2015, 29, 617–623. [Google Scholar] [CrossRef]
- Ming, R.; Wang, W.; Draye, X.; Moore, H.; Irvine, E.; Paterson, H. Molecular dissection of complex traits in autopolyploids: Mapping QTLs affecting sugar yield and related traits in sugarcane. Theor. Appl. Genet. 2002, 105, 332–345. [Google Scholar] [CrossRef]
- Qiu, L.H.; Fan, Y.G.; Luo, H.M.; Huang, X.; Chen, R.F.; Yang, R.Z.; Wu, J.M.; Li, Y.R. Advances of regulation study on tillering formation and stem forming from available tillers in sugarcane (Saccharum officinarum). J. Plant Physiol. 2018, 54, 192–202. [Google Scholar]
- Winfield, M.O.; Allen, A.M.; Burridge, A.J.; Barker, G.L.; Benbow, H.R.; Wilkinson, P.A.; Coghill, J.; Waterfall, C.; Davassi, A.; Scopes, G.; et al. High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 2016, 14, 1195–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giomi, G.M.; Sampietro, D.A.; Velazco, J.G.; Iglesias, J.; Fernández, M.; Oviedo, M.S.; Presello, D.A. Map overlapping of QTL for resistance to fusarium ear rot and associated traits in maize. Euphytica 2021, 217, 81. [Google Scholar] [CrossRef]
- Tang, W.; Wu, T.; Ye, J.; Sun, J.; Jiang, Y.; Yu, J.; Tang, J.; Chen, G.; Wang, C.; Wan, J. SNP-based analysis of genetic diversity reveals important alleles associated with seed size in rice. BMC Plant Biol. 2016, 16, 128. [Google Scholar]
- You, Q.; Sood, S.; Luo, Z.; Liu, H.; Sariful Islam, M.; Zhang, M.; Wang, J. Identifying genomic regions controlling ratoon stunting disease resistance in sugarcane (Saccharum spp.) clonal F1 population. Crop J. 2020, 9, 1070–1078. [Google Scholar] [CrossRef]
- Wang, Z.; Lu, G.; Wu, Q.; Li, A.; Que, Y.; Xu, L. Isolating QTL controlling sugarcane leaf blight resistance using a two-way pseudo-testcross strategy. Crop J. 2021, 10, 1131–1140. [Google Scholar] [CrossRef]
- Lu, G.; Pan, Y.B.; Wang, Z.; Xu, F.; Cheng, W.; Huang, X.; Ren, H.; Pang, C.; Que, Y.; Xu, L. Utilization of a sugarcane 100K single nucleotide polymorphisms microarray-derived high-density genetic map in quantitative trait loci mapping and function role prediction of genes related to Chlorophyll content in sugarcane. Front. Plant Sci. 2021, 12, 817875. [Google Scholar] [CrossRef]
- Daugrois, J.H.; Grivet, L.; Roques, D.; Hoarau, J.Y.; Lombard, H.; Glaszmann, J.C.; D’Hont, A. A putative major gene for rust resistance linked with a RFLP marker in sugarcane cultivar ‘R570’. Theor. Appl. Genet. 1996, 92, 1059–1064. [Google Scholar] [CrossRef]
- Asnaghi, C.; Roques, D.; Ruffel, S.; Kaye, C.; Hoarau, J.Y.; Télismart, H.; Girard, J.C.; Raboin, L.M.; Risterucci, A.M.; Grivet, L.; et al. Targeted mapping of a sugarcane rust resistance gene (Bru1) using bulked segregant analysis and AFLP markers. Theor. Appl. Genet. 2004, 108, 759–764. [Google Scholar] [CrossRef]
- Costet, L.; Le Cunff, L.; Royaert, S.; Raboin, L.M.; Hervouet, C.; Toubi, L.; Telismart, H.; Garsmeur, O.; Rousselle, Y.; Pauquet, J.; et al. Haplotype structure around Bru1 reveals a narrow genetic basis for brown rust resistance in modern sugarcane cultivars. Theor. Appl. Genet. 2012, 125, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Balsalobre, T.W.; Da Silva Pereira, G.; Margarido, G.R.; Gazaffi, R.; Barreto, F.Z.; Anoni, C.O.; Cardoso-Silva, C.B.; Costa, E.A.; Mancini, M.C.; Hoffmann, H.P.; et al. GBS-based single dosage markers for linkage and QTL mapping allow gene mining for yield-related traits in sugarcane. BMC Genom. 2017, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Islam, M.S.; Sood, S.; Maya, S.; Hanson, E.A.; Comstock, J.; Wang, J. Identifying quantitative trait loci (QTLs) and developing diagnostic markers linked to orange rust resistance in sugarcane (Saccharum spp.). Front. Plant Sci. 2018, 9, 350. [Google Scholar] [CrossRef] [PubMed]
- Aljanabi, S.M.; Parmessur, Y.; Kross, H.; Dhayan, S.; Saumtally, S.; Ramdoyal, K.; Autrey, L.C.; Dookun-Saumtally, A. Identification of a major quantitative trait locus (QTL) for yellow spot (Mycovellosiella koepkei) disease resistance in sugarcane. Mol. Breed. 2007, 19, 1–14. [Google Scholar] [CrossRef]
- Da Silva, J.; Honeycutt, R.J.; Burnquist, W.; Al-Janabi, S.M.; Sorrells, M.E.; Tanksley, S.D.; Sobral, B.W.S. Saccharum spontaneum L. ‘SES 208’ genetic linkage map combining RFLP- and PCR-based markers. Mol. Breed. 1995, 1, 165–179. [Google Scholar] [CrossRef]
- Mudge, J.; Andersen, W.R.; Kehrer, R.; Fairbanks, D. A RAPD genetic map of Saccharum officinarum. Crop Sci. 1996, 36, 1362–1366. [Google Scholar] [CrossRef]
- You, Q.; Yang, X.; Peng, Z.; Islam, M.S.; Sood, S.; Luo, Z.; Comstock, J.; Xu, L.; Wang, J. Development of an Axiom Sugarcane 100K SNP array for genetic map construction and QTL identification. Theor. Appl. Genet. 2019, 132, 2829–2845. [Google Scholar] [CrossRef]
- Singh, R.K.; Singh, S.P.; Tiwari, D.K.; Srivastava, S.; Singh, S.B.; Sharma, M.L.; Singh, R.; Mohapatra, T.; Singh, N.K. Genetic mapping and QTL analysis for sugar yield-related traits in sugarcane. Euphytica 2013, 191, 333–353. [Google Scholar] [CrossRef]
- Raihan, M.S.; Liu, J.; Huang, J.; Guo, H.; Pan, Q.; Yan, J. Multi-environment QTL analysis of grain morphology traits and fine mapping of a kernel-width QTL in Zheng58 × SK maize population. Theor. Appl. Genet. 2016, 129, 1465–1477. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, L.; Song, G.; Huang, T.; Wu, G.; Tan, J.; Wang, P.; Cheng, Q.; Li, C.; Zhong, Q.; et al. Localization of salt-tolerant QTL in rice germination stage under different salinity concentrations. Euphytica 2022, 218, 78. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, X.; Hao, Y.; Zhang, Y.; Liu, Y.; Pu, Z.; Tian, Y.; Xu, D.; Xia, X.; He, Z.; et al. QTL mapping for grain zinc and iron concentrations in bread wheat. Front. Nutr. 2021, 8, 680391. [Google Scholar] [CrossRef]
- Che, Y.; He, Y.; Song, N.; Yang, Y.; Wei, L.; Yang, X.; Zhang, Y.; Zhang, J.; Han, H.; Li, X.; et al. Four-year and five-developing-stage dynamic QTL mapping for tiller number in the hybrid population of Agropyron Gaertn. Front. Plant Sci. 2022, 13, 835437. [Google Scholar] [CrossRef] [PubMed]
- Vom Endt, D.; Kijne, J.W.; Memelink, J. Transcription factors controlling plant secondary metabolism: What regulates the regulators? Phytochemistry 2002, 61, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Seale, M.; Bennett, T.; Leyser, O. BRC1 expression regulates bud activation potential but is not necessary or sufficient for bud growth inhibition in Arabidopsis. Development 2017, 144, 1661–1673. [Google Scholar] [PubMed] [Green Version]
- Cui, P.; Liu, H.; Ruan, S.; Ali, B.; Gill, R.A.; Ma, H.; Zheng, Z.; Zhou, W. A zinc finger protein, interacted with cyclophilin, affects root development via IAA pathway in rice. J. Integr. Plant Biol. 2017, 59, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Bolle, C. The role of GRAS proteins in plant signal transduction and development. Planta 2004, 218, 683–692. [Google Scholar] [CrossRef]
- Waseem, M.; Nkurikiyimfura, O.; Niyitanga, S.; Jakada, B.H.; Shaheen, I.; Aslam, M.M. GRAS transcription factors emerging regulator in plants growth, development, and multiple stresses. Mol. Biol. Rep. 2022, 49, 9673–9685. [Google Scholar] [CrossRef]
- Lei, L.; Zheng, H.L.; Wang, J.G.; Liu, H.L.; Sun, J.; Zhao, H.W.; Yang, L.M.; Zou, D.T. Genetic dissection of rice (Oryza sativa L.) tiller, plant height, and grain yield based on QTL mapping and metaanalysis. Euphytica 2018, 214, 109. [Google Scholar] [CrossRef]
- Li, Z.Y.; Qiu, L.; Yan, H.F.; Zhou, H.W.; Chen, R.F.; Fan, Y.; Liang, H.; Wu, J.M. Effect of exogenous gibberellin signal on sugarcane tillering and its endogenous hormones. Chin. J. Trop. Crops 2021, 42, 2942–2951. [Google Scholar]
- Moore, P.H. Additive and nonadditive effects of serial applications of gibberellic acid on sugarcaneinternode growth. Physiol. Plant. 1980, 49, 271–276. [Google Scholar] [CrossRef]
- Nguyen, C.; Dang, L.H.; Nguyen, D.T.; Tran, K.P.; Giang, B.L.; Tran, N.Q. Effect of GA3 and Gly plant growth regulators on productivity and sugar content of sugarcane. Agriculture 2019, 9, 136. [Google Scholar] [CrossRef]
- Zhao, D.D.; Park, J.R.; Jang, Y.H.; Kim, E.G.; Du, X.X.; Farooq, M.; Yun, B.J.; Kim, K.M. Identification of one major QTL and a novel gene OsIAA17q5 associated with tiller number in rice using QTL analysis. Plants 2022, 11, 538. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Yamagami, D.; Umehara, M.; Hanada, A.; Yoshida, S.; Sasaki, Y.; Yajima, S.; Kyozuka, J.; Ueguchi-Tanaka, M.; Matsuoka, M.; et al. Regulation of strigolactone biosynthesis by gibberellin signaling. Plant Physiol. 2017, 174, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Gachomo, E.W.; Jimenez-Lopez, J.C.; Baptiste, L.J.; Kotchoni, S.O. GIGANTUS1 (GTS1), a member of transducin/WD40 protein superfamily, controls seed germination, growth and biomass accumulation through ribosome-biogenesis protein interactions in Arabidopsis thaliana. BMC Plant Biol. 2014, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Shu, K.; Yang, W. E3 ubiquitin ligases: Ubiquitous actors in plant development and abiotic stress responses. Plant Cell Physiol. 2017, 58, 1461–1476. [Google Scholar] [CrossRef] [Green Version]
- Hussain, F.; Sarwar, M.; Ahmad, S.; Ali, M.; Fiaz, N.; Chattha, A. Effect of stale and fresh cane setts (SEED) on yield and quality of suarcane (Saccharum officinarum L.). Int. J. Agric. Appl. Sci. 2011, 3, 35–37. [Google Scholar]
- Chumphu, S.; Jongrungklang, N.; Songsri, P. Association of physiological responses and root distribution patterns of ratooning ability and yield of the second ratoon cane in sugarcane elite clones. Agronomy 2019, 9, 200. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J. 2015, 3, 269–283. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, L.; Wu, W.; Wang, J. GACD: Integrated software for genetic analysis in clonal F1 and double cross populations. J. Hered. 2015, 106, 741–744. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environments | Parents | F1 Population | ||||||
|---|---|---|---|---|---|---|---|---|
| YT93-159(%) | ROC22(%) | Max(%) | Min(%) | Mean (%) | CV (%) | K-s (sig) | ||
| The plant cane | 2020DH | 100.0 a | 66.7 b | 303.3 | 28.1 | 117.1 | 39.9 | 0.589 |
| 2020BS | 104.3 a | 58.3 b | 325.0 | 3.3 | 131.1 | 40.3 | 0.656 | |
| 2021ZJ | 103.5 a | 56.3 b | 316.7 | 11.4 | 155.0 | 38.5 | 0.607 | |
| The first ratoon cane | 2021DH | 109.1 a | 122.2 a | 445.6 | 69.6 | 195.8 | 37.7 | 0.296 |
| 2021BS | 155.6 a | 154.2 a | 672.7 | 51.9 | 208.6 | 38.1 | 0.144 | |
| 2022ZJ | 126.8 a | 112.5 a | 512.5 | 78.1 | 215.5 | 31.8 | 0.842 | |
| The second ratoon cane | 2022DH | 118.2 a | 144.4 a | 620.0 | 44.4 | 203.9 | 48.0 | 0.118 |
| 2022BS | 152.2 a | 133.3 a | 586.7 | 29.6 | 202.5 | 50.7 | 0.214 | |
| Environment | QTL Name | LG | GD (cM) | Left Marker | Right Marker | LOD | PVE (%) | Effect F | Effect M |
|---|---|---|---|---|---|---|---|---|---|
| 2020DH P | qPCTR-R9 | 9 | 2.2 | AX-171327125 | AX-171310449 | 3.8035 | 5.8797 | 0.1134 | −0.0048 |
| qPCTR-R20 | 20 | 29.3 | AX-171272742 | AX-171293480 | 3.7847 | 9.498 | 0.0889 | −0.0137 | |
| qPCTR-R62 | 62 | 11.9 | AX-171318447 | AX-171292913 | 3.4595 | 5.3478 | −0.0491 | −0.025 | |
| qPCTR-R63 | 63 | 9.7 | AX-171319734 | AX-171359264 | 4.1141 | 8.1183 | −0.0124 | −0.0201 | |
| qPCTR-Y2 | 2 | 9.4 | AX-171302044 | AX-171244192 | 3.0729 | 4.2714 | 0.0437 | 0.0813 | |
| qPCTR-Y8-1 | 8 | 3.7 | AX-171329699 | AX-171239513 | 7.7142 | 11.8968 | 0.0141 | −0.0475 | |
| qPCTR-Y8-2 | 8 | 1.5 | AX-171262083 | AX-171274601 | 15.6779 | 25.6955 | 0.0052 | −0.0483 | |
| qPCTR-Y21 | 21 | 4.7 | AX-171321699 | AX-171270548 | 3.1302 | 4.3498 | −0.0396 | −0.0478 | |
| qPCTR-Y24-1 | 24 | 1.1 | AX-171283687 | AX-171367948 | 3.5537 | 4.8965 | 0.0625 | 0.0559 | |
| qPCTR-Y41 | 41 | 8.1 | AX-171250323 | AX-171273872 | 3.6921 | 5.4547 | 0.0409 | 0.0993 | |
| qPCTR-Y45 | 45 | 4.3 | AX-171306380 | AX-171313333 | 3.4984 | 4.8351 | 0.0796 | 0.0542 | |
| 2020BS P | qPCTR-R1 | 1 | 1.1 | AX-171351615 | AX-171293624 | 3.0308 | 6.2062 | 0.0835 | −0.0816 |
| qPCTR-Y6 | 6 | 8.7 | AX-171257841 | AX-171273471 | 3.1402 | 5.9345 | −0.0309 | −0.0951 | |
| qPCTR-Y8-3 | 8 | 9.4 | AX-171324757 | AX-171308215 | 2.9123 | 5.002 | −0.099 | 0.0519 | |
| qPCTR-Y24-2 | 24 | 9.4 | AX-171283359 | AX-171249399 | 3.558 | 6.924 | 0.0231 | 0.1043 | |
| qPCTR-Y28 | 28 | 0.7 | AX-171326575 | AX-171286677 | 2.914 | 5.2792 | −0.043 | 0.0097 | |
| qPCTR-Y60 | 60 | 0.7 | AX-171257349 | AX-171272905 | 4.7622 | 8.8548 | 0.1576 | 0.0033 | |
| 2021ZJ P | qPCTR-R9 | 9 | 2.2 | AX-171327125 | AX-171310449 | 3.0126 | 5.7327 | 0.1097 | −0.033 |
| qPCTR-R12 | 12 | 8.7 | AX-171247255 | AX-171245486 | 3.449 | 7.7469 | 0.0904 | −0.1005 | |
| qPCTR-R29 | 29 | 17.0 | AX-171323413 | AX-171256547 | 3.9984 | 9.3369 | −0.0513 | −0.0944 | |
| qPCTR-R32-1 | 32 | 18.7 | AX-171357712 | AX-171244747 | 3.893 | 7.6342 | 0.0834 | 0.1306 | |
| qPCTR-R72 | 72 | 7.6 | AX-171281899 | AX-171351113 | 3.3203 | 6.2522 | 0.0937 | 0.0537 | |
| qPCTR-Y7 | 7 | 11.8 | AX-118065381 | AX-171315846 | 3.6844 | 6.994 | −0.1538 | 0.0151 | |
| qPCTR-Y24-3 | 24 | 2.5 | AX-171367948 | AX-171365860 | 3.5175 | 6.0727 | 0.0961 | 0.1057 | |
| qPCTR-Y28 | 28 | 0.7 | AX-171326575 | AX-171286677 | 2.9923 | 5.1918 | −0.0946 | −0.0355 | |
| qPCTR-Y48-2 | 48 | 3.2 | AX-171357778 | AX-171366781 | 3.3065 | 6.2547 | −0.1493 | 0.0242 |
| Environment | QTL Name | LG | GD (cM) | Left Marker | Right Marker | LOD | PVE (%) | Effect F | Effect M |
|---|---|---|---|---|---|---|---|---|---|
| 2021DH Ⅰ | qRSR-R51 | 51 | 20.8 | AX-171263257 | AX-117188968 | 4.3298 | 13.5387 | 0.1165 | −0.1086 |
| qRSR-Y8 | 8 | 3.7 | AX-171329699 | AX-171239513 | 3.7314 | 7.8827 | 0.1537 | 0.0250 | |
| 2021BS Ⅰ | qRSR-Y43-1 | 43 | 1.4 | AX-171352848 | AX-171281749 | 3.1239 | 6.1969 | −0.0471 | 0.1666 |
| 2022Zl Ⅰ | qRSR-R21 | 21 | 9.1 | AX-171350534 | AX-171287731 | 3.1185 | 7.3514 | −0.0233 | 0.1449 |
| qRSR-R25 | 25 | 1.1 | AX-171255139 | AX-171288321 | 3.6249 | 8.5039 | −0.1671 | 0.0768 | |
| qRSR-Y7 | 7 | 9.3 | AX-171313995 | AX-171351245 | 3.8632 | 8.6699 | −0.0769 | 0.0434 | |
| qRSR-Y43-2 | 43 | 29.8 | AX-171313964 | AX-171238129 | 3.3784 | 11.7340 | −0.0796 | 0.1491 | |
| 2022DH Ⅱ | qRSR-R84 | 84 | 11.5 | AX-171312460 | AX-171238357 | 3.2971 | 7.0035 | 0.0014 | −0.2769 |
| qRSR-Y2 | 2 | 5.7 | AX-171350044 | AX-171252982 | 3.8197 | 8.9052 | −0.0218 | −0.1243 | |
| qRSR-Y60 | 60 | 0.7 | AX-171257349 | AX-171272905 | 3.3165 | 6.6547 | 0.2756 | −0.0384 | |
| 2022BS Ⅱ | qRSR-R32-2 | 32 | 15.3 | AX-171261438 | AX-171357712 | 5.6301 | 9.5406 | 0.2679 | −0.1295 |
| QTL Name | Candidate Genes | Gene Description |
|---|---|---|
| qPCTR-Y8-1/(qRSR-Y8) | Soffic.03G0018200-4Ea | Gibberellin-ACID insensitive, repressor of gai1-3 and scarecrow (GRAS) |
| Soffic.03G0018550-2Pa | C2H2 zinc-finger protein (C2H2-ZFP) transcription factors (C2H2) | |
| Soffic.03G0018550-1P a | C2H2 zinc-finger protein (C2H2-ZFP) transcription factors (C2H2) | |
| Soffic.05G0000090-4Pa | ethylene responsive factor (ERF) | |
| Soffic.09G0002040-4Ea | Cysteine3Histidine (C3H) gene family (C3H) | |
| qPCTR-Y8-2 | Soffic.02G0010890-1P b | basic region leucine zipper (bZIP) |
| Soffic.03G0019220-2Eb | Trihelix transcription factors (Trihelix) | |
| Soffic.09G0002240-5Pb | NAC transcription factor (NAC) |
| QTL Name | Candidate Genes | Gene ID in NCBI | E Value | Gene Description |
|---|---|---|---|---|
| qPCTR-Y60/(qRSR-Y60) | Soffic.09G0011210-7Pa | XP_025804889.1 | 0.0 | Protein NRT1/ PTR FAMILY 2.3-like [Panicum hallii] |
| qPCTR-Y8-1/(qRSR-Y8) | Soffic.03G0018620-2E a | NP_001146962.1 | 7 × 10−106 | Ubiquitin-conjugating enzyme E2 5A [Zea mays] |
| Soffic.08G0000260-3Ea | XP_022678687.1 | 4 × 10−43 | zinc finger CCCH domain-containing protein 43-like [Setaria italica] | |
| Soffic.03G0018530-1Pa | XP_002456072.1 | 0.0 | long chain acyl-CoA synthetase 4 [Sorghum bicolor] | |
| Soffic.03G0018510-3Ea | XP_008656712.1 | 0.0 | zinc finger CCCH domain-containing protein 13 isoform X2 [Z. mays] | |
| Soffic.03G0018500-3Ea | NP_001348564.1 | 0.0 | Histone-lysine N-methyltransferase ATX4 [Z. mays] | |
| Soffic.03G0018500-2Ta | AQK97044.1 | 0.0 | Histone-lysine N-methyltransferase ATX3 [Z. mays] | |
| Soffic.03G0018460-3E a | AQK97027.1 | 0.0 | Transducin/WD40 repeat-like superfamily protein [S. bicolor] | |
| Soffic.03G0018330-3Ea | ONM39830.1 | 0.0 | GDSL esterase/lipase [Z. mays] | |
| Soffic.03G0038370-1Ea | XP_002458232.2 | 0.0 | DELLA protein DWARF8 [S. bicolor] | |
| qPCTR-Y8-2 | Soffic.03G0047110-1Eb | XP_002489071.1 | 5 × 10−97 | universal stress protein A-like protein [S. bicolor] |
| Soffic.09G0002260-3P b | NP_001334869.1 | 1 × 10−102 | ubiquitin-conjugating enzyme E2 36 [Z. mays] | |
| Soffic.03G0019180-3E b | PWZ32250.1 | 0.0 | E3 ubiquitin-protein ligase RLIM [Z. mays] | |
| Soffic.03G0019260-3Eb | XP_002458305.1 | 0.0 | tubby-like F-box protein 1 [S. bicolor] | |
| qPCTR-Y28 | Soffic.04G0019040-3Cb | XP_002452590.1 | 0.0 | MACPF domain-containing protein At1g14780 [S. bicolor] |
| Soffic.04G0018650-2Cb | XP_002454299.1 | 0.0 | RING-H2 finger protein ATL46 [S. bicolor] | |
| qPCTR-R9 | Soffic.04G0013030-6F b | XP_002453932.1 | 9 × 10-142 | Germin-like protein 2-4 [S. bicolor] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Xu, F.; Wang, Z.; Wu, Q.; Cheng, W.; Que, Y.; Xu, L. Mapping of QTLs and Screening Candidate Genes Associated with the Ability of Sugarcane Tillering and Ratooning. Int. J. Mol. Sci. 2023, 24, 2793. https://doi.org/10.3390/ijms24032793
Wang T, Xu F, Wang Z, Wu Q, Cheng W, Que Y, Xu L. Mapping of QTLs and Screening Candidate Genes Associated with the Ability of Sugarcane Tillering and Ratooning. International Journal of Molecular Sciences. 2023; 24(3):2793. https://doi.org/10.3390/ijms24032793
Chicago/Turabian StyleWang, Ting, Fu Xu, Zhoutao Wang, Qibin Wu, Wei Cheng, Youxiong Que, and Liping Xu. 2023. "Mapping of QTLs and Screening Candidate Genes Associated with the Ability of Sugarcane Tillering and Ratooning" International Journal of Molecular Sciences 24, no. 3: 2793. https://doi.org/10.3390/ijms24032793