Abstract
The emergence of cancer resistance to targeted therapy represents a significant challenge in cancer treatment. Therefore, identifying new anticancer candidates, particularly those addressing oncogenic mutants, is an urgent medical demand. A campaign of structural modifications has been conducted to further optimize our previously reported 2-anilinoquinoline-diarylamides conjugate VII as a B-RAFV600E/C-RAF inhibitor. Considering the incorporation of a methylene bridge between the terminal phenyl and cyclic diamine, focused quinoline-based arylamides have been tailored, synthesized, and biologically evaluated. Among them, the 5/6-hydroxyquinolines 17b and 18a stood out as the most potent members, with IC50 values of 0.128 µM, 0.114 µM against B-RAFV600E, and 0.0653 µM, 0.0676 µM against C-RAF. Most importantly, 17b elicited remarkable inhibitory potency against the clinically resistant B-RAFV600K mutant with an IC50 value of 0.0616 µM. The putative binding mode of 17b and 18a were studied by molecular docking and molecular dynamics (MD). Moreover, the antiproliferative activity of all target compounds has been examined over a panel of NCI-60 human cancer cell lines. In agreement with cell-free assays, the designed compounds exerted superior anticancer impact over the lead quinoline VII against all cell lines at a 10 µM dose. Notably, both 17b and 18b showed highly potent antiproliferative activity against melanoma cell lines with growth percent under −90% (SK-MEL-29, SK-MEL-5, and UACC-62) at a single dose, while 17b maintained potency with GI50 values of 1.60–1.89 µM against melanoma cell lines. Taken together, 17b, a promising B-RAFV600E/V600K and C-RAF kinase inhibitor, may serve as a valuable candidate in the arsenal of anticancer chemotherapeutics.
1. Introduction
Cancer is a primary life-threatening disease, coming to the fore as the second leading cause of global mortality with more than 600,000 estimated deaths in the United States according to the Centers for Disease Control and Prevention (CDC) in 2021 [1], and ranked sixth worldwide according to the World Health Organization (WHO) in 2019 [2]. Protein kinases and their relevant signaling pathways are essential in regulating cellular proliferation, differentiation, and apoptosis. However, the dysregulation of protein kinase activity due to genetic changes, such as translocations and mutations, along with overexpression, is implicated in the pathogenesis of various types of neoplasms. In this line, identifying new chemical entities targeting protein kinases constitutes a promising therapeutic approach for cancer treatment [3,4].
The RAF family kinases are the key components of the mitogen-activated protein kinase (MAPK) pathway, which encompasses the RAS-RAF-MEK-ERK cascade [5]. RAF has three isomers: A-, B-, and C-RAF. Among them, B-RAF is known as the primary contributor to phosphorylating the downstream MEK kinase. It has been reported that mutations of B-RAF were observed in about 30% of all cancer types. It accounts for ~70% of malignant melanomas, 36–53% of thyroid carcinomas, 30% of ovarian tumors, ~3% of lung cancers, 5–22% of colon cancers, and 1–4% of various other cancer types [6,7,8,9]. B-RAFV600E is the most clinically observed mutant in melanoma, followed by the B-RAFV600K mutant form, which exists in 10–30% of melanoma cases [10]. Dysregulation of either B-RAFV600E or B-RAFV600K promotes excessive cancer cell proliferation, and the role of B-RAFV600K in inhibiting the apoptosis pathway has been reported [11]. C-RAF plays an important role in the phosphorylation of its N-terminal, providing RAF dimerization necessary for pathway progression [12]. However, A-RAF exhibits the weakest influence among the other RAF isoforms toward MEK activation [13,14]. Therefore, medicinal chemistry research on RAF kinase inhibitors is focused on designing new molecules that can inhibit such oncogenic proteins, particularly the mutant forms [15,16,17].
To date, five drugs have been approved by FDA as RAF inhibitors for cancer treatment. (Figure 1) Sorafenib, a diarylurea containing picolinamide [18], and its fluoro congener regorafenib [19] were approved in 2005 and 2013, respectively, as multi-kinase inhibitors targeting VEGFR, PDGFR, B-RAF, RET, and c-KIT [20]. However, they exhibited weak antiproliferative activity in melanoma cells harboring B-RAFV600E mutation [21]. Such a drawback was overcome by the development of the 7-azaindole derivative vemurafenib [22] and the thiazole derivative dabrafenib [23]. These difluorophenyl sulfonamide drugs showed remarkably improved selectivity against the oncogenic B-RAFV600E than its wild type by 100- and 500-fold, respectively [23,24]. However, vemurafenib and dabrafenib trigger a paradoxical activation of the MAPK pathway upon B-RAF inhibition, which results in specific adverse side effects and the emergence of resistance in many patients [14,23]. Therefore, they are clinically used with MEK inhibitors to achieve a more sustained inhibition of the MAPK pathway [24]. Most recently, the diarylpyrazole sulfonamide encorafenib was approved by the FDA in 2018 in combination with the MEK inhibitor binimetinib for the treatment of B-RAFV600E/K-mutant melanoma [25,26]. Encorafenib elicited better potency and had a longer residence time than vemurafenib and dabrafenib against B-RAFV600E cells [21,27].
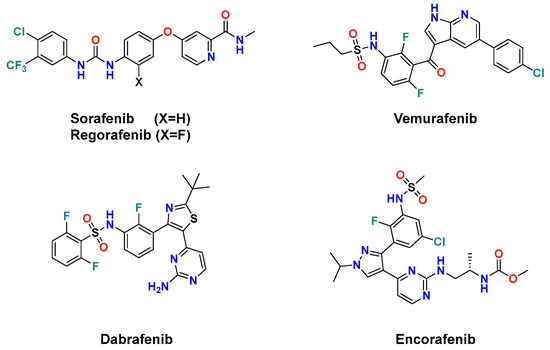
Figure 1.
FDA-approved anticancer B-RAF/C-RAF inhibitors.
Among the plethora of small molecules targeting RAF kinase, diarylamides represent one of the substantial structural features (Figure 2) because of their favorable impact on kinase binding affinity and cellular potency. GNF-7 was reported as a potent AblT315I/B-RAFV600E inhibitor, with IC50 values of 61 and 6.1 nM, respectively [28,29]. The diarylamide tethered pyridine II was designed as a sorafenib analog, and moderately suppressed B-RAF activity (IC50 = 0.531 µM) [30]. The pyrimidine aminoethylpiperazine III was discovered as a pan-RAF inhibitor with two digits nanomolar IC50 values [31]. On the other hand, compounds IV and V, with 4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)pheny moiety, were identified as multiple kinase targeting agents. Compound IV was reported by Cheng et al. [32] as a dual B-RAFV600E/EGFR inhibitor with IC50 values of 51 and 8 nM, respectively. At the same time, the pyrazolopyrimidine V elicited significant inhibitory potency against Src, B-RAFWT/V600E and C-RAF with IC50 values of 0.9, 15, 92, and 27 nM, respectively [33]. The diarylamides quinoline ether VI was reported to possess moderate selective potency toward C-RAF (77% inhibition at 10 µM) [34]. Structural analysis of those diarylamides discloses their similarity, as all feature meta-CF3 group at the terminal phenyl group which contribute to the inhibition activity of cancer-relevant RAF kinases.
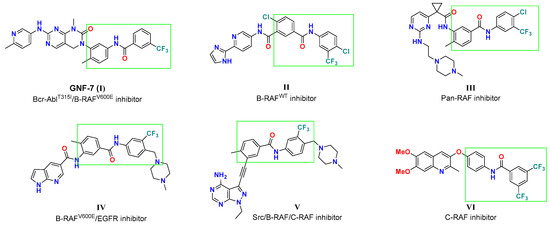
Figure 2.
Representative examples of various diarylamides as RAF kinase inhibitors.
In a previous report, our group identified compound VII (Figure 3), diarylamides aminoquinoline, as a B-RAFV600E/C-RAF inhibitor with promising anti-proliferative activity against NCI-60 cancer cell lines, including melanoma [35]. However, VII exerted moderate enzymatic potency against B-RAFV600E and C-RAF, with IC50 values of 1.46 and 0.370 µM, respectively. In the course of our ongoing endeavors to discover novel potent oncogenic kinase inhibitors [35,36,37,38], in the current investigation we planned to pursue a concise structure–activity relationship (SAR) analysis based on VII as a lead compound to improve its inhibitory potency against B-RAFV600E/C-RAF.
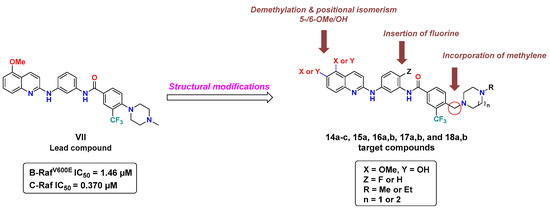
Figure 3.
Structural modifications of lead compound VII for the discovery of novel RAF kinase inhibitors.
As revealed from the docking study of compound VII with B-RAFV600E, the 5-OMe group of quinoline engages with the kinase hinge region by hydrogen bonding, the terminal phenyl substituted with N-methyl piperazine and trifluoromethyl moieties share multiple hydrophobic interactions with DFG motif. In addition, the amide linker participates in a hydrogen bonding interaction with catalytic Glu501 [35]. Considering the role of these structural features in binding interactions, in this study we conducted a campaign of structural modifications to scout their impact on the activity. These molecular alterations of VII included three main aspects addressing the three essential binding regions in the catalytic kinase domain of RAF (Figure 3): (1) the introduction of a methylene bridge between the terminal phenyl ring and the cyclic diamine (N-methyl/N-ethylpiperazine, or N-methyl homopiperazine) that extends to occupy the allosteric pocket, encouraged by the potency of the abovementioned compounds (IV and V), (2) the introduction of fluorine in the central phenyl ring at the ortho-position of amide and the para-position of amine (similar to regorafenib), (3) exploring the role of methoxy and its corresponding hydroxyl group (hinge binding motifs) at both the 5- and 6-position of quinoline. We hypothesized that such structural changes would influence the hydrophobic-, halogen-, and hydrogen-bonding interactions and therefore make a difference in kinase inhibitory potency and cellular activity.
2. Results
2.1. Chemistry
To synthesize the target compounds, a set of three meta-trifluoromethylbenzoic acid derivatives have been synthesized as illustrated in Scheme 1. Fischer esterification of the commercially available 4-methyl-3-(trifluoromethyl)benzoic acid with methanol using a catalytic amount of H2SO4 afforded the benzoate ester 1. Next, the treatment of 1 with N-bromosuccinimide (NBS) and azobisisobutyronitrile (AIBN) in acetonitrile at reflux temperature afforded the corresponding bromobenzyl derivative 2, which then underwent SN2 reaction with the appropriate cyclic diamines in dichloromethane (DCM) using K2CO3 as a base [39] to yield 3a–c. Lastly, base-catalyzed ester hydrolysis of 3a–c was performed to furnish the desired 3,4-disubstitutedbenzoic acid derivatives 4a–c [40].
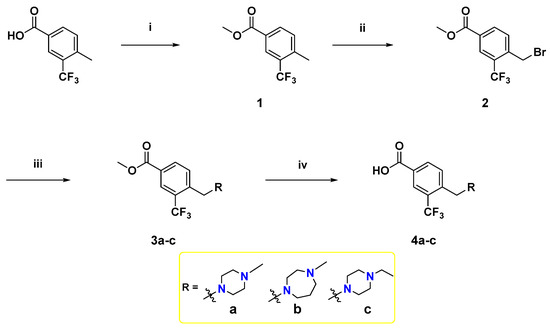
Scheme 1.
Reagents and reaction conditions: (i) methanol, H2SO4 (cat.), reflux, 3 h, 90%; (ii) NBS, AIBN, acetonitrile, reflux, overnight, 70%; (iii) appropriate N-alkyl cyclic diamine, K2CO3, DCM, rt, 2 h, 64%–89%; (iv) NaOH, methanol, 60 °C, 2 h, acid work-up, 76–100%.
On the other hand, 5- or 6-methoxy quinoline-based diamine intermediates 12, 12′, and 13 were synthesized as shown in Scheme 2. Starting from 5-methoxyquinoline (6), which was synthesized following our previous study [41], or its corresponding 6-methoxy isomer 6′, N-oxide intermediates 7 and 7′ were synthesized through oxidation with meta-chloroperoxybenzoic acid (m-CPBA). Treatment of N-oxide 7 with p-tosyl chloride and K2CO3 in DCM afforded the lactam form of quinoline 8. Treatment of 7′ and 8 with POCl3 furnished the 2-chloroquinoline derivatives 9 and 9′. Nucleophilic aromatic substitution of 9 and 9′ with meta-nitroaniline derivative produced the corresponding nitroanilines 10, 10′, and 11. Reduction of these nitro derivatives using palladium on carbon catalyst under hydrogen atmosphere [42] yielded 5- or 6-methoxy quinoline-based diamine intermediates 12, 12′, and 13.
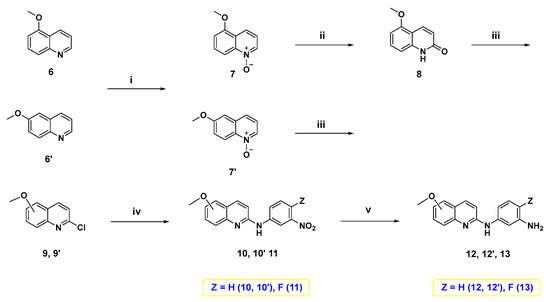
Scheme 2.
Reagents and reaction conditions: (i) m-CPBA, DCM, 0 °C to rt, 3–6 h; (ii) p-TsCl, 10% aq. K2CO3, DCM, rt, 18 h, 65%; (iii) POCl3, reflux, 1 h, 41–86%; (iv) 3-nitroaniline or 4-fluoro-3-nitroaniline, neat, 160 °C, 3 h; (v) Pd/C, H2, methanol, rt, 12 h, 69–81%.
Finally, the target compounds were prepared following Scheme 3. The first six target compounds 14a–c, 15a, 16a, and 16b were obtained by an amide coupling reaction between diamine 12, 12′, and 13, and the proper benzoic acid derivative 4a–c with HATU and N,N-diisopropylethylamine (DIPEA) in N,N-dimethylformamide (DMF) under inert atmosphere. Demethylation of 14a,b, and 16a,b was conducted using BBr3 to afford their corresponding hydroxyl counterparts, 17a,b, and 18a,b.
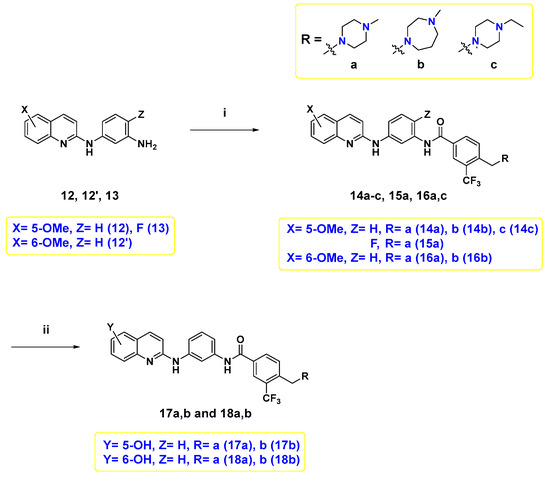
Scheme 3.
Reagents and reaction conditions: (i) 4a–c, HATU, DIPEA, DMF, rt, overnight, 9.4–44%; (ii) 1.0 M BBr3 in DCM, 0 °C to rt, overnight, 4.5–46%.
2.2. In Vitro Kinase Inhibition Assay
The inhibitory effects of all synthesized compounds against B-RAFV600E and C-RAF kinases were investigated using the HotSpotTM kinase assay, at Reaction Biology Corporation (RBC), with GW5074 (structure is shown in Supplementary Materials) as a positive control, and the results are listed in Table 1.

Table 1.
In vitro kinase inhibitory activity (IC50, µM) of the lead compound VII and target compounds against B-RAFV600E and C-RAF a,b,c.
As disclosed from the biochemical assay results, all tested compounds showed promising activities with sub-micromolar IC50 values, which is about 2–10-fold superior to the lead compound VII (Table 1). Comparing the IC50 of VII (B-RAFV600E; 1.46 µM, C-RAF; IC50 = 0.370 µM) and 14a (B-RAFV600E; IC50 = 0.498 µM, C-RAF; IC50 = 0.151 µM), it was found that the incorporation of a methylene bridge between the terminal phenyl and piperazine resulted in improved inhibitory potency against both kinases. The same conclusion applies upon inspecting the activity of 14c and 14b (14b, B-RAFV600E; IC50 = 0.355 µM, C-RAF; IC50 = 0.192 µM; 14c, B-RAFV600E; IC50 = 0.619 µM, C-RAF; IC50 = 0.182 µM), which further emphasized the favorable contribution of the methylene moiety in strengthening the binding interactions with DFG motif of B-RAFV600E and C-RAF kinases. Regarding 15a, in which the hydrogen at the ortho-position to the amide of the central phenyl ring was substituted with fluorine from 14a, inhibition was slightly increased against B-RAFV600E (14a, 15a: B-RAFV600E; IC50 = 0.498, 0.314 µM, respectively), which may stem from the enhanced halogen interaction of fluorine with B-RAFV600E.
Meanwhile, when the connection of the methoxy groups was shifted from 5- to the 6-position of quinoline, there was slight tendency to improve the activity. For example, the 6-hydoxyquinoline 16a (B-RAFV600E; IC50 = 0.193 µM, C-RAF; IC50 = 0.103 µM) exerted better inhibitory potency than its corresponding 6-methyoxy congener 14a (B-RAFV600E; IC50 = 0.498 µM, C-RAF; IC50 = 0.151 µM). This finding might be attributed to the ability of the -OH group to form a tight hydrogen bond interaction with the hinge region of the kinase backbone. Particularly, the substitution of a hydroxyl group at the 5-position of quinoline achieved considerably improved potency towards both B-RAFV600E and C-RAF by 2.8–3.6- and 1.6–2.9-fold, respectively. (14a, 17a: B-RAFV600E; IC50 = 0.498, 0.137 µM, C-RAF; IC50 = 0.151, 0.0950 µM.) Grafting the hydroxyl group of 17a with N-methylpiperazine (B-RAFV600E; IC50 = 0.137 µM, C-RAF; IC50 = 0.0950 µM) to 6-position 18a was favorable for activity (B-RAFV600E; IC50 = 0.114 µM, C-RAF; IC50 = 0.0653 µM); however, this observation was reversed in the case of N-methylhomopiperazine derivatives 17b and 18b. At last, for the change in the R moiety, it was more favorable for the activity to install N-methylhomopiperazine in the case of 5-OMe or 5-OH, and N-methylpiperazine in the case of 6-OMe or 6-OH, but the difference was marginal. A summary of SAR is illustrated in Figure 4.
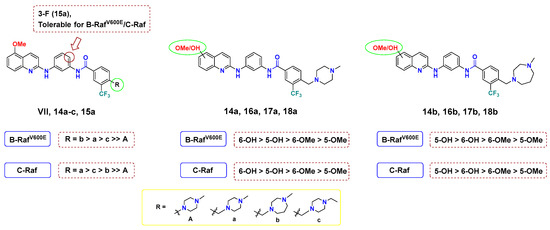
Figure 4.
SAR analysis of final compounds 14a–c, 15a, 16a,b, 17a,b and 18a,b for B-RAFV600E/C-RAF.
Taken together, it is thought that a change in the interaction with B-RAFV600E and C-RAF kinases, caused by additional methylene and a hydroxyl group, greatly influenced activity. Accordingly, two highly potent B-RAFV600E and C-RAF inhibitors, 17b and 18a, have been discovered. To gain insight into the inhibitory mode of this new array of quinolines, 18a was tested at 1 µM concentration of ATP to compare it with its IC50 at 10 µM (Table 2). As shown in Table 2, 18a showed IC50 values of 0.0843 and 0.0293 µM against B-RAFV600E and C-RAF, respectively, at 1 µM ATP. This biochemical outcome points out that the IC50 values vary depending on the ATP concentration, which indicates that 18a acts as a competitive ATP kinase inhibitor. Moreover, the inhibitory activity of 17b and 18a was tested against the clinically resistant B-RAFV600K, for which many RAF inhibitors are inactive. Interestingly, both 17b and 18a elicited promising inhibitory potency against B-RAFV600K, with IC50 values of 61.6 and < 0.5 nM, respectively.
Table 2.
In vitro kinase inhibitory activity (IC50, µM) of the most potent compounds, 17b and 18a, against B-RAFV600E/V600K and C-RAF a,b,c.
2.3. In Vitro Screening of the Anticancer Activities
2.3.1. Single Dose Testing against NCI-60 Cell Line Panel
Encouraged by the promising results in the cell-free assay, the antiproliferative activity of all final compounds was evaluated by the National Cancer Institute (NCI) in a single dose of 10 µM over a library of 60 human cancer cell lines representing nine cancer types; leukemia, non-small cell lung (NSCL), colon, central nervous system (CNS), melanoma, ovarian, renal, prostate, and breast cancer.
Generally, all target compounds, except 18b, showed significantly improved antiproliferative effects in almost all cancer cell lines (Figure 5). Compared with the lead compound Vll, 14a–c with methylene between the bridge of the terminal phenyl ring and the R groups were endowed with broad growth inhibitory activity. In particular, the antiproliferative activity against melanoma, which is mostly driven by RAF mutations, was greatly improved as indicated by −100 up to −70 growth percent (minus growth percent indicates cell lethal effect). Such cellular outcomes were in agreement with the findings observed in the biochemical kinase assay (Table 1). Furthermore, 14a–c exhibited cell growth percents below −90 in some cell lines belonging to NSCL, colon, CNS, ovarian, renal, and breast cancer, and even caused more sound lethal effects towards some cell lines with growth percent −100, including NCI-H322M (NSCL cancer), HCT-15 (colon cancer), SF-539, SNB-19 (CNS cancer), LOX IMVI, MDA-MB-435, SK-MEL-28, SK-MEL-5 (melanoma), OVCAR-5 (ovarian cancer), ACHN and CAKI-1 (renal cancer).
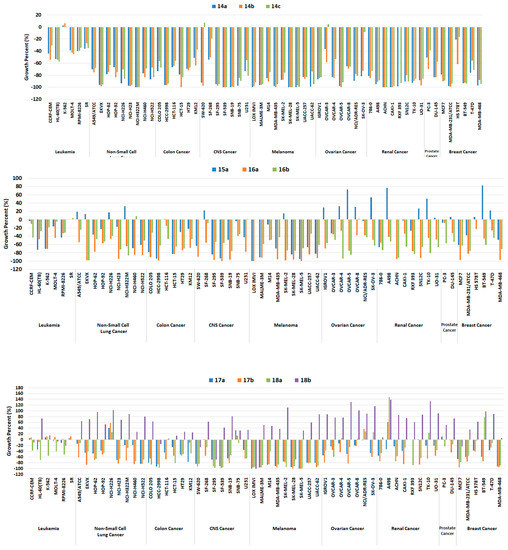
Figure 5.
Growth percentage of the NCI-60 human tumor cell lines upon treatment with 10 μM of compounds 14a–c (upper panel), 15a, 16a, and 16b (middle panel), 17a, 17b, 18a, and 18b (lower panel).
As for 15a, overall antiproliferative activity was relatively decreased compared to 14a–c; in particular, although the inhibition ability against B-RAFV600E of 15a was slightly greater, it did not inhibit the growth of melanoma cells, except LOX IMVI and UACC-62 in a better fashion. Additionally, both 6-methoxyquinoline-based diarylamides 16a and 16b showed considerable worthy growth inhibitory ability over all cell lines. In particular, 16a was slightly better than 16b against all melanoma cells, which is the same trend observed with the kinase assay findings.
Meanwhile, the growth percent of compounds 17a, 17b, and 18a, in which the methoxy group was replaced with the hydroxyl group, was moderately decreased for most cells; however, it maintained at a high level for melanoma cell lines (Figure 5). They were lethal to some melanoma cells against LOX IMVI, SK-MEL-28, and SK-MEL-5 with growth percent −100, and against MALME-3M, MDA-MB-435, SK-MEL-2, and UACC-62 cell lines, the growth percent values were less than -90. On the other hand, unlike other related compounds, the growth percentage of 18b was greatly reduced for all cells, and although it had a lethal effect against the LOX IMVI cell line, it lost the antiproliferative ability to other melanoma cell lines greatly. This finding may be attributed to the decreased RAF inhibitory potency of 18b.
2.3.2. Five-Dose Testing against NCI-60 Cell Line Panel
To deeply investigate the antiproliferative activity of final compounds, the further assay was processed in a five-dose testing mode, using sorafenib as a reference compound (Table 3 and Table 4). In a similar fashion to that observed in the single dose assay, compounds 14a–c demonstrated distinct antiproliferative ability while showing excellent GI50 spanning the range of 1.20–11.8 µM for all cancer cell lines. Towards melanoma cell lines, except SK-MEL-2, GI50 values of 14a–c were less than 2.0 µM concentration, while over the majority of the other cell lines, including leukemia, NSCL, colon, CNS, renal, and breast cancer, 14a–c showed a similar level of GI50.
Table 3.
GI50 (μM) of compounds 14a–c, 15a and 16a against NCI-60 human tumor cell lines in five-dose test a,b.
Table 4.
GI50 (μM) of compounds 17a, 17b, 18a, 18b, and Sorafenib against NCI-60 human tumor cell lines in five-dose test a,b,c.
Contrary to the single-dose mode, the antiproliferative property of 15a significantly reduced GI50 values of 3.61–18.8 µM for all cells and 11.7–16.3 µM for melanoma cells, respectively, despite showing slightly better inhibition of B-RAFV600E when compared with 14a. We speculate that 15a may be promising at the molecular level, yet has a poor growth inhibitory potency at the cellular level, which may be due to low solubility/or permeability endowed by fluorine. This discrepancy seems to be observed in the case of 16a as well. Comparing the results of 14a and 16a, the antiproliferative ability of 16a decreased considerably against all cancer cell lines, except 12-cell lines (leukemia; K-562, SR, NSCL; HOP-92, NCI-H226, colon; HCT-15, HT29, CNS; SNB-75, melanoma; LOX IMVI, ovarian; SK-OV-3, breast; MCF7, BT-549, MDA-MB-468), only by the change of the methoxy group substitution position, from 5 to 6, of quinoline. Specifically, for melanoma cells, antiproliferative activity potency was maintained for only LOX IMVI cells (GI50 = 1.55 µM), but not for the other cells. Likewise, 16a showed better kinase inhibition activity compared to 14a for B-RAFV600E and C-RAF (2.6-fold and 1.5-fold, respectively); we expected that the result of 16a will also have a similar reason as 15a.
Referring to the hydroxyquinolines, the antiproliferative potency of 17a,b tended to exert lower potency as indicated by GI50 values over 2.0 µM against all leukemia cell lines, except MOLT-4 (GI50 = 1.86 µM). These observations were similar against NSCL cancer cell lines, and in particular, for NCI-H226, the cellular potency was decreased by about 3-fold at least (14a, 17a; GI50 = 4.63, 19.2 µM, respectively). For colon and CNS cancer cells, the moderate GI50 values were maintained at a maximum of 3.81 µM (17a; SF-268 of CNS) or less. Fortunately, against melanoma cells, the potent antiproliferative activity was maintained. In particular, against SK-MEL-2, 17a and 17b elicited slightly better antiproliferative ability (14a, 17a: GI50 = 3.52, 1.92 µM, respectively), although against the M14 cell line, the GI50 of 17a was decreased slightly (14a, 17a: GI50 = 1.76, 2.21 µM, respectively). For the rest of the cancer cell lines, a similar trend of GI50 values was noticed. However, the results of representative compounds 18a and 18b with the 6-OH group turned out to be disappointing. These compounds generally showed reduced antiproliferative activity compared to 17a and 17b, and a noticeably modest effect against NSCL and colon cell lines (GI50 against NSCL and colon: 3.39–17.8 µM and 2.09–18.5 µM, respectively.). Additionally, for CNS cancer cell lines, antiproliferative activity was decreased by at least 2.7-fold; however, 18a showed significant potency with a submicromolar GI50 value of 0.656 µM against SNB-75. While the GI50 values of 18a against the LOX IMVI, MALME-3M, and SK-MEL-28 cell lines remained at a similar level to 17a, maintaining GI50 1.59–1.79 µM, the GI50 values were increased by 1.37–9.11-fold against other melanoma cell lines. Strangely, the cellular potency of 18b was somehow modest, with a GI50 range 10.4–17.2 µM, and both 18a and 18b also exhibited less efficacy in inhibiting cell growth against the rest of the cancer cell lines. Based on the findings of the kinase assay, 18b could be expected to have a reduced antiproliferative ability against melanoma cells, but although 18a was the most potent against B-RAFV600E, its antiproliferative ability against melanoma cells was shown to be insufficient. Therefore, the 6-OMe group of quinoline may be influential at kinase inhibition but is associated with a decrease in antiproliferative activity in the cells. Based on these findings, we identified 17b as the most potent member showing excellent inhibitory ability even on cancer cells, and its broad antiproliferative spectrum offered the possibility that it can target other molecular targets in the cellular context.
While comparing with sorafenib, the first FDA-approved RAF inhibitor, 17b was more potent than sorafenib in some cell lines. Against colon cancer, 17b exerted slightly better potency than sorafenib, except KM12 cells, with GI50 values less than 2.0 μM. A similar finding was also noticed for melanoma cells. Except for SK-MEL-5 cells, 17b had better GI50 values of less than 2.0 μM over all cell lines. Interestingly, 17b elicited superior antiproliferative activity than sorafenib against all renal cancer cell lines, except A498 cells. Since sorafenib is clinically used for the treatment of renal cell carcinoma, these findings suggested hope that 17b may be a promising candidate for renal cancer. On the other hand, the antiproliferative potency of 17b was reduced against ovarian cancer cell lines, except IGROV1, OVCAR-3, and OVCAR-8. At the same time, its anticancer activity was comparable to sorafenib over the remaining tested cell lines.
2.4. Molecular Docking Study
In order to rationalize the observed RAF kinase inhibitory results from a 3D structural perspective, 14b, 16a, 17b, and 18a were docked in the catalytic kinase domains of B-RAFV600E (PDB code: 1UWJ) [44] and C-RAF homology models. Molecular docking studies were performed by inserting quinoline-based diarylamide compounds into the sorafenib binding sites of B-RAFV600E and C-RAF homology models. Both B-RAFV600E and C-RAF homologous proteins bound well and showed similar binding modes (Figure 6 and Figure 7, Figures S1–S5).
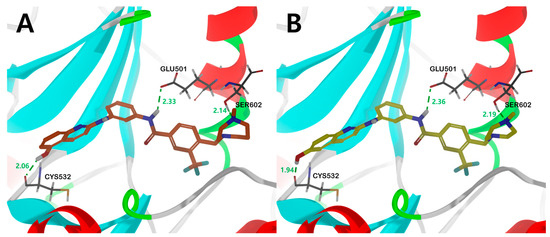
Figure 6.
The binding mode of compound (A) 17b and (B) 18a in the catalytic kinase domain of B-RAFV600E model. For clarity purposes, only key residues having interactions were shown. Compounds 17b and 18a are shown in orange and yellow colored stick models, respectively. Fundamental hydrogen bonding interactions are shown in green dashes. The number near the dashes indicates the bonding distance in units of Å.
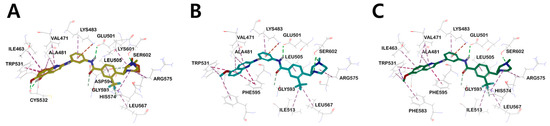
Figure 7.
(A) Three-dimensional structural overlay of compounds 17b (orange) and 18a (yellow) inside B-RAFV600E protein. The binding mode of compound (B) 14b (sky blue), (C) 16a (green) in the B-RAFV600E protein. Inhibitors are shown in the stick model, and surrounding residues are shown in the line models. Various interactions are shown in dashes.
Compounds 17b and 18a, with sound kinase effects, were calculated to have a nearly similar binding mode to the B-RAFV600E protein. The three significant hydrogen bonds are (1) between the hydrogens of the amine moiety in the central amide bond and the side-chain carboxylate of the catalyst Glu501, (2) between the hydroxyl hydrogens of the quinoline moiety and the amino carbonyl oxygens of Cys532 in the hinge region, and (3) between the hydroxyl group of Ser602 and the nitrogen of N-methylpiperazine or N-methyl-1,4-diazepane (Figure 6).
Interestingly, in the case of 14b and 16a, the methoxy derivatives of 17b and 18a, no major hydrogen bond with the Cys532 residue was found. Through this, it was suggested that the substituent providing hydrogen that can form a hydrogen bond with Cys532 plays an essential role in kinase efficacy (Figure 6).
Similar to the results of docking simulations in previous studies, these quinoline moieties consistently occupy the ATP adenine binding site of B-RAFV600E through π–alkyl interactions with surrounding hydrophobic moieties in all compounds. The aromatic residue of quinoline π–π interacts with Trp531 of the hinge region and quinoline π–alkyl interacts with the side-chain alkyl group of Val471, Ile463, and Ala481 of the P-loop in B-RAFV600E. The central phenyl ring of the inhibitors interacts with the aliphatic side chain of Lys483. The carbon–hydrogen bonds were formed between the hydrogen of the carboxyl group in the central amide bond and the hydrogen of Cαin Gly593. The trifluoromethyl phenyl ring shared π–alkyl interactions with the side-chain alkyl group of Leu505. For heterocyclic ring sizes, such as N-methylpiperazine and N-methyl-1,4-diazepane, no major binding mode that could affect the efficacy with B-RAF was found, and the alkyl interaction between the heterocyclic ring and Arg575 was common. Therefore, through docking studies, it can be suggested that the hydroxyl group at the 5- and 6-positions of quinoline has a greater effect on RAF kinase efficacy than the effect of the bulky heterocyclic ring. The mode of binding between C-RAF kinase and inhibitors 17b and 18a is mainly similar to that of B-RAFV600E. However, among the three primary hydrogen bonds, only two hydrogen bonds between the hydrogen of the amine residue in the central amide bond and the side chain carboxylate of the catalyst Glu393, and between the hydroxyl hydrogen of the quinoline residue and the amino carbonyl oxygen of Cys424 were shown. Interestingly, the distances of major hydrogen bonds were generally shorter in C-RAF than in B-RAFV600E, suggesting a tighter bond between inhibitor and kinase in C-RAF than in B-RAFV600E (Figure S3).
2.5. Molecular Dynamics
Molecular simulations were performed to investigate the stability of 17b and 18a binding to C-RAF and B-RAFV600E, respectively. Both compounds interacted with each protein maintaining relative stability (RMSD value < 2.5 Å, Figure 8). Even when the MD simulation execution time was increased to 120 ns, the fluctuation values of the RMSD values of the two compounds 17b and 18a were not large (Figure S6). Through this, it can be seen that the two compounds and the B-RAFV600E protein reached a relatively stable interaction state.
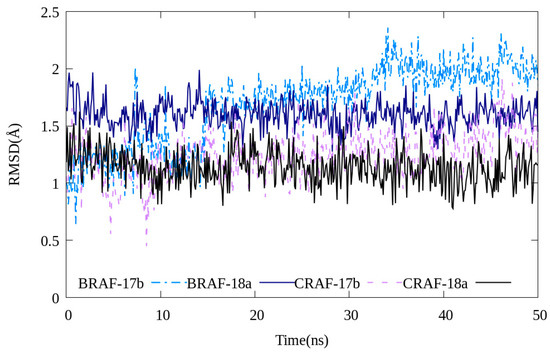
Figure 8.
RMSD values of compounds 17b and 18a during 50 ns of molecular dynamics simulations.
In addition, the bonding distances of the three major hydrogen-bonding interactions between ligands and proteins presented in molecular docking studies (Figure 6) were monitored by MD simulations (Figures S7 and S8). Among them, the Cys532 and Glu501 residues showed very small distance changes, confirming that these two hydrogen bonds are the main interaction sites of ligand–protein bonding. In the case of the Ser602 residue, the distance change was calculated to be large compared to the other two residues. This is interpreted as a less stable bond than the other two hydrogen bonds because the Ser602 residue was modeled as a missing residue using the GalaxyFill algorithm and used for calculation and is also located in a ring with relatively many tertiary structural changes.
Table 5 lists the calculated binding free energies of the most potent compounds 17b and 18a. Based on the RMSD plot, it was confirmed that the docking pose of each inhibitor did not deviate significantly from the initial state during the simulation process. In addition, the molecular dynamics simulation results show good agreement with the IC50 results. In both proteins, 18a produces a smaller binding free energy, indicating a higher binding affinity. In addition, all of the selected compounds obtained a smaller binding free energy to C-RAF than to B-RAFV600E, indicating that the compounds showed stronger binding to CRAF.
Table 5.
Calculated binding free energy of compounds 17b and 18a against B-RAFV600E and C-RAF.
3. Conclusions
In this report, a concise set of ten new quinoline-based diarylamides have been designed and synthesized to improve the RAF inhibitory potency of our previously reported lead VII. Herein, multidimensional structural modifications of lead compound Vll have been performed, including the introduction of methylene between the terminal phenyl ring and cyclic diamine, the substitution of fluorine and installing hydroxyl groups at the central phenyl ring, and the 5-/6-position of quinoline, respectively. All the target compounds have been tested against B-RAFV600E and C-RAF at a 10 µM concentration of ATP. The SAR study pointed out that the additional methylene and hydroxyl group resulted in notable increased inhibitory activity. Among the newly designed members, 17b and 18a emerged as the most potent compounds against B-RAFV600E and C-RAF. Moreover, 17b elicited nanomolar potency against the clinically resistant mutant B-RAFV600K. Compound 18a showed a better IC50 value at 1 µM concentration of ATP, indicating that these compounds are ATP-competitive kinase inhibitors. Single-dose assay of all compounds over a panel of NCI-60 human cancer cell lines indicated that, consistent with the kinase assay, additional methylene led to a broad antiproliferative advantage. In the five-dose testing mode, 17b maintained highly potent antiproliferative activity against melanoma cell lines. Given both cell-free and cell-based assay findings, 17b was discovered as a promising RAF-inhibitor candidate with the potential for cancer treatment, especially oncogenic RAF-derived melanoma.
4. Materials and Methods
4.1. Chemistry
General: All reagents and anhydrous solvents were purchased from Sigma-Aldrich Korea, TCI, and Combi-blocks and used without further purification. Unless otherwise noted, the reactions were carried out under an argon atmosphere and monitored on thin-layer chromatography (TLC) plates (Merck, silica gel 60 F254). TLC plates were observed using UV light (254 nm and 365 nm) and ninhydrin or phosphomolybdic acid (PMA) staining solution. Silica gel (Merck, 230–400 mesh) was used for flash column chromatography. 1H and 13C nuclear magnetic resonance (NMR) spectra were obtained by measuring with Bruker Advance III 400 MHz spectrometers, and CDCl3-d, methanol-d4, DMSO-d6 and acetone-d6 from Cambridge Isotope laboratories were used as NMR solvents. Chemical shift (δ) was recorded in ppm [45] using tetramethylsilane (TMS) as an internal standard, and the abbreviations s (single), d (doublet), t (triplet), m (multiplet), br. s (broad singlet), dd (doublet of doublet), or dt (doublet of triplet). The coupling constant (J) was recorded in hertz (Hz). High-resolution mass spectra (HRMS) were obtained by measuring with Waters SYNAPT G2 mass spectrometer ESI-TOF mode. Compounds 6, 9, and 12 were prepared following the reported procedures references [35,37].
4.2. Synthesis
4.2.1. Methyl 4-methyl-3-(trifluoromethyl)benzoate (1)
To a solution of 4-methyl-3-(trifluoromethyl)benzoic acid (5.00 g, 24.5 mmol) in anhydrous methanol (80.0 mL), H2SO4 (8.50 mL) was added dropwise. The reaction mixture was stirred under reflux overnight, cooled down, and the solvent was evaporated under reduced pressure. NaHCO3 (aq.) was added dropwise to the residue in an ice bath until slightly basic (pH = 8), then extracted with ethyl acetate (20 × 3 mL). The combined organic layer was washed with brine, and dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure to afford the titled product 1 (4.83 g, 90% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 8.07 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 3.92 (s, 3H), 2.53 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 165.89, 142.06 (d, J = 1.2 Hz), 132.64, 132.20, 129.24 (q, J = 30 Hz), 128.26, 127.10 (q, J = 5.6 Hz), 124.18 (q, J = 272 Hz), 52.22, 19.39 (d, J = 1.9 Hz).
4.2.2. Methyl 4-(bromomethyl)-3-(trifluoromethyl)benzoate (2)
To a solution of 1 (4.39 g, 20.1 mmol) in anhydrous acetonitrile (15.0 mL), NBS (4.30 g, 24.1 mmol) and AIBN (1.30 g, 7.92 mmol) were added and stirred under reflux overnight. The reaction mixture was cooled, and distilled H2O was added to quench the reaction, then extracted with ethyl acetate (20 × 3 mL). The combined organic layer was washed with brine solution, and dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure to afford residue 2 (4.18 g, 70% yield) as a yellow oil. The residue was used in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.20 (dd, J = 8.1, 1.4 Hz, 1H), 7.69 (d, J = 8.1 Hz, 1H), 4.64 (s, 2H), 3.96 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 165.35, 140.91, 133.34, 133.11, 130.57, 128.64 (q, J = 31 Hz), 127.63 (q, J = 5.7 Hz), 123.71 (q, J = 273 Hz), 52.68, 27.48 (d, J = 2.6 Hz).
4.2.3. General Procedure for the Synthesis of Benzoate 3a–c
To a solution of 2 (1.0 eq.) and appropriate cyclic amine (3.0 eq.) in anhydrous DCM (20 mL), K2CO3 (3.0 eq) was added. The reaction mixture was stirred at room temperature for 2 h, then extracted with distilled water (20 × 3 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2SO4, and filtered. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel flash column chromatography using proper eluent solvent to get the 3a–c.
- Methyl 4-((4-methylpiperazin-1-yl)methyl)-3-trifluoromethyl) (3a)
The silica gel flash column chromatography was conducted using MeOH:28% NH3(aq.):EA = 0.5:1:98.5 to get the 3a (1.90 g, 64% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 8.15 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 3.93 (s, 3H), 3.69 (s, 2H), 2.51–2.45 (m, 8H), 2.29 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 166.09, 143.52, 132.82, 130.63, 129.09, 129.07 (q, J = 31 Hz), 127.32 (q, J = 5.9 Hz), 124.14 (d, J = 272 Hz), 58.20 (d, J = 1.5 Hz), 55.38, 53.44, 52.55, 46.25.
- Methyl 4-((4-methyl-1,4-diazepan-1-yl)methyl)-3-(trifluoromethyl)benzoate (3b)
The silica gel flash column chromatography was conducted using MeOH:28% NH3(aq.): EA = 3:1:96 to get the 3c (1.05 g, 83% yield) as a yellow oil. H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 8.16 (dd, J = 8.1, 1.1 Hz, 1H), 7.97 (d, J = 8.1 Hz, 1H), 3.93 (s, 3H), 3.93 (s, 2H), 2.75–2.72 (m, 6H), 2.68–2.65 (m, 2H), 2.40 (s, 3H), 1.87 (quintet, J = 6.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 165.93, 144.34, 132.67, 130.53, 128.88, 128.67 (q, J = 31 Hz), 127.16 (q, J = 5.9 Hz), 124.00 (q, J = 272 Hz), 58.38, 58.36, 58.29, 56.63, 54.66, 54.58, 52.38, 46.86, 27.37.
- Methyl 4-((4-ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)benzoate (3c)
The silica gel flash column chromatography was conducted using hexane:ethyl acetate = 1:1 to obtain 3b (1.59 g, 89% yield) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.16 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H), 3.94 (s, 3H), 3.70 (s, 2H), 2.53–2.41 (m, 10H), 1.09 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.05, 143.51, 132.76, 130.60, 129.02 (q, J = 31 Hz), 129.01, 127.26 (q, J = 5.9 Hz), 124.09 (q, J = 272 Hz), 58.19, 53.42, 53.00, 52.50, 52.44, 12.16.
4.2.4. General Procedure for the Synthesis of Benzoic Acid (4a–c)
A suspension of 3a, 3b, or 3c (1.0 eq.) in anhydrous methanol (20 mL) was cooled in an ice bath. 2.0 N NaOH (aq.) (25 mL) was added dropwise for 15 min. The reaction mixture was stirred and heated at 60 °C overnight, then cooled down. The solvent was evaporated under reduced pressure, then 1 N HCl (aq.) was added dropwise to the residue in the ice bath until a slightly acidic pH = 5–6 value. The solvent was evaporated under reduced pressure, then methanol (10 mL) and DCM (10 mL) were added. The precipitate was filtered, and the filtrate was evaporated to obtain the 4a–c as a light yellow (4a and 4b) or white solid (4c) produt.
- 4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)benzoic acid (4a)
94% yield; 1H NMR (400 MHz, Methanol) δ 8.16 (s, 1H), 8.03 (d, J = 8.0 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H), 3.66 (s, 2H), 3.10 (br. s, 4H), 2.70 (s, 3H), 2.63 (br. s, 4H); 13C NMR (100 MHz, methanol) δ 172.56, 140.36, 137.50, 133.98, 131.78, 129.45 (q, J = 30 Hz), 128.07 (q, J = 5.6 Hz), 125.92 (q, J = 272 Hz), 58.50, 54.94, 51.24, 43.72.
- 4-((4-methyl-1,4-diazepan-1-yl)methyl)-3-(trifluoromethyl)benzoic acid (4b)
100% yield; 1H NMR (400 MHz, methanol) δ 8.27 (s, 1H), 8.15 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 3.90 (s, 2H), 3.43 (t, J = 5.4 Hz, 2H), 3.28 (methanol-d4 overlap, 2H), 2.91–2.89 (m, 5H), 2.79 (t, J = 6.2 Hz, 2H), 2.07 (quintet, J = 5.8 Hz, 2H); 13C NMR (100 MHz, methanol-) δ 170.60, 142.70, 134.80, 134.13, 131.96, 129.40 (q, J = 30 Hz), 128.03 (q, J = 5.8 Hz), 125.74 (d, J = 272 Hz), 59.22, 58.44, 56.35, 55.01, 50.96, 49.88, 45.06, 25.70.
- 4-((4-ethylpiperazin-1-yl)methyl)-3-(trifluoromethyl)benzoic acid (4c)
76% yield; 1H NMR (400 MHz, Methanol) δ 7.92 (s, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 3.50 (s, 2H), 3.03–2.90 (m, 6H), 2.51 (br. s, 4H), 1.07 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, methanol) δ 172.54, 140.10, 137.85, 133.89, 131.68, 129.99, 129.44 (q, J = 30 Hz), 128.04 (q, J = 6.0 Hz), 125.92 (d, J = 271 Hz), 58.54, 52.99, 52.85, 51.38, 9.82.
4.2.5. 2-Chloro-6-methoxyquinoline (9′)
A solution of 6-methoxyquinoline (5.0 g, 30.8 mmol) in anhydrous DCM (20.0 mL) was cooled in an ice bath, then m-CPBA (77%, 10.4 g, 46.2 mmol) was added. After the reaction mixture was stirred at room temperature for 3 h, saturated NaHCO3 aqueous solution was added to render the solution basic pH = 8. The aqueous layer was extracted with ethyl acetate (20 × 3 mL), and the combined organic layers were washed with brine, dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure to obtain a residue for the next step. After the residue was cooled in an ice bath, POCl3 (9.0 mL) was added, heated at 110 °C, and stirred for 1 h. To the reaction mixture, ammonium hydroxide solution was added to alkalinize (pH = 8), and the aqueous layer was extracted with DCM (20 × 3 mL). The combined organic layers were washed with brine solution, dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure to afford a residue which was purified by silica gel flash column chromatography (Hexane:DCM = 1:3) to obtain the 9′ (2.42 g, 41% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 8.6 Hz, 1H), 7.91 (d, J = 9.2 Hz, 1H), 7.38 (dd, J = 9.2, 2.8 Hz, 1H), 7.33 (d, J = 8.6 Hz, 1H), 7.06 (d, J = 2.8 Hz, 1H), 3.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 158.07, 148.00, 143.78, 137.61, 129.90, 127.89, 123.04, 122.46, 105.25, 55.59.
4.2.6. 4-Fluoro-N1-(5-methoxyquinolin-2-yl)benzene-1,3-diamine (12′)
A mixture of 2-chloro-5-methoxyquinoline (300 mg, 1.55 mmol) and 4-fluoro-3-nitroaniline (242 mg, 1.55 mmol) was heated at 160 °C. After 3 h, the reaction mixture was cooled, then anhydrous methanol (20.0 mL) and palladium on carbon (10%, 33.0 mg, 0.03 mmol) were added and stirred under H2 atmosphere at room temperature. After 12 h, the reaction mixture was filtered over celite, and distilled water was added to the filtrate. The aqueous layer was extracted with ethyl acetate (20 × 3 mL), and the combined organic layer was washed with brine, dried over anhydrous Na2SO4, and evaporated under reduced pressure to afford a residue which was purified by silica gel flash column chromatography (hexane:ethyl acetate = 3:1, then switching to 1:1) to obtain the 12′ (335 mg, 69% yield) as a dark brown color solid. 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 8.6 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.35 (d, J = 7.7 Hz, 1H), 6.96–6.88l (m, 3H), 6.68 (d, J = 7.7 Hz, 2H), 3.95 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 155.54, 155.05, 148.39 (d, J = 234 Hz), 147.80, 136.21 (d, J = 2.6 Hz), 135.11 (d, J = 14 Hz), 132.98, 130.17, 118.27, 115.60 (d, J = 20 Hz), 115.57, 111.65 (d, J = 6.7 Hz), 110.26 (d, J = 3.3 Hz), 102.03, 55.63.
4.2.7. N1-(6-Methoxyquinolin-2-yl)benzene-1,3-diamine (13)
A mixture of 9′ (500 mg, 2.58 mmol) and 3-nitroaniline (357 mg, 2.58 mmol) was heated at 160 °C overnight. After 3 h, the reaction mixture was cooled, then anhydrous MeOH (20.0 mL) and palladium on carbon (10%, 137 mg, 0.129 mmol) were added and stirred under H2 atmosphere at room temperature. After 22 h, the reaction mixture was filtered over celite, and distilled water was added to the filtrate. The aqueous layer was extracted with ethyl acetate (20 × 3 mL), and the combined organic layer was washed with brine, dried over anhydrous Na2SO4, and filtered. The solvent was evaporated under reduced pressure to afford a residue which was purified by silica gel flash column chromatography (hexane:ethyl acetate = 3:1, then switching to 1:1) to obtain the 13 (552 mg, 81% yield) as a yellow color solid. 1H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 9.0 Hz, 1H), 7.63 (d, J = 9.1 Hz, 1H), 7.21 (dd, J = 9.1, 2.8 Hz, 1H), 7.07 (t, J = 7.9 Hz, 1H), 7.02 (d, J = 9.0 Hz, 1H), 6.95 (d, J = 2.8 Hz, 1H), 6.87 (t, J = 2.1 Hz, 1H), 6.38 (dd, J = 7.6, 1.8 Hz, 1H), 3.84 (s, 3H), 2.81 (br. s, 2H); 13C NMR (100 MHz, CDCl3) δ 155.44, 153.27, 147.26, 142.80, 141.35, 136.83, 129.93, 127.46, 124.47, 121.37, 112.14, 110.77, 110.05, 107.07, 106.30, 55.47.
4.2.8. General Procedure for Synthesis 14a–c, 15a, 16a, and 16b
To a solution of 12, 12′ or 13 (1.0 eq.), 4a–c (1.1 eq.), and HATU (1.5 eq.) in anhydrous DMF (1.0 mL), DIPEA (4.0 eq.) was added, then the reaction mixture was stirred at room temperature overnight. The reaction was quenched by adding a saturated NaHCO3 aqueous solution. The aqueous layer was extracted with ethyl acetate (20 × 3 mL), then the combined organic layer was washed with brine, dried over anhydrous Na2SO4, and filtered. The solvent was evaporated under reduced pressure to afford a residue which was purified by silica gel flash column chromatography using proper eluent to obtain the desired 14a–c, 15a, 16a, and 16b.
- 4-((4-methylpiperazin-1-yl)methyl)-N-(3-((5-methoxyquinolin-2-yl)amino)phenyl)-3-(trifluoromethyl)benzamide (14a)
The silica gel flash column chromatography was conducted using MeOH:DCM = 1:19 eluent to obtain the desired 14a (116 mg, 80% yield) as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.52 (s, 1H), 8.29 (d, J = 9.2 Hz, 1H), 8.13 (s, 1H), 8.02–7.99 (m, 2H), 7.88 (d, J = 8.0 Hz, 1H), 7.47–7.27 (m, 6H), 6.90 (d, J = 9.2 Hz, 1H), 6.64 (d, J = 7.6 Hz, 1H), 3.94 (s, 3H), 3.67 (s, 2H), 2.50 (br. s, 8H), 1.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.94, 164.62, 155.46, 154.38, 148.46, 142.05, 141.16, 138.47, 133.69, 132.52, 130.77, 130.30, 129.81, 129.68, 129.17, 125.32, 124.83 (d, J = 5.5 Hz), 119.19, 116.24, 115.94, 114.61, 111.88, 110.95, 102.07, 57.92, 55.60, 55.17, 53.17, 46.03; HRMS (ESI-TOF) m/z calcd for C30H31F3N5O2 [M + H]+: 550.2430, found: 550.2427.
- N-(3-((5-methoxyquinolin-2-yl)amino)phenyl)-4-((4-methyl-1,4-diazepan-1-yl)methyl)-3-(trifluoromethyl)benzamide (14b)
The silica gel flash column chromatography was conducted using MeOH:DCM = 2:98 eluent to obtain the desired 14b (36.7 mg, 20% yield) as a light yellow solid. 1H NMR (400 MHz, methanol) δ 8.37 (s, 1H), 8.32 (d, J = 8.8 Hz, 1H), 8.28 (s, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.56–7.54 (m, 1H), 7.47 (t, J = 8.2 Hz, 1H), 7.36–7.32 (m, 3H), 6.98 (d, J = 9.2 Hz, 1H), 6.76 (d, J = 8.0 Hz, 1H), 3.97 (s, 3H), 3.95 (s, 2H), 3.46 (t, J = 5.4 Hz, 2H), 3.35–3.33 (m, 2H), 2.94–2.92 (m, 5H), 2.80 (t, J = 6.1 Hz, 2H), 2.07 (quintet, J = 5.7 Hz, 2H);13C NMR (100 MHz, methanol) δ 167.10, 156.96, 156.24, 149.26, 142.95, 142.89, 140.21, 135.82, 133.36, 132.51, 132.25, 131.04, 130.21, 130.02, 129.86 (d, J = 31 Hz), 126.65,119.79, 117.16, 116.78, 116.18, 113.59, 113.36, 103.36, 59.29, 58.63, 56.51, 56.26, 55.07, 51.13, 45.18, 25.93; HRMS (ESI-TOF) m/z calcd for C31H33F3N5O2 [M + H]+: 564.2586, found: 564.2593.
- 4-((4-ethylpiperazin-1-yl)methyl)-N-(3-((5-methoxyquinolin-2-yl)amino)phenyl)-3-(trifluoromethyl)benzamide (14c)
The silica gel flash column chromatography was conducted using MeOH:DCM = 3:97 eluent to obtain the desired 14c (44.7 mg, 38% yield) as a light yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 9.0 Hz, 1H), 8.20 (s, 1H), 8.11 (s, 1H), 8.07 (s, 1H), 8.00 (d, J = 8.2 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.48–7.42 (m, 2H), 7.38 (d, J = 8.4 Hz, 1H), 7.32–7.28 (m, 2H), 6.92 (d, J = 9.1 Hz, 1H), 6.65 (d, J = 7.7 Hz, 1H), 3.95 (s, 3H), 3.68 (s, 2H), 2.55–2.44 (m, 10H), 1.11 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 164.51, 155.41, 154.22, 148.35, 141.84, 141.09, 138.38, 133.67, 132.50, 130.81, 130.18, 129.79, 129.65, 128.91, 124.87, 119.19, 116.17, 115.90, 114.55, 117.74, 110.96, 102.07, 57.87, 55.56, 52.78, 52.67, 52.24, 11.68; HRMS (ESI-TOF) m/z calcd for C31H33F3N5O2 [M + H]+: 564.2586, found: 564.2587.
- N-(2-fluoro-5-((5-methoxyquinolin-2-yl)amino)phenyl)-4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)benzamide (15a)
The silica gel flash column chromatography was conducted using MeOH:DCM = 5:95 eluent to obtain the desired 15a (15.2 mg, 9.4% yield) as a light brown solid. 1H NMR (400 MHz, CDCl3) δ 8.60–8.58 (m, 1H), 8.33 (d, J = 9.0 Hz, 1H), 8.16 (s, 1H), 8.09 (br. s, 1H), 8.01–7.95 (m, 2H), 7.65–7.63 (m, 1H), 7.48 (t, J = 8.1 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 7.12 (dd, J = 10.5, 9.0 Hz, 1H), 6.89 (d, J = 9.1 Hz, 1H), 6.66 (d, J = 7.4 Hz, 1H), 3.95 (s, 3H), 3.72 (s, 2H), 2.55 (br. s, 8H), 2.33 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.13, 164.04, 155.44, 154.26, 148.51 (d, J = 238 Hz), 148.13, 142.46, 136.86, 132.22, 132.75, 130.94, 129.98, 129.95, 127.90 (q, J = 31 Hz), 126.28, 126.17, 125.01 (d, J = 5.9 Hz), 123.86 (d, J = 273 Hz), 119.05, 116.36, 115.83, 115.25, 115.05, 113.41, 110.62, 102.12, 57.89, 55.60, 55.09, 53.00, 45.89; HRMS (ESI-TOF) m/z calcd for C30H30F4N5O2 [M + H]+: 536.2274, found: 536.2278.
- N-(3-((6-methoxyquinolin-2-yl)amino)phenyl)-4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)benzamide (16a)
The silica gel flash column chromatography was conducted using MeOH:ethyl acetate = 5:95 eluent to obtain the desired 16a (49.8 mg, 27% yield) as a light brown solid. 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 1H), 8.10 (s, 1H), 8.02 (s, 1H), 7.97 (d, J = 8.1 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.78 (d, J = 8.9 Hz, 1H), 7.70 (d, J = 9.1 Hz, 1H), 7.38 (d, J = 7.8 Hz, 1H), 7.28–7.20 (m, 4H), 7.03 (br. s, 1H), 6.94 (d, J = 2.8 Hz, 1H), 6.91 (d, J = 8.9 Hz, 1H), 3.86 (s, 3H), 3.67 (s, 2H), 2.50 (br. s, 8H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 164.65, 155.69, 152.47, 142.72, 142.02, 141.41, 138.38, 136.84, 133.66, 130.77, 130.23, 129.67, 129.02 (q, J = 31 Hz), 128.00, 124.81 (q, J = 5.8 Hz), 124.65, 123.93 (q, J = 273 Hz), 121.41, 115.85, 114.30, 112.66, 111.43, 106.27, 57.90, 55.51, 55.13, 53.09, 45.97; HRMS (ESI-TOF) m/z calcd for C30H31F3N5O2 [M + H]+: 550.2430, found: 550.2436.
- N-(3-((6-methoxyquinolin-2-yl)amino)phenyl)-4-((4-methyl-1,4-diazepan-1-yl)methyl)-3-(trifluoromethyl)benzamide (16b)
The silica gel flash column chromatography was conducted using MeOH:DCM = 10:90 eluent to obtain the desired 16b (108 mg, 44% yield) as a light brown solid. 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 8.09 (s, 1H), 8.05 (br. s, 1H), 7.96 (br. s, 2H), 7.78 (d, J = 8.9 Hz, 1H), 7.70 (d, J = 9.1 Hz, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.28–7.20 (m, 4H), 6.94 (d, J = 2.7 Hz, 1H), 6.91 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 3.80 (s, 2H), 2.72–2.60 (m, 8H), 2.37 (s, 3H), 1.82 (quintet, J = 5.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 164.70, 155.65, 152.49, 143.22, 142.93, 141.51, 138.39, 136.70, 133.52, 130.75, 130.18, 129.64, 128.73 (d, J = 31 Hz), 128.19, 124.80 (d, J = 5.8 Hz), 124.69, 123.97 (q, J = 273 Hz), 121.34, 115.75, 114.20, 112.74, 111.34, 106.25, 58.27, 58.20, 56.73, 55.51, 54.98, 54.59, 47.01, 27.60; HRMS (ESI-TOF) m/z calcd for C31H33F3N5O2 [M + H]+: 564.2586, found: 564.2597.
4.2.9. General Procedure for Synthesis 17a,b and 18a,b
The crude of 14a–c and 16a,b were prepared as starting material. To the crude mixture in an ice bath, 1.0 M BBr3 in DCM (6.0 eq.) was added dropwise, then the reaction mixture was stirred at 0 °C for 30 min at room temperature overnight. After the reaction mixture was cooled down, a saturated NaHCO3 aqueous solution was added to render the solution basic, pH = 8. The aqueous layer was extracted with ethyl acetate (20 × 3 mL) and the combined organic layer was washed with brine, dried over anhydrous Na2SO4, and filtered. The solvent was evaporated under reduced pressure; then, the residue was purified by silica gel flash column chromatography using proper eluent to obtain the desired target compounds 17a,b and 18a,b.
- N-(3-((5-hydroxyquinolin-2-yl)amino)phenyl)-4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)benzamide (17a)
The silica gel flash column chromatography was conducted using MeOH:DCM (6:94, then switching to 10:90) eluent to obtain the desired 17a (97.5 mg, 46% yield) as a brown solid. 1H NMR (400 MHz, DMSO) δ 10.55 (s, 1H), 9.56 (s, 1H), 8.36–8.17 (m, 4H), 7.94 (d, J = 7.8 Hz, 1H), 7.60–7.43 (m, 4H), 7.23 (d, J = 8.0 Hz, 1H), 7.07 (d, J = 8.7 Hz, 1H), 6.77 (s, 1H), 3.80 (s, 2H), 3.16 (s, 1H), 3.09–2.91 (m, 6H), 2.81 (s, 3H), 2.46–2.43 (m, 2H); 13C NMR (100 MHz, Methanol) δ 167.00, 155.55, 154.85, 144.28, 141.60, 140.76, 140.05, 136.91, 135.53, 133.11, 132.47, 132.22, 130.94, 130.13, 126.89, 126.60 (d, J = 5.7 Hz), 119.28, 118.71, 115.80, 115.38, 114.14, 112.45, 108.99, 58.19, 54.99, 51.06, 43.61.; HRMS (ESI-TOF) m/z calcd for C29H29F3N5O2 [M + H]+: 536.2274, found: 536.2281.
- N-(3-((5-hydroxyquinolin-2-yl)amino)phenyl)-4-((4-methyl-1,4-diazepan-1-yl)methyl)-3-(trifluoromethyl)benzamide (17b)
The silica gel flash column chromatography was conducted using MeOH:DCM (10:90) eluent to obtain the desired 17b (19.5 mg, 9.4%) as a yellow solid. 1H NMR (400 MHz, methanol) δ 8.38 (d, J = 9.2 Hz, 1H), 8.28 (t, J =1.9 Hz, 1H), 8.25 (br. s, 1H), 8.19 (d, J = 8.2 Hz, 1H), 8.02 (d, J = 8.2 Hz, 1H), 7.47 (dt, J = 7.6, 1.7 Hz, 1H), 7.41–7.34 (m, 3H), 7.23 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 9.2 Hz, 1H), 6.69 (d, J = 7.5 Hz, 1H), 3.91 (s, 2H), 3.43 (t, J = 5.4 Hz, 2H), 3.34 (s, 1H), 3.32 (methanol overlap, 1H), 2.91 (d, J = 5.1 Hz, 2H), 2.89 (s, 3H), 2.76 (t, J = 6.1 Hz, 2H), 2.05 (quintet, J = 5.7 Hz, 2H); 13C NMR (100 MHz, methanol) δ 167.07, 155.49, 155.21, 142.91, 141.55, 140.46, 135.57, 135.31, 132.45, 132.21, 132.08, 130.53, 129.79 (d, J = 30 Hz), 126.56 (d, J = 5.9 Hz), 118.16, 117.36, 116.28, 115.84, 114.64, 112.66, 108.15, 59.18, 58.53, 56.42, 54.99, 51.04, 45.10, 25.83.; HRMS (ESI-TOF) m/z calcd for C30H31F3N5O2 [M + H]+: 550.2430, found: 550.2444.
- N-(3-((6-hydroxyquinolin-2-yl)amino)phenyl)-4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)benzamide (18a)
The silica gel flash column chromatography was conducted using MeOH:DCM (5:95, then switching to 10:90) eluent to obtain the desired 18a (15.0 mg, 9.4%) as a brown solid. 1H NMR (400 MHz, methanol) δ 8.23 (s, 1H), 8.10 (s, 1H), 8.02 (s, 1H), 7.97 (d, J = 8.1 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.78 (d, J = 8.9 Hz, 1H), 7.70 (d, J = 9.1 Hz, 1H), 7.38 (d, J = 7.8 Hz, 1H), 7.28–7.20 (m, 4H), 7.03 (br. s, 1H), 6.94 (d, J = 2.8 Hz, 1H), 6.91 (d, J = 8.9 Hz, 1H), 3.86 (s, 3H), 3.67 (s, 2H), 2.50 (br. s, 8H), 2.30 (s, 3H); 13C NMR (100 MHz, methanol) δ 164.65, 155.69, 152.47, 142.72, 142.02, 141.41, 138.38, 136.84, 133.66, 130.77, 130.23, 129.67, 129.02 (q, J = 31 Hz), 128.00, 124.81 (q, J = 5.8 Hz), 124.65, 123.93 (q, J = 273 Hz), 121.41, 115.85, 114.30, 112.66, 111.43, 106.27, 57.90, 55.51, 55.13, 53.09, 45.97; HRMS (ESI-TOF) m/z calcd for C29H29F3N5O2 [M + H]+: 536.2273, found: 536.2279.
- N-(3-((6-hydroxyquinolin-2-yl)amino)phenyl)-4-((4-methyl-1,4-diazepan-1-yl)methyl)-3-(trifluoromethyl)benzamide (18b)
The silica gel flash column chromatography was conducted using MeOH:DCM (10:90, then switching to 15:95) eluent to obtain the desired 18a (19.1 mg, 4.5%) as yellow solid. 1H NMR (400 MHz, methanol) δ 8.27 (s, 1H), 8.23–8.22 (m, 2H), 8.08 (d, J = 8.2 Hz, 1H), 8.03 (d, J = 9.1 Hz, 1H), 7.67 (d, J = 9.0 Hz, 1H), 7.45–7.40 (m, 3H), 7.21 (dd, J = 9.0, 2.7 Hz, 1H), 7.08 (dd, J = 5.9, 3.2 Hz, 2H), 3.96 (s, 2H), 3.48 (t, J = 5.3 Hz, 2H), 3.35 (s, 2H), 2.96–2.93 (m, 5H), 2.81 (t, J = 6.1 Hz, 2H), 2.09 (quintet, J = 5.7 Hz, 2H); 13C NMR (100 MHz, Methanol) δ 165.71, 154.11, 151.90, 141.57, 139.44, 139.29, 138.99, 134.13, 131.09, 130.88, 129.41,128.42 (d, J = 30 Hz), 125.16 (d, J = 5.8 Hz), 124.46, 123.91, 121.47, 117.19, 116.55, 113.66, 113.49, 109.89, 57.80, 57.15, 55.07, 53.60, 49.66, 48.46, 43.76, 24.48; HRMS (ESI-TOF) m/z calcd for C30H31F3N5O2 [M + H]+: 550.2430, found: 550.2441.
4.3. In Vitro Kinase Screening
Reaction Biology Corporation (RBC) Kinase HotSpotSM service was utilized for cell-free biochemical kinase evaluation of the target compounds according to the reported assay protocol [46].
4.4. Anticancer Screening at NCI
The anticancer screening of target compounds over a full panel of 60 human cancer cell lines was carried out using Sulforhodamine B (SRB) assay at the National Cancer Institute (NCI), Bethesda, Maryland, USA, employing the standard protocol [47].
4.5. Molecular Docking
The crystal structure of B-RAFV600E protein in its DFG-out conformation, co-crystal structure with Sorafenib was taken from Protein Data Bank (PDB code: 1UWJ). Homology modeling of C-RAF (Uniprot: p04049) was built from B-RAFV600E structure after sequence alignment using Discovery Studio 2022 (DS). C-RAF homology model was selected with the lowest PDF energy. The protein structure of B-RAFV600E and C-RAF were prepared for docking by employing the protocol “prepare protein” by removing water molecules, and ligands were prepared through protonation at pH 7.4 and energy minimization. The inhibitors were docked into B-RAFV600E and C-RAF models, respectively, using the CDOCKER in DS software.
4.6. Molecular Dynamcis Simulation
Molecular dynamics (MD) simulations were conducted using the same crystal structure of the B-RAFV600E and the C-RAF used in molecular docking calculations. Missing residues were additionally modeled using GalaxyFill [48]. The initial systems were prepared using CHARMM-GUI [49]. AMBER20 software [50] was used to carry out MD simulation. The topology and parameters were modeled using ff19SB force field [51] for the protein and the generalized AMBER force field (GAFF) [52] for the ligands. Each simulation system was solvated using TIP3P water model, and extra NaCl ions were added to the box to neutralize the systems. Temperature and pressure were kept at 300 K and 1 atm, respectively. A SHAKE algorithm was used to constrain all hydrogen atom bonds [53]. Long-range electrostatic interactions were calculated using the particle mesh Ewald (PME) method [54]. Each system was first minimized using 500 steps of steepest descent, and another 20 ps simulation was performed for the equilibration of the system at 300 K in the NVT ensemble. Then, 10 ns equilibration runs were performed to fully relax the systems in NPT ensemble. Finally, we ran a production run for 50 ns and 120 ns (for Figure S6), and last, 20 ns was used for the MM-GBSA calculation.
For MM-GBSA analyses, snapshots were taken every 100 ps of 20 ns MD production runs, resulting in two hundred snapshots per compound. The energies were obtained using the MM_GBSA module of Amber20 [55].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24043216/s1, The supporting information associated with this article is available.
Author Contributions
Conceptualization, A.K.E.-D. and G.K.; synthesis and characterization of target compounds, H.J.K. and A.K.E.-D.; in silico study, J.W.P., S.S. and K.-H.C.; validation and data curation, M.M.A., E.-K.B., J.W.P. and A.K.E.-D.; writing—original draft preparation, H.J.K. and A.K.E.-D.; writing—review and editing, A.K.E.-D. and G.K.; supervision, A.K.E.-D. and G.K.; project administration, G.K.; funding acquisition, A.K.E.-D., E.-K.B. and G.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Research Council of Science & Technology (NST) grant by the Korean government (MSIT) (No. CAP-20-01-KRIBB) and the Institutional Program grant by the Korea Institute of Science and Technology (2E32212). A.K. El-Damasy was supported by the Korea Research Fellowship Program grant through NRF funded by the Ministry of Science and ICT (2019H1D3A1A0107088214). This research was supported by the Researchers Supporting Project number (RSPD2023R628), King Saud University, Riyadh, Saudi Arabia.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
We would like to express our gratitude and appreciation to the National Cancer Institute (NCI, Bethesda, Maryland, USA) for conducting the anticancer evaluation of the new compounds. This work was supported by the National Supercomputing Center of KISTI with supercomputing resources including technical support. The authors extend their appreciation to the Researchers Supporting Project number (RSPD2023R628), King Saud University, Riyadh, Saudi Arabia for supporting this research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ahmad, F.B.; Cisewski, J.A.; Miniño, A.; Anderson, R.N. Provisional Mortality Data—United States, 2020. MMWR Surveill. Summ. 2021, 70, 519–522. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 30 November 2022).
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine Kinase—Role and Significance in Cancer. Int. J. Med. Sci. 2012, 1, 101–115. [Google Scholar] [CrossRef]
- Cohen, P. Protein Kinases—The Major Drug Targets of the Twenty-First Century? Nat. Rev. Drug Discov. 2002, 1, 309. [Google Scholar] [CrossRef]
- Li, N.; Batt, D.; Warmuth, M. B-Raf Kinase Inhibitors for Cancer Treatment. Curr. Opin. Investig. Drugs 2007, 8, 452–456. [Google Scholar]
- Leicht, D.T.; Balan, V.; Kaplun, A.; Singh-Gupta, V.; Kaplun, L.; Dobson, M.; Tzivion, G. Raf Kinases: Function, Regulation and Role in Human Cancer. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1196–1212. [Google Scholar] [CrossRef]
- Durrant, D.E.; Morrison, D.K. Targeting the Raf Kinases in Human Cancer: The Raf Dimer Dilemma. Nat. Publ. Gr. 2017, 118, 3–8. [Google Scholar] [CrossRef]
- Mercer, K.; Giblett, S.; Green, S.; Lloyd, D.; Dias, S.D.R.; Plumb, M.; Marais, R.; Pritchard, C. Expression of Endogenous Oncogenic V600EB-Raf Induces Proliferation and Developmental Defects in Mice and Transformation of Primary Fibroblasts. Cancer Res. 2005, 65, 11493–11500. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Li, Y.; Umbach, D.M.; Li, L. Putative Genomic Characteristics of BRAF V600K versus V600E Cutaneous Melanoma. Melanoma Res. 2017, 27, 527–535. [Google Scholar] [CrossRef]
- Nepote, A.; Avallone, G.; Ribero, S.; Cavallo, F.; Roccuzzo, G.; Mastorino, L.; Conforti, C.; Paruzzo, L.; Poletto, S.; Schianca, F.C.; et al. Current Controversies and Challenges on BRAF V600K-Mutant Cutaneous Melanoma. J. Clin. Med. 2022, 11, 828. [Google Scholar] [CrossRef]
- Takahashi, M.; Li, Y.; Dillon, T.J.; Kariya, Y.; Stork, P.J.S. Phosphorylation of the C-Raf N Region Promotes Raf Dimerization. Mol. Cell. Biol. 2017, 37, e00132-17. [Google Scholar] [CrossRef]
- An, S.; Yang, Y.; Ward, R.; Liu, Y.; Guo, X.X.; Xu, T.R. A-Raf: A New Star of the Family of Raf Kinases. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 520–531. [Google Scholar] [CrossRef]
- Khan, P.S.; Rajesh, P.; Rajendra, P.; Chaskar, M.G.; Rohidas, A.; Jaiprakash, S. Recent Advances in B-RAF Inhibitors as Anticancer Agents. Bioorg. Chem. 2022, 120, 105597. [Google Scholar] [CrossRef]
- Okaniwa, M.; Hirose, M.; Arita, T.; Yabuki, M.; Nakamura, A.; Takagi, T.; Kawamoto, T.; Uchiyama, N.; Sumita, A.; Tsutsumi, S.; et al. Discovery of a Selective Kinase Inhibitor (TAK-632) Targeting Pan-RAF Inhibition: Design, Synthesis, and Biological Evaluation of C -7-Substituted 1,3-Benzothiazole Derivatives. J. Med. Chem. 2013, 56, 6478–6494. [Google Scholar] [CrossRef]
- Jung, H.; Kim, J.; Im, D.; Moon, H.; Hah, J.M. Design, Synthesis, and In Vitro Evaluation of N-(3-(3-Alkyl-1H-Pyrazol-5-Yl) Phenyl)-Aryl Amide for Selective RAF Inhibition. Bioorg. Med. Chem. Lett. 2019, 29, 534–538. [Google Scholar] [CrossRef]
- Gong, Z.H.; Yao, J.; Ji, J.F.; Yang, J.; Xiang, T.; Zhou, C.K. Synthesis and Biological Evaluation of Novel N-(5-Phenyl-1H-Pyrazol-3-Yl)Benzenesulfonamide Derivatives as Potential BRAFV600E Inhibitors. Med. Chem. Res. 2017, 26, 2583–2591. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and Development of Sorafenib: A Multikinase Inhibitor for Treating Cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Wilhelm, S.; Adnane, L.; Hirth-Dietrich, C.; Ehrlich, P.; Lynch, M. Preclinical Characterization of BAY 73-4506: A Kinase Inhibitor with Broad Spectrum Antitumor Activity Targeting Oncogenic and Angiogenic Kinases. Mol. Cancer Ther. 2007, 6, B260. [Google Scholar]
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer. Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef]
- Pan, J.H.; Zhou, H.; Zhu, S.B.; Huang, J.L.; Zhao, X.X.; Ding, H.; Pan, Y.L. Development of Small-Molecule Therapeutics and Strategies for Targeting RAF Kinase in BRAF-Mutant Colorectal Cancer. Cancer Manag. Res. 2018, 10, 2289–2301. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The First Drug Approved for BRAF-Mutant Cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Rheault, T.R.; Stellwagen, J.C.; Adjabeng, G.M.; Hornberger, K.R.; Petrov, K.G.; Waterson, A.G.; Dickerson, S.H.; Mook, R.A.; Laquerre, S.G.; King, A.J.; et al. Discovery of Dabrafenib: A Selective Inhibitor of Raf Kinases with Antitumor Activity against B-Raf-Driven Tumors. ACS Med. Chem. Lett. 2013, 4, 358–362. [Google Scholar] [CrossRef]
- Huestis, M.P.; Dela Cruz, D.; Dipasquale, A.G.; Durk, M.R.; Eigenbrot, C.; Gibbons, P.; Gobbi, A.; Hunsaker, T.L.; La, H.; Leung, D.H.; et al. Targeting KRAS Mutant Cancers via Combination Treatment: Discovery of a 5-Fluoro-4-(3H)-Quinazolinone Aryl Urea Pan-RAF Kinase Inhibitor. J. Med. Chem. 2021, 64, 3940–3955. [Google Scholar] [CrossRef]
- Sun, J.; Zager, J.S.; Eroglu, Z. Encorafenib/Binimetinib for the Treatment of BRAF-Mutant Advanced, Unresectable, or Metastatic Melanoma: Design, Development, and Potential Place in Therapy. Onco. Targets. Ther. 2018, 11, 9081–9089. [Google Scholar] [CrossRef]
- Shirley, M. Encorafenib and Binimetinib: First Global Approvals. Drugs 2018, 78, 1277–1284. [Google Scholar] [CrossRef]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef]
- Choi, H.G.; Ren, P.; Adrian, F.; Sun, F.; Lee, H.S.; Wang, X.; Ding, Q.; Zhang, G.; Xie, Y.; Zhang, J.; et al. A Type-Ii Kinase Inhibitor Capable of Inhibiting the T315I “Gatekeeper” Mutant of Bcr-Abl. J. Med. Chem. 2010, 53, 5439–5448. [Google Scholar] [CrossRef]
- Choi, S.H.; Shin, I.; Kim, N.; Nam, Y.; Sim, T. The First Small Molecules Capable of Strongly Suppressing Proliferation of Cancer Cells Harboring BRAF Class I/II/III Mutations. Biochem. Biophys. Res. Commun. 2020, 532, 315–320. [Google Scholar] [CrossRef]
- Jiao, Y.; Xin, B.T.; Zhang, Y.; Wu, J.; Lu, X.; Zheng, Y.; Tang, W.; Zhou, X. Design, Synthesis and Evaluation of Novel 2-(1H-Imidazol-2-Yl) Pyridine Sorafenib Derivatives as Potential BRAF Inhibitors and Anti-Tumor Agents. Eur. J. Med. Chem. 2015, 90, 170–183. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, G.; Zhang, Q.; Duan, C.; Zhang, Y.; Zhang, Z.; Zhou, Y.; Lu, T.; Tang, W. Rational Design, Synthesis, and Biological Evaluation of Pan-Raf Inhibitors to Overcome Resistance. Org. Biomol. Chem. 2017, 15, 3455–3465. [Google Scholar] [CrossRef]
- Cheng, H.; Chang, Y.; Zhang, L.; Luo, J.; Tu, Z.; Lu, X.; Zhang, Q.; Lu, J.; Ren, X.; Ding, K. Identification and Optimization of New Dual Inhibitors of B-Raf and Epidermal Growth Factor Receptor Kinases for Overcoming Resistance against Vemurafenib. J. Med. Chem. 2014, 57, 2692–2703. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Zheng, M.W.; Li, Y.P.; Lin, X.D.; Huang, M.; Zhong, L.; Li, G.B.; Zhang, R.J.; Lin, W.T.; Jiao, Y.; et al. Design, Synthesis, and Structure-Activity Relationship Studies of 3-(Phenylethynyl)-1H-Pyrazolo [3,4-d]Pyrimidin-4-Amine Derivatives as a New Class of Src Inhibitors with Potent Activities in Models of Triple Negative Breast Cancer. J. Med. Chem. 2015, 58, 3957–3974. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Khan, M.A.; Abdel-Maksoud, M.S.; Gamal El-Din, M.M.; Oh, C.H. A New Series of Diarylamides Possessing Quinoline Nucleus: Synthesis, In Vitro Anticancer Activities, and Kinase Inhibitory Effect. Eur. J. Med. Chem. 2014, 87, 484–492. [Google Scholar] [CrossRef] [PubMed]
- El-Damasy, A.K.; Haque, M.M.; Park, J.W.; Shin, S.C.; Lee, J.S.; EunKyeong Kim, E.; Keum, G. 2-Anilinoquinoline Based Arylamides as Broad Spectrum Anticancer Agents with B-RAFV600E/C-RAF Kinase Inhibitory Effects: Design, Synthesis, In Vitro Cell-Based and Oncogenic Kinase Assessments. Eur. J. Med. Chem. 2020, 208, 112756. [Google Scholar] [CrossRef] [PubMed]
- El-Damasy, A.K.; Lee, J.-H.; Seo, S.H.; Cho, N.C.; Pae, A.N.; Keum, G. Design and synthesis of new potent anticancer benzothiazole amides and ureas featuring pyridylamide moiety and possessing dual B-RafV600E and C-Raf kinase inhibitory activities. Eur. J. Med. Chem. 2016, 115, 201–216. [Google Scholar] [CrossRef]
- El-Damasy, A.K.; Cho, N.C.; Pae, A.N.; Kim, E.E.; Keum, G. Novel 5-Substituted-2-Anilinoquinolines with 3-(Morpholino or 4-Methylpiperazin-1-Yl)Propoxy Moiety as Broad Spectrum Antiproliferative Agents: Synthesis, Cell Based Assays and Kinase Screening. Bioorg. Med. Chem. Lett. 2016, 26, 3307–3312. [Google Scholar] [CrossRef]
- El-Damasy, A.K.; Seo, S.H.; Cho, N.C.; Pae, A.N.; Kim, E.E.; Keum, G. Design and Synthesis of New 2-Anilinoquinolines Bearing N-Methylpicolinamide Moiety as Potential Antiproliferative Agents. Chem. Biol. Drug Des. 2017, 89, 98–113. [Google Scholar] [CrossRef]
- Yasri, A.; Cheve, G.; Bories, C.; Delon, L. Derivatives of Azaindoles as Inhibitors of Protein Kinase ABL and SRC. WIPO (PCT) WO2010092489A1, 19 August 2010. [Google Scholar]
- Song, S.; Li, X.; Guo, J.; Hao, C.; Feng, Y.; Guo, B.; Liu, T.; Zhang, Q.; Zhang, Z.; Li, R.; et al. Design, Synthesis and Biological Evaluation of 1-Phenanthryl-Tetrahydroisoquinoline Derivatives as Novel P21-Activated Kinase 4 (PAK4) Inhibitors. Org. Biomol. Chem. 2015, 13, 3803–3818. [Google Scholar] [CrossRef]
- El-Damasy, A.K.; Seo, S.H.; Cho, N.C.; Kang, S.B.; Pae, A.N.; Kim, K.S.; Keum, G. Design, Synthesis, in-Vitro Antiproliferative Activity and Kinase Profile of New Picolinamide Based 2-Amido and Ureido Quinoline Derivatives. Eur. J. Med. Chem. 2015, 101, 754–768. [Google Scholar] [CrossRef]
- Perzyna, A.; Dal Zotto, C.; Durand, J.O.; Granier, M.; Smietana, M.; Melnyk, O.; Stará, I.G.; Starý, I.; Klepetářová, B.; Šaman, D. Reaction of Isocyanate-Functionalised Silicon Wafers with Complex Amino Compounds. Eur. J. Org. Chem. 2007, 2007, 4032–4037. [Google Scholar] [CrossRef]
- Farag, A.K.; Hassan, A.H.E.; Chung, K.S.; Lee, J.H.; Gil, H.S.; Lee, K.T.; Roh, E.J. Diarylurea Derivatives Comprising 2,4-Diarylpyrimidines: Discovery of Novel Potential Anticancer Agents via Combined Failed-Ligands Repurposing and Molecular Hybridization Approaches. Bioorg. Chem. 2020, 103, 104121. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Babij, N.R.; McCusker, E.O.; Whiteker, G.T.; Canturk, B.; Choy, N.; Creemer, L.C.; Amicis, C.V.D.; Hewlett, N.M.; Johnson, P.L.; Knobelsdorf, J.A.; et al. NMR Chemical Shifts of Trace Impurities: Industrially Preferred Solvents Used in Process and Green Chemistry. Org. Process Res. Dev. 2016, 20, 661–667. [Google Scholar] [CrossRef]
- Reaction Biology Corporation. Available online: http://www.reactionbiology.com/webapps/site/Kinase_Assay_Protocol.aspx (accessed on 30 November 2022).
- DTP Human Tumor Cell Line Screen Process. Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 30 November 2022).
- Coutsias, E.A.; Seok, C.; Jacobson, M.P.; Dill, K.A. A Kinematic View of Loop Closure. J. Comput. Chem. 2004, 25, 510–528. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Sahakyan, H. Improving virtual screening results with mm/gbsa and mm/pbsa rescoring. J. Comput. Aided Mol. Des. 2021, 35, 731–736. [Google Scholar] [CrossRef] [PubMed]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).