Abstract
Store-operated Ca2+ entry (SOCE) is activated in response to the inositol-1,4,5-trisphosphate (InsP3)-dependent depletion of the endoplasmic reticulum (ER) Ca2+ store and represents a ubiquitous mode of Ca2+ influx. In vascular endothelial cells, SOCE regulates a plethora of functions that maintain cardiovascular homeostasis, such as angiogenesis, vascular tone, vascular permeability, platelet aggregation, and monocyte adhesion. The molecular mechanisms responsible for SOCE activation in vascular endothelial cells have engendered a long-lasting controversy. Traditionally, it has been assumed that the endothelial SOCE is mediated by two distinct ion channel signalplexes, i.e., STIM1/Orai1 and STIM1/Transient Receptor Potential Canonical 1(TRPC1)/TRPC4. However, recent evidence has shown that Orai1 can assemble with TRPC1 and TRPC4 to form a non-selective cation channel with intermediate electrophysiological features. Herein, we aim at bringing order to the distinct mechanisms that mediate endothelial SOCE in the vascular tree from multiple species (e.g., human, mouse, rat, and bovine). We propose that three distinct currents can mediate SOCE in vascular endothelial cells: (1) the Ca2+-selective Ca2+-release activated Ca2+ current (ICRAC), which is mediated by STIM1 and Orai1; (2) the store-operated non-selective current (ISOC), which is mediated by STIM1, TRPC1, and TRPC4; and (3) the moderately Ca2+-selective, ICRAC-like current, which is mediated by STIM1, TRPC1, TRPC4, and Orai1.
1. Introduction
The endothelial monolayer forms the innermost lining of all blood vessels and of the cardiac chambers, where it is known as endocardium. Endothelial cells provide a widespread interface that continuously perceives and integrates signals delivered by blood-borne humoral cues and by changes in shear stress and/or pulsatile stretch. Additionally, endothelial cells may sense even subtle alterations in local tissue microenvironment and/or in the signalling activity of neighbouring cells [1,2,3,4,5]. Endothelial dysfunction is, therefore, involved in the pathogenesis of life-threatening cardiovascular disorders, such as coronary artery disease, acute myocardial infarction, atherosclerosis, hypertension, diabetes mellitus, stroke, and, more, recently COVID-19 [3,6,7]. Endothelial colony forming cells (ECFCs) can be mobilized from vascular stem cell niches to replace senescent/injured endothelial cells or reconstruct the vascular network damaged by an ischemic insult, thereby attempting to preserve cardiovascular homeostasis [8]. Insufficient or excessive vascularization by capillary endothelial cells and circulating ECFCs has been recognized as a common denominator of multiple debilitating or deadly diseases, including age-related blindness, acute myocardial infarction, and cancer [9,10].
A spatio-temporal increase in intracellular Ca2+ concentration ([Ca2+]i) is crucial to select the most appropriate endothelial response to the multiple inputs that are simultaneously delivered either from blood flow or from adjoining tissue [2,11,12,13]. Endothelial Ca2+ signals can adopt distinct waveforms, e.g., biphasic Ca2+ elevation or intracellular Ca2+ oscillations, to regulate a plethora of functions [2,14], such as vascular tone and blood pressure [15,16], vascular permeability [17,18], and repair [19,20], neurovascular coupling [4,21], platelet aggregation and blood coagulation [22], leukocyte infiltration [23], and angiogenesis [24]. Furthermore, an increase in [Ca2+]i drives ECFCs’ angiogenic activity both in vitro [7,25] and in vivo [26,27]. Typically, the endothelial Ca2+ response to humoral stimulation is triggered by Ca2+ release from the endoplasmic reticulum (ER), i.e., the largest endogenous Ca2+ reservoir, through inositol-1,4,5-trisphosphate (InsP3) receptors (InsP3Rs) [24]. This may cause a dramatic drop in ER Ca2+ concentration ([Ca2+]ER), which results in the activation of a Ca2+-permeable pathway in the plasma membrane (PM), known as store-operated Ca2+ entry (SOCE) [28,29,30]. SOCE fulfils the primary role to refill the ER with Ca2+ and thereby maintains the intracellular Ca2+ response over time. Additionally, SOCE recruits the variety of Ca2+-dependent decoders that effect the endothelial response to extracellular stimulation [28,29,30]. Therefore, SOCE is regarded as the most important Ca2+ entry pathway in both vascular endothelial cells [28,29,30] and in circulating ECFCs [31].
Traditionally, it is assumed that depletion of the endothelial ER Ca2+ store may lead to the activation of two distinct inward currents that exhibit distinct electrophysiological features and are mediated by different cation channels on the PM. Store depletion in vascular endothelial cells could activate a Ca2+-selective and inwardly rectifying current that resembles the Ca2+ release-activated Ca2+ (CRAC) current (ICRAC) originally described in mast cells [32] and is, therefore, referred to as ICRAC [33,34,35]. Alternately, the reduction in [Ca2+]ER could result in the development of a non-selective cation current that displays a slightly linear current-to-voltage (IV) relationship and is known as store-operated current (ISOC) [36,37]. A flurry of investigations published over the last twenty years paved the way towards the general agreement that, in vascular endothelial cells, Stromal Interaction Molecule 1 (STIM1) and Orai1 proteins mediate the ICRAC [38,39,40], whereas members of the Transient Receptor Potential (TRP) Canonical (TRPC) subfamily of non-selective cation channels underlie the ISOC [41,42,43]. A careful scrutiny of the literature, however, reveals that depletion of the ER Ca2+ store can activate a third endothelial Ca2+-entry pathway, which is more Ca2+ selective than the ISOC but displays a weaker inward rectification and has a more negative reversal potential (Erev) as respect to the ICRAC [44,45,46,47]. This current has been termed as ISOC [48] or ICRAC-like [46,49]. In our opinion, the latter definition more closely depicts the electrophysiological properties of this store-dependent current, whose molecular architecture has been recently unveiled and consists of both Orai and TRP channel proteins [50,51]. Therefore, herein, we aim at bringing order to the distinct mechanisms whereby endothelial SOCE can occur and modulate different functions in the vascular tree from multiple species (e.g., human, mouse, rat, and bovine).
2. How to Activate Endothelial SOCE: Physiological vs. Pharmacological Reduction in [Ca2+]ER
Endothelial SOCE can be activated either by physiological cues that stimulate membrane receptors to liberate ER stored Ca2+ via InsP3Rs or by pharmacological compounds that mobilize intraluminal Ca2+ and thereby increase the membrane permeability to Ca2+ through receptor-independent mechanisms [49,52].
2.1. The Physiological Activation of SOCE by InsP3-Dependent ER Ca2+ Release
Physiologically, the endothelial Ca2+ store is depleted upon extracellular stimulation by humoral agonists, such as hormones, neurotransmitters, and growth factors, which bind to their specific Gq/11 protein-coupled receptors (GqPCRs) or tyrosine kinase receptors (TKRs). GqPCRs and TKRs, respectively, engage phospholipase Cβ (PLCβ) and PLCγ that cleave phosphatidylinositol 4,5-bisphosphate (PIP2), a minor phospholipid residing in the inner leaflet of the PM, in diacylglycerol (DAG) and InsP3 [24,53]. InsP3 diffuses in the cytoplasm and primes ER-embedded InsP3Rs towards ambient Ca2+, thereby mobilizing ER Ca2+ through the mechanism of Ca2+-induced Ca2+ release (CICR) [24,53]. The local Ca2+ pulse that is required by InsP3 to engage the Ca2+-dependent InsP3Rs can be provided by the closely apposed two-pore channels (TPCs), that release endolysosomal Ca2+ in response to nicotinic acid adenine dinucleotide phosphate (NAADP) [54,55]. Conversely, ryanodine receptors (RyRs) play a minor role in ER Ca2+ release in endothelial cells, as recently discussed in [14].
The mechanisms whereby the stimulation of GqPCRs leads to the InsP3-dependent depletion of the ER Ca2+ store have been finely dissected in calf pulmonary artery endothelial (CPAE) cells [28]. Agonist-induced elevation in [Ca2+]i arises in the form of spatially restricted InsP3-dependent ER Ca2+ release events, known as Ca2+ puffs, that arise in the cell periphery and subsequently propagate as repetitive Ca2+ waves to engulf the whole cytoplasm through CICR [56,57]. Notably, focal stimulation with an InsP3-producing agonist (i.e., ATP) may result in local depletion of the InsP3-sensitive ER Ca2+ store, which leads to SOCE activation both at the stimulated site and, following repetitive stimulations, at more distant regions where no Ca2+ release events has occurred [58]. An increase in ATP concentration caused a dose-dependent reduction in [Ca2+]ER that induced a gradual increase in SOCE activity; furthermore, SOCE underwent periodic cycles of rapid activation and slow deactivation during the slow intracellular Ca2+ oscillations that can be elicited by prolonged stimulation of CPAE cells with a submaximal concentration of ATP [59]. These observations were recently confirmed in human umbilical vein endothelial cells (HUVECs), where local PLC activity generates spatially-restricted Ca2+ pulses at the front edge of migrating cells, thereby causing local ER Ca2+ depletion and SOCE activation [60].
2.2. The Pharmacological Activation of SOCE by ER Ca2+ Mobilization
SOCE can also be activated by the pharmacological depletion of the ER Ca2+ by means of multiple drugs. The most common strategy to evoke SOCE in endothelial cells (and in circulating ECFCs) is represented by the blockade of Sarco-Endoplasmic Reticulum Ca2+ (SERCA) activity with cyclopiazonic acid (CPA) [61,62,63,64], a mycotoxin and a fungal neurotoxin produced by the moulds Aspergillus and Penicillium and by thapsigargin [38,39,51,63], a plant-derived lactone, whereas 2,5-di-tert-butylhydroquinone (tBHQ) was mostly exploited in earlier studies [65,66,67]. The blockade of SERCA activity results in passive ER Ca2+ efflux from leakage channels, whose molecular identity in endothelial cells remains to be unveiled [68]. The following reduction in [Ca2+]ER is sufficient to activate SOCE on the PM in an InsP3-independent manner [52]. Extracellular application of thapsigargin (whose inhibitory effect on SERCA is irreversible) has routinely been exploited to detect SOCE by whole-cell patch-clamp electrophysiology [38,39,43,46,69], while both CPA and thapsigargin may be used to elicit SOCE during Ca2+ imaging recordings [38,39,61,62,63,64]. An alternative strategy to induce store-operated inward currents is through the intracellular infusion of InsP3 (to induce ER Ca2+ depletion) and/or a high concentration (e.g., 20 mM) of BAPTA (to buffer Ca2+ leaking out from the ER) through the patch pipette [35,38,43]. The Ca2+ ionophore, ionomycin, can also activate endothelial SOCE [70,71,72], but its use has been limited by the evidence that this drug can increase the Ca2+ permeability of other organelles, such as mitochondria [73].
2.3. The Search for the Coupling Mechanism between ER Ca2+ Depletion and SOCE Activation in Vascular Endothelial Cells
The mechanisms whereby a reduction in [Ca2+]ER leads to SOCE activation in endothelial cells [28,30], as well as in both non-excitable [52] and excitable cells [74], have been object of intense scrutiny over the years. A groundbreaking discovery was published in 2005, when Roos et al. demonstrated that stromal interaction molecule 1 (STIM1) was crucial to elicit SOCE [75]. Shortly thereafter, Liou et al. showed that STIM1 served as the sensor of ER Ca2+ concentration ([Ca2+]ER) that is activated upon ER Ca2+ emptying [76]. Subsequent investigations rapidly demonstrated that STIM1 could interact with the newly discovered Orai channels to mediate the classical ICRAC [77,78,79] or with members of the TRPC sub-family of non-selective cation channels, e.g., TRPC1, to mediate the ISOC [80,81,82]. Interestingly, STIM1, Orai1, and TRPC1 can interact either directly, i.e., by forming supermolecular ternary complexes [80,83,84], or functionally, e.g., through Orai1-dependent insertion of TRPC1 channels on the PM [85]. The potential interaction of the STIM1-gated Ora1 and TRPC1 channels, which display distinct Ca2+ permeabilities and Erev (see below), expands the repertoire of electrophysiological features that can be presented by store-operated currents. These findings were rapidly reproduced in endothelial cells, in which it became quickly evident that STIM1 could associate with both Orai1 [38,39,40] and TRPC1 channels to mediate SOCE [41,86,87].
3. Ca2+-Selective vs. Non-Selective Cation Currents Activated by ER Ca2+ Store Depletion
As anticipated in Section 2.3, SOCE can be mediated by at least two distinct cation channels, which display clearly different electrophysiological features, i.e., ICRAC and ISOC (Figure 1) [88,89]. An additional ICRAC-like current, which is mediated by STIM1, TRPC1, TRPC4, and Orai1, has also been reported (Figure 1) [51,90,91].
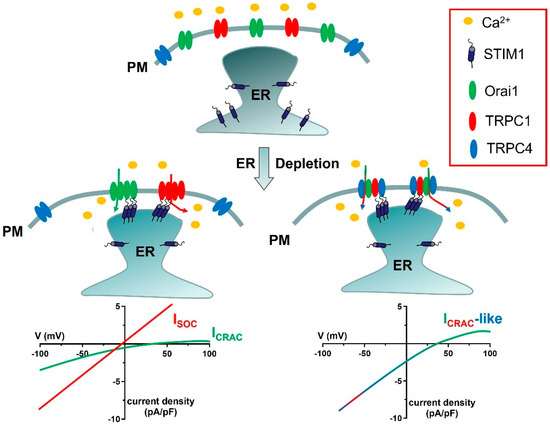
Figure 1.
Proposed models for ICRAC, ISOC, and ICRAC−like currents. In the absence of extracellular stimulation, STIM1 is homogeneously distributed within ER cisternae, whereas Orai1, TRPC1, and TRPC4 are located on the PM. Upon depletion of the ER Ca2+ store, STIM1 aggregates and translocates in close apposition to the PM, thereby recruiting Orai1 hexamers into spatially confined puncta and activating the ICRAC. Orai1−mediated extracellular Ca2+ entry can cause TRPC1 insertion into the plasma membrane (shown in Figure 2), thereby enabling TRPC1 activation by STIM1 and activating the ISOC. Finally, STIM1 can determine the assembly of a complex ion channel signalplex consisting also of Orai1, TRPC1, and TRPC4 and responsible for the development of ICRAC−like currents. As explained in Section 6.1, this supermolecular channel complex includes 1 TRPC1 subunit and 2 TRPC4 subunits. The lower current density of the ICRAC as compared to the ISOC and the ICRAC-like current reflects the single-channel conductance of Orai1 channels, which is 1000-fold lower as compared to TRPC channels. The current density is defined by the ratio between the magnitude of an ion current, in pA, and the cell membrane capacitance, in pF.
3.1. STIM and Orai Proteins Mediate the ICRAC
The ICRAC has been initially described and characterized in studies carried out in Jurkat T cells [92,93] and mast cells [32]. The ICRAC displays a clearly discernible electrophysiological fingerprinting, including high Ca2+-selectivity (≈1000-fold more conducive for Ca2+ over Na+), low single-channel conductance (≈20 fS, i.e., 1000-fold lower than most ion channels), Na+ conduction upon removal of extracellular Ca2+ and Mg2+, inward-rectification (i.e., the property to preferentially conduct the current into the cell), fast Ca2+-dependent inactivation (CDI), and no clear Erev up to +60 mV [32,93,94]. Large scale RNA interference (RNAi) screens—one using HeLa cells and four using Drosophila S2 cells—provided the straightforward evidence that the ICRAC is mediated by the physical interaction between STIM proteins on the ER and Orai channels on the PM (Figure 1) [75,77,78,79].
Studies conducted on immune cells showed that, in the absence of extracellular stimulation, STIM1 and Orai1 proteins are uniformly distributed, respectively, in the ER and PM. Following agonist stimulation, InsP3-dependent reduction in [Ca2+]ER stimulates STIM1 proteins to oligomerize and relocalize to ER-PM junctions, known as puncta, where peripheral ER cisternae come in close contact (10–25 nm) with the PM. Within these junctions, STIM1 binds to and gates hexamers of Orai1 channels through a trap-diffusion mechanism, thereby activating SOCE [94,95]. The mechanistic coupling between ER Ca2+ release and ICRAC/SOCE activation is strengthened by the tonic inhibition of InsP3Rs by inactivated STIM proteins [96]. Subsequent evidence confirmed that STIM1 and Orai1 support SOCE in virtually all cell types [52,74,94,95]. Orai1 presents a shorter splicing variant, known as Orai1β, which lacks 64 a.a. in the NH2-terminal tail but is still activated by ER Ca2+ depletion and can therefore mediate the ICRAC [97]. However, Orai1β displays faster mobility in the PM [98] and a less pronounced CDI [97]. Orai1 presents two paralogue proteins, namely Orai2 and Orai3, which present different electrophysiological features [99,100]. For instance, the fast CDI is more pronounced in Orai3 as compared to Orai2, while Orai1 presents the slowest fast CDI among the three Orai isoforms [100]. Therefore, Orai1 proteins encode CRAC channels that exhibit larger ICRAC and SOCE as compared to either Orai2 or Orai3 [95,100]. However, Orai2 and Ora3 can assemble with and negatively regulate Orai1 function [95]. Global knockout mice and CRISPR/Cas9 gene knockout in HEK-293 cells revealed that Orai1 can heteromerize with Orai2 and Orai3 in naive CRAC channels; these may, therefore, comprise different stoichiometric assortments of Orai isoforms and present distinct electrophysiological profiles as compared to Orai1 hexamers, thereby enhancing the bandwidth of intracellular Ca2+ signals [96,101].
STIM1 has a paralogue protein, namely STIM2 [76], which has a lower Ca2+ affinity (~500 µM vs. ~200 µM) and has long been thought to regulate resting Ca2+ entry and maintain [Ca2+]ER [102]. In accord, a recent investigation showed that constitutive InsP3-mediated ER Ca2+ release promotes STIM2 pre-clustering at ER-PM junctions, thereby recruiting Orai1 channels and ensuring basal Ca2+ influx even in the absence of agonist stimulation [103,104]. Furthermore, STIM2 supports SOCE activation across the whole spectrum of agonist concentrations [96] and is indispensable to recruiting STIM1 to Orai1 channels also in response to weak agonist stimulation [103,104]. STIM2 presents alternative splice variants, such as STIM2.1, which inhibits SOCE by preventing Orai1 clustering in several cell types [105]; a long variant, known as STIM1L, which is mainly expressed in skeletal muscle [106]; and a short splice variant, termed STIM1B, which is located in neurons and results in slower ICRAC kinetics and inactivation [107].
3.2. TRPC Channels Mediate the ISOC: Molecular Interaction with STIM1 and Functional Interplay with Orai1
The term ISOC encompasses a variety of store-operated non-selective cation currents that present a different electrophysiological fingerprinting as compared to the highly Ca2+-selective CRAC channels (Figure 1) [88,108]. The ISOC is featured by an Erev ranging from 0 to ~+10 mV; slightly linear (or outwardly rectifying) IV relationship; permeability to K+ (outgoing) and Na+, Cs+, and Ca2+ (ingoing); and single-channel conductance in the pS range (~5–23 pS) [88,108]. These electrophysiological properties are quite different from those exhibited by CRAC channels but are strongly reminiscent of those presented by TRPC channels, which are all activated in response to PLC activation [108,109]. Six TRPC proteins (TRPC1, TRPC3, TRPC4, TRPC5, TRPC6, and TRPC7) have been identified in humans [108]. TRPC isoforms have been subdivided into four subgroups, i.e., TRPC1, TRPC2, TRPC4/TRPC5, and TRPC3/TRPC6/TRPC7, based upon their sequence homology and similarities in gating mechanisms [108,110]. The available data concur to support a major role for TRPC1 in mediating the ISOC [88,89,111]. The Ambudkar group has provided solid evidence that, in human salivary gland (HSG) cells, InsP3-mediated ER Ca2+ depletion stimulates the ICRAC via the formation of a STIM1-Orai1 complex, thereby triggering extracellular Ca2+ entry. The subsequent elevation in submembrane Ca2+ concentration promotes the exocytosis of TRPC1-containing vesicles, thereby inserting TRPC1 channels on the PM (Figure 2). Herein, TRPC1 is directly gated by STIM1 to mediate the ISOC [85,112], and assembles into a signalling complex that already contains Orai1 and is located within ER-PM junctions [80,84]. The following studies confirmed that the dynamic STIM1/Orai1/TRPC1 ternary complex could be formed and mediate SOCE in other cell types, including human platelets [83] and megakaryocytes [113]. In agreement with these observations, Orai1-mediated extracellular Ca2+ entry can activate endogenous TRPC1 channels in HEK293 cells [114,115]. It is still disputed whether Orai1α and Orai1β can interchangeably serve to recruit and activate TRPC1 [97] or whether only Orai1α is required [116]. Conversely, STIM1L is more prone to gate TRPC1 than STIM1 [115]. The two distinct STIM1/Orai1 and STIM1/TRPC1 complexes could finely tune distinct Ca2+-dependent functions in HSG cells, in which the ICRAC elicits a local Ca2+ signal that stimulates the nuclear translocation of the transcription factor, nuclear factor of activated T-cells (NFAT), while the ISOC leads to a global increase in [Ca2+]i that engages a different transcription factor, i.e., nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) [85,112].
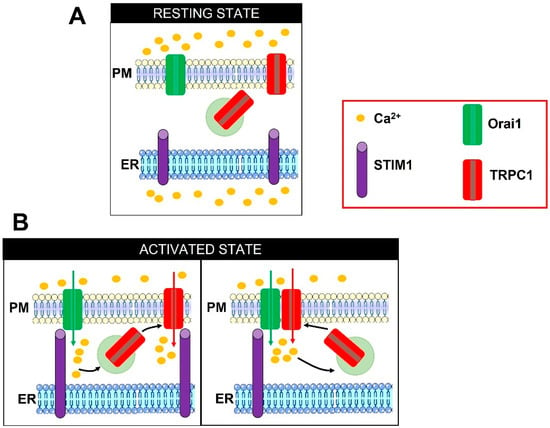
Figure 2.
Illustrations describing the two proposed models of Orai1-dependent TRPC1 activation. In the absence of extracellular stimulation, TRPC1 is located both on the PM and on submembrane vesicles (A). Depletion of the ER Ca2+ store prompts STIM1 to oligomerize, extend the cytosolic COOH-terminal domain towards the PM, translocate to ER-PM junction, and physically engage Orai1 to mediate the ICRAC (B, left panel). The following influx of Ca2+ can induce the exocytosis of TRPC1-containing vesicles. TRPC1 is inserted into the PM in close apposition to Orai1 and is thereafter activated by STIM1 to mediate the ISOC (B, left panel). Alternately, TRPC1 can physically interact with Orai1 and indirectly become store-operated (B, right panel). Adapted from [88].
The model presented above can be further implemented by recalling that STIM1 can also bind to and directly activate other TRPC isoforms, i.e., TRPC4 and TRPC5, and that STIM1-regulated TRPC channels can confer store dependency to their heteromeric partners, such as TRPC3 and TRPC6 [81,117]. Furthermore, TRPC1, TRPC4 and TRPC5 can assemble with each other into STIM1-gated heteromultimers, such as TRPC1/TRPC4 in human myoblasts [115,118] and TRPC1/TRPC4/TRPC5 in rat neonatal right ventricular cardiomyocytes [119] and mouse hippocampal neurons [120].
3.3. Do or Do Not Orai1 and TRPC1 form Heteromeric Channels? An Ongoing Controversy
The evidence that Orai1 and TRPC1 form a ternary complex including STIM1 in several cellular models led some authors to suggest that that these Ca2+-permeable channels do not only mediate distinct store-dependent currents but could also assemble into a heteromeric channel (Figure 1) [121,122,123]. According to this hypothesis, Orai1 and TRPC1 could both contribute to form the channel pore, thereby supporting a cation current with mixed properties between ICRAC and ISOC (e.g., Erev ranging between 0 and +60 mV, larger Na+ selectivity, weak inward rectification or quasi-linearity). Alternatively, Orai1 has been suggested to serve as regulatory subunit by conferring store sensitivity to TRPC1 activation [83,124,125,126]. The prediction that TRPC1 and Orai1 might assemble and form heteromeric SOCs that exhibit electrophysiological features distinct from those mediated by TRPC1 and Orai1 alone has been largely discounted [108,121], with a few notable exceptions [123,124,125]. Nevertheless, in hypertrophied right ventricular cardiomyocytes, the integration of Orai1 in the STIM1/TRPC1/TRPC4 complex turns the ISOC into an inwardly-rectifying non-selective cation current that reverses at around 0 mV [90]. These features resemble the mixed non-selective cation current that has been reported in aldosterone-treated rat left ventricular adult cardiomyocytes: this current is activated by depletion of the sarcoplasmic reticulum (SR), reverses at around 0 mV, presents a slight inward-rectification, and is significantly reduced by a dominant-negative Orai1 [119]. Interestingly, depletion of the SR Ca2+ store in these cells promoted the formation of a macromolecular complex including STIM1, TRPC1, TRPC4, and Orai1 [119]. Similarly, ER Ca2+ store depletion induced a non-selective cation current, which presented an inwardly-rectifying IV relationship, but with an Erev ranging between–5 and 0 mV, in human macrophages [91] and in mouse Müller glia cells [127]. These currents were mediated by the synergistic activation of STIM1, Orai1, and TRPC1, although these studies did not document the formation of supermolecular ternary complex. A scrutiny of the literature reveals that additional store-operated currents with mixed electrophysiological features between ISOC and ICRAC have been reported. In human gingival keratinocytes [128] and bovine adrenal cells [129], SOCE involves the early activation of an ICRAC-like current that shows an Erev ranging between +30 and +40 mV and is suppressed by the genetic deletion of TRPC4. TRPC4 presents a Ca2+/Na+ permeability ratio (PCa/PNa) around 1.1, while the shape of the IV relationship depends on its physical interaction with other TRPC subunits. For instance, heterotetrameric TRPC4 channels present a doubly-rectifying IV relationship [130], while heteromeric combinations of TRPC1 with TRPC4 result in a non-selective cation current that reverses at around 0 mV and is outwardly-rectifying [131]. These observations strongly suggest that the ICRAC-like current recorded in human gingival keratinocytes [128] and bovine adrenal cells [129] could involve an endogenous subunit, e.g., Orai1, that had not been identified at that time and conferred to TRPC4 (maybe combined with TRPC1) the ability to inwardly-rectify and conduct Ca2+ better than Na+ [132].
4. The Endothelial ICRAC Is Mediated by STIM1 and Orai1
The first evidence that SOCE is expressed in vascular endothelial cells has been provided in HUVECs by Ron Jacob and coworkers, who found a Ca2+ influx pathway that was “controlled by the degree of fullness of the internal (Ca2+) store”, i.e., the ER [133,134]. After this seminal discovery, the quest for the endothelial ICRAC immediately started [49] and was based upon the electrophysiological fingerprinting of CRAC channels in mast cells [32].
4.1. Electrophysiological Properties of the Endothelial ICRAC
The first attempts to measure the ICRAC by using the whole-cell patch-clamp technique in endothelial cell types failed whatever the chemical agonist employed to reduce the [Ca2+]ER and the intracellular Ca2+-buffering conditions (i.e., introduction or not of Ca2+ buffers, such as EGTA or BAPTA, in the pipette solution), as shown in Table 1.

Table 1.
Unsuccessful attempts to measure the endothelial ICRAC.
The first electrophysiological demonstration that vascular endothelial cells present the ICRAC was provided by Cristina Fasolato and Bernd Nilius in 1998 [35]. The authors found that, in CPAE cells, depletion of the ER Ca2+ store evoked by tBHQ induced an inward current bearing the same electrophysiological properties of the ICRAC recorded in Jurkat cells as the positive control (Table 2): (1) inward rectification; (2) Erev > +40 mV; (3) small current density (−0.5 pA/pF at −80 mV, 20 mM Ca2+ in the external solution); (4) high Ca2+ selectivity and sensitivity to external La3+ (10 µM) and poor Ba2+ permeability; and (5) large permeability to monovalent cations in the presence of a divalent-free medium [35]. A decade later, these findings were confirmed by Mohamed Trebak and coworkers in HUVECs [38]. The ICRAC recorded in these cells was evoked by intracellular dialysis of BAPTA or extracellular perfusion of thapsigargin displayed a rather small current density (around −0.3 pA/pF at −100 mV, 20 mM Ca2+ in the external solution), an inwardly-rectifying IV relationship, and an Erev > +40 mV [38] (Table 2). Furthermore, this CRAC channel was also sensitive to extracellular lanthanides (10 µM Gd3+) and conducted large and fast-inactivating inward Na+ currents upon removal of extracellular divalent cations [38]. A subsequent study failed to measure a detectable ICRAC in HUVECs [39], but the latter investigation exploited VEGF to stimulate the ICRAC and this agonist could induce a smaller reduction in [Ca2+]ER, thereby resulting in the activation of a sub-pA ionic current [39]. Alternately, VEGF could target a sub-compartment of the ER that presents a weaker coupling to the CRAC channels on the PM as compared to thapsigargin [136]. These pioneering investigations demonstrated that vascular endothelial cells express an ICRAC that displays similar electrophysiological properties to that recorded from mast cells.
Table 2.
Electrophysiological features of the endothelial ICRAC.
4.2. STIM1 and Orai1 Mediate the Endothelial ICRAC
The ICRAC recorded in HUVECs by Mohamed Trebak and coworkers was abolished by the genetic deletion of either STIM1 or Orai1 with specific small interfering RNAs (siSTIM1 and siOrai1, respectively) [38]. Conversely, this ICRAC was not affected by the genetic suppression of TRPC1 and TRPC4 with a similar strategy [38]. As widely illustrated in other cell types, STIM1 showed a distributed microtubule-like distribution pattern in the presence of a replete ER, whereas it was stimulated by the depletion of ER Ca2+ to relocalize in subplasmalemmal clusters [39]. A study conducted on the HUVECs-derived cell line, EA.hy926, showed that intracellular Ca2+ overload may prevent STIM1-Orai1 interaction even when the ER Ca2+ pool is depleted [137]. However, mitochondrial Ca2+ uptake can prevent the Ca2+-dependent inhibition of STIM1-Orai1-dependent SOCE [138]. Intriguingly, the small current density of the ICRAC in HUVECs was associated with the low basal levels of STIM1 expression: in accord, STIM1 overexpression produced an ICRAC whose current density was similar to that measured in mast cells [38]. This feature might explain the difficulty in detecting a measurable ICRAC in different endothelial cell types (Table 1) or in the same endothelial cell types (e.g., HUVECs), but provided by different sources [38,39]. Three additional mechanisms might contribute to reduce the current density of the endothelial ICRAC below the resolution limit of conventional patch-clamp amplifiers (Table 1). First, ultrastructural analysis revealed that the distance between ER and PM in microvascular endothelial cells (87 nm) is significantly longer as compared to macrovascular endothelial cells (8 nm) [139]. If the minimum distance between the membranes that is required to accommodate STIM1- and Orai1-contaning puncta is 10–25 nm [52], this could explain why a true ICRAC has never been recorded and SOCE is often lower [140] or even undetectable [141] in microvascular endothelial cells. Second, Orai2 may negatively modulate Orai1-mediated SOCE, as shown in the bovine brain capillary endothelial cell line, t-BBEC117 [142]. Therefore, changes in the expression levels of Orai2 (and, possibly, Orai3) in endothelial cells from different vascular districts and species could underlie the difficulties in recording the endothelial ICRAC. Third, Orai1 activity can ben modulated by multiple intracellular signalling pathways, which could thereby finely tune ICRAC amplitude.
Agonist-evoked SOCE was compromised in HUVECs lacking STIM1 or Orai1 [38,39]. Therefore, SOCE was then exploited as readout of Orai1 activation to investigate the regulatory mechanisms of the endothelial ICRAC, which include: (1) tetraspanin Tspan18, which regulates Orai1 clustering and activity on the PM [22]; (2) SOCE-associated regulatory factor (SARAF) [143,144], which colocalizes with Orai1 and modulates the interaction between STIM1 and Orai1 during the early steps of SOCE activation [145]; (3) membrane cholesterol, which is required for STIM1-Orai1 interaction and whose reduction can significantly attenuate SOCE [146]; (4) redox signalling, which can directly inhibit Orai1 activity [147]; (5) the endocannabinoid N-arachidonoyl glycine (NAGly) [148] and the G protein-coupled estrogen receptor 1 (GPER) [149], which prevent STIM1-Orai1 interaction (6) STIM1 phosphorylation at Y361 via proline rich kinase 2 (Pyk2), which enables Orai1 recruitment to STIM1 puncta [150]; and (7) STIM1 phosphorylation by p38β mitogen-activated protein kinase (p38β MAPK), which inhibits SOCE activation [151]. Subtle changes in the extent of STIM1 or Orai1 modulation by any of these signalling pathways could affect ICRAC activation and, therefore, favour or oppose ICRAC recording depending not only upon the endothelial cell type, but also upon the experimental conditions.
4.3. The Role of STIM and Orai Proteins in Endothelial Function
The seminal discovery by the Trebak group [38] stimulated a flurry of studies that employed genetic tools or rather specific Orai1 inhibitors, such as low micromolar doses of trivalent cations and the pyrazole derivatives, YM-58483 (also known as BTP-2), Synta66 (S66), GSK-7975A (GSK) or Pyr6 [52,152,153], to unveil the endothelial functions modulated by the ICRAC (Table 3).
The primary function fulfilled by STIM and Orai in endothelial cells is to refill the ER with Ca2+ (Table 3). A landmark study conducted on Ea.hy926 cells revealed that ER Ca2+ replenishment after InsP3-evoked ER Ca2+ mobilization is contributed by Orai1, as well as by the reverse-mode of the Na+/Ca2+ exchanger, and is physically supported by specific ER-PM junctions [154]. Furthermore, endothelial STIM1 and Orai1 support angiogenesis (Table 3) [24,30,155]. Genetic silencing of STIM1, STIM2, or Orai1 inhibited proliferation by causing cell cycle arrest at S and G2/M phase in HUVECs [38]. In the same endothelial cell type, genetic knockdown or pharmacological blockade with S66 or GSK abolished VEGF-induced extracellular Ca2+ entry, proliferation, and tube formation [39,143]. In addition, genetic silencing of Orai1 interfered with parathyroid hormone-induced HUVEC proliferation and migration [156] and with VEGF-induced Ca2+ entry and migration in human aortic endothelial cells (HAECs) [157]. In agreement with these in vitro findings, GSK inhibited aorta sprouting and the development of retinal vasculature in mice [143]. Intriguingly, RTK activation and PLCγ signalling were spatially restricted at the front of the migrating leader cell in HUVEC monolayers; herein, PLCγ promotes local InsP3-induced ER Ca2+ pulses that recruit STIM1 and selectively activate SOCE to accelerate cell-matrix adhesion and stimulate forward migration [60]. In addition, Orai1-mediated SOCE could promote angiogenesis by stimulating the nuclear translocation of NFAT (Table 3) [24,156].
Endothelial STIM1 and/or Orai1 can also control blood pressure by driving NO release (Table 3) [158]. Intriguingly, Lothar Blatter and his colleagues have provided evidence that the Ca2+ source responsible for the Ca2+-dependent recruitment of the eNOS is provided by SOCE rather than by InsP3-induced ER Ca2+ release [28,159]. An early investigation showed that genetic suppression of STIM1 inhibited thrombin-induced extracellular Ca2+ entry and NO release in porcine aortic endothelial cells [160]. A subsequent study generated a transgenic mouse model selectively lacking the endothelial STIM1, which showed impaired endothelium-dependent relaxation due to a robust reduction in acetylcholine-induced NO release [161]. These findings were confirmed by another study using endothelial cell-specific STIM1 knockout mice, which further showed a significant elevation in blood pressure [162]. Recent findings suggest that Orai1 mediates endothelium-dependent NO production also at the neurovascular unit, where NO is crucial to increasing cerebral blood flow in response to neuronal activity [4,163,164]. The pharmacological blockade of Orai1 inhibited NO release induced in human cerebrovascular endothelial cells by multiple neurotransmitters, such as glutamate [165,166], GABA [167,168], and acetylcholine [62], and neuromodulators, including histamine [169] and arachidonic acid [170]. Interestingly, Orai2 is the only isoform expressed in mouse cerebrovascular endothelial cells [21], which is consistent with the crucial role played by Orai2 in neuronal SOCE in mice [74]. The pharmacological inhibition of Orai2 also attenuated endothelium-derived NO production at the mouse neurovascular unit [21,171]. Orai3 has been shown to mediate VEGF-induced, arachidonic acid-dependent extracellular Ca2+ entry in HUVECs [172], but the role of this Orai isoform in endothelial SOCE is still unknown.
Interestingly, the Trebak group showed that STIM1 could control endothelial permeability in a SOCE-dependent manner. STIM1 was found to interact with the thrombin receptor, thereby recruiting the guanosine triphosphatase RhoA to stimulate myosin light chain phosphorylation and actin stress fiber formation [161]. A follow-up investigation confirmed that SOCE is also dispensable for histamine-induced formation of inter-endothelial gaps in microvascular endothelial cell monolayers [173]. In addition, the Hu group reported that, in HUVECs, a Leu-to-Pro substitution in the signal peptide enabled STIM1 to translocate to the nuclear membrane in response to a reduction in [Ca2+]ER [174]. The mutated STIM1 was found to amplify the RyRs-mediated increase in nuclear Ca2+ concentration, thereby enhancing the subsequent cAMP responsive element binding protein activity, matrix metalloproteinase-2 (MMP-2) gene expression, and endothelial tube formation. It is noteworthy that the nuclear Ca2+ elevation was independent on SOCE activation [174]. These findings further expand the versatility of STIM1 signalling and place the question as to whether also Orai1 can signal in a non-canonical manner in endothelial cells.
Table 3.
Endothelial functions modulated by the ICRAC.
Table 3.
Endothelial functions modulated by the ICRAC.
| Endothelial Cell Type | Evidence of ICRAC Involvement | Function | Reference |
|---|---|---|---|
| Ea.hy926 | Orai1DN | ER Ca2+ refilling | [154] |
| HUVECs | siSTIM1 and siOrai1, OraiDN, Synta66, BTP-2, GSK | Proliferation, cell cycle control (S and G2/M phase), tube formation | [38,39,143] |
| HUVECs | siSTIM1, STIM1over, BTP-2 | Migration | [60] |
| Rat aortic endothelial cells | 10–100 µM GSK (IC50 = 34.22 µM) | Aorta sprouting | [143] |
| Mouse retinal vasculature | 2.6–31.8 mg/kg GSK (IC50 = 18.4 mg/kg) | Neovessel formation | [143] |
| Porcine aortic endothelial cells | siSTIM1 | NO release | [160] |
| Endothelial cells from mesenteric resistance and thoracic arteries | STIM1EC-/- mice | NO release and vasorelaxation | [161,162] |
| Mouse cerebrovascular endothelial cells | 20 µM BTP-2, 10 µM La3+ | NO release | [175] |
| Human cerebrovascular endothelial cells | 10 µM Pyr6 | NO release | [62,169] |
Abbreviations: Orai1DN: expression of dominant negative Orai1; siOrai1: small interfering RNA selectively targeting Orai1; siSTIM1: small interfering RNA selectively targeting STIM1; STIM1EC-/- mice: endothelial cell-specific knockout mice; STIM1over: STIM1 overexpression.
4.4. The Role of STIM and Orai Proteins in Endothelial Dysfunction
STIM1 and/or Orai1 drive endothelial cell activation during the inflammatory response to noxious stimuli and infection (Table 4). Early investigations revealed that the genetic silencing of STIM1 or Orai1 inhibited NFAT-dependent gene expression in HUVECs exposed to the inflammatory mediators, histamine [176], and tumor necrosis factor-α (TNF-α) [177]. Furthermore, genetic suppression of Orai1 attenuated TNF-α-induced expression of the adhesion molecules, VCAM-1 and ICAM-1, in mouse aorta and reduced the amount of pro-inflammatory cytokines, such as MCP-1, IL-6, and IL-8, in the serum (Table 4) [177]. Consistently, Lipopolysaccharide (LPS), which is derived from bacterial endotoxin, failed to induce intracellular Ca2+ oscillations, to stimulate the nuclear translocation of NFAT, and to increase vascular permeability in a transgenic mouse model selectively lacking the endothelial STIM1 (Table 4) [178]. These protective actions against systemic inflammation and acute lung injury (ALI) were mimicked by the small-molecule drug, BTP-2 [178]. In addition, BTP-2 alleviated endothelial hyperpermeability and lung oedema in a mouse model of ventilation-induced lung injury Orai1 may also exacerbate endothelial dysfunction under hyperglycaemic conditions, e.g., in diabetes [14]. The expression levels of STIM1-2 and Orai1-3 increased in HAECs during chronic exposure to high glucose (25 mM) and were also enhanced in the aorta of diabetic patients and of diabetic mice [179]. Furthermore, high glucose induced the up-regulation of Ora1-3 proteins and enhanced SOCE activity in human coronary artery endothelial cells (HCAECs), thereby inducing endothelial hyperpermeability and hyperproliferation [180]. In agreement with these observations, the genetic silencing of STIM1 and Orai1 alleviated the increase in permeability induced by the pro-inflammatory mediator, High-Mobility Group Box 1 protein (HMGB1), in Ea.hy926 monolayers [181], whereas a selective siOrai1 alleviated tunicamycin-induced ER stress and unfolded protein response, which are a feature of atherosclerosis, were shown to be alleviated by a selective siOrai1 in HUVECs [182]. Thus, the mechanisms by which chronic inflammation or hyperglycaemia lead to atherosclerosis involve an increase in endothelial permeability and proliferation that depend on STIM1 and Orai1 (Table 4) [183]. This evidence led to the hypothesis that Orai1 could contribute to inflammation-induced pulmonary endothelial cell injury during SARS-CoV-2 infection [184]. Moreover, Orai1-mediated extracellular Ca2+ entry mediates histamine- or thrombin-induced release of von Willebrand factor, thereby favouring platelet aggregation and thrombi formation in mouse models of deep vein thrombosis and ischemia-reperfusion cardiac injury [22]. As shown for angiogenesis and inflammation, Orai1-mediated extracellular Ca2+ influx stimulates the nuclear translocation of NFAT to induce the expression of the gene encoding for von Willebrand factor [22].
Table 4.
Role of the ICRAC in endothelial dysfunction.
5. The Endothelial ISOC Is Mediated by STIM1, TRPC1, and/or TRPC4
The search for the endothelial ICRAC led to the discovery that ER Ca2+ depletion by stimuli that do not involve InsP3-induced Ca2+ release, i.e., the SERCA inhibitors CPA, thapsigargin, and tBHQ, induced either an ISOC [47,187,188] or an ICRAC-like current [44,189]. In the present section, we discuss the evidence supporting the notion that the endothelial ISOC is mediated by STIM1, TRPC1, and TRPC4, while in Section 6 we illustrate the findings indicating that the incorporation of Orai1 into this supermolecular complex turns the ISOC into an ICRAC-like current.
5.1. Electrophysiological Properties of the Endothelial ISOC
The quest for the endothelial ICRAC led to discovery of slightly different currents, which were activated by a reduction in [Ca2+]ER but displayed the electrophysiological features of an ISOC (Table 5) [49]. Depletion of the ER Ca2+ store with CPA in CPAE cells evoked a non-selective cation current that was carried by Na+, K+, Ca2+ and, possibly, the organic cation, tetraethylammonium, displayed a rather linear IV relationship and reversed close to 0 mV [187]. Furthermore, both CPA and the intracellular infusion of InsP3 caused the Ca2+-dependent recruitment of Ca2+-dependent K+ channels that induced endothelial hyperpolarization [187]. A membrane current bearing electrophysiological features similar to the ISOC was then recorded in HUVECs [37,43,188,190] and in human and rat PAECs (hPAECs and rPAECs, respectively) [191,192].
Table 5.
Electrophysiological features of the endothelial ISOC.
5.2. STIM1, TRPC1, and TRPC4 Mediate the Endothelial ISOC
Groschner et al. were the first to suggest that the endothelial ISOC was associated with an endogenous TRP channel likely belonging to the TRPC subfamily [37]. In agreement with this proposal, the Malik group reported that the intracellular infusion of InsP3 in HUVECs induced an ISOC that was inhibited by a selective antibody targeting an extracellular epitope of TRPC1 and enhanced by TRPC1 overexpression [43]. Subsequently, CPA-evoked ISOC was found to increase following TRPC4 overexpression in hPAECs [192].
The emerging evidence that TRPC1 and/or TRPC4 were both able to mediate SOCE and thereby control endothelial permeability, NO release, and gene expression [193,194,195,196,197], led to hypothesizing the assembly of a heterotetramer consisting of both subunits and recruitable by STIM1 in response to ER Ca2+ depletion (Figure 3). In agreement with this model, it was first revealed that TRPC1 and TRPC4 form a heterotetrameric channel in BAECs and mediate extracellular Ca2+ entry downstream of TKR activation. The evidence that neither subunit was phosphorylated suggested that the heterotetrameric complex was engaged upon ER Ca2+ depletion [198]. Subsequently, the Malik group showed that Ca2+ influx secondary to the physiological or pharmacological depletion of the ER Ca2+ store in mouse and human pulmonary artery microvascular endothelial cells (mPMECs and hPMECs, respectively) was inhibited by the genetic blockade of STIM1, TRPC1, and TRPC4 [41]. Coimmunoprecipitation studies revealed that STIM1 associates to TRPC1 and TRPC4 upon a reduction in [Ca2+]ER [41] and suggested that the physical interaction between TRPC1 and TRPC4 is favoured by caveolin-1 (Figure 3) [195,199,200]. Intriguingly, caveolin-1 may also promote the association of TRPC1 and TRPC4 on the PM with InsP3Rs on the ER [195,199,201]. The co-localization of InsP3Rs, TRPC1, TRPC4, and STIM1 [202], in the same signalplex could accelerate SOCE activation by promoting the large drop in [Ca2+]ER peripheral cisternae that is required to activate endothelial TRPC1 and TRPC4 channels, as recently shown for Orai1 in HeLa cells (Figure 3).
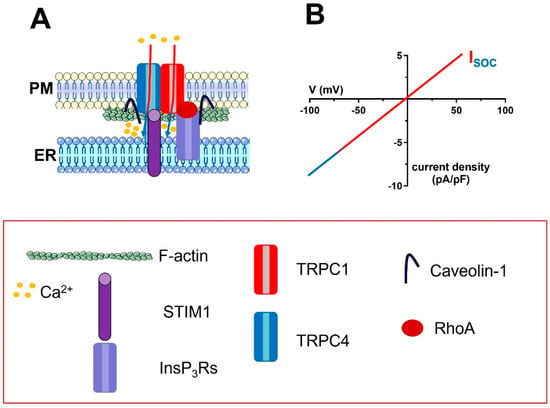
Figure 3.
The molecular architecture of the ISOC in vascular endothelial cells. (A), the endothelial ISOC is mediated by a complex ion channel signalplex that is located on plasmalemmal caveolae. The ion channel pore is contributed by TRPC1 and TRPC4 channels, which are informed about changes in [Ca2+]ER by STIM1. The signalling microdomain is enriched with InsP3Rs, which interact with the TRPC1 and TRPC4 subunits via caveolin-1. For sake of clarity, only the interaction between InsP3Rs and TRPC1 has been shown. The monomeric GTP−binding protein, RhoA, also supports the interaction between InsP3Rs and TRPC1 via F−actin polymerization (two bundles of F-actin were drawn beneath the plasma membrane). (B), the IV relationship of the endothelial ISOC.
The endothelial TRPC1 and TRPC4 channels can be modulated by multiple signalling pathways [5,203,204], but only a few of them were shown to modulate both channels. For instance, TNF-alpha Receptor Ubiquitous Signaling and Scaffolding protein (TRUSS) and tumor necrosis factor receptor-1 (TNF-R1) interact with TRPC1 and TRPC4 to enhance SOCE and promote ER refilling after agonist evoked InsP3-induced Ca2+ mobilization [205]. Furthermore, protein kinase Cα phosphorylated TRPC1 and increased the ISOC in HUVECs [43], whereas the monomeric GTP-binding protein RhoA favoured the association of InsP3Rs with TRPC1 after store depletion by promoting F-actin polymerization in hPAECs [201].
5.3. The Role of the ISOC in Endothelial Function and Dysfunction
The Malik group has provided a major contribution to unveiling the pleiotropic role played by the endothelial ISOC in endothelial cells (Table 6). The overexpression of TRPC1 increased vascular endothelial growth factor (VEGF)-induced extracellular Ca2+ influx and hyperpermeability in HUVECs and human dermal microvascular endothelial cells (HMECs) (Table 6) [193]. This effect was counteracted by angiopoietin-1, which interfered with the incorporation of InsP3Rs into the TRPC1-containing signalplex [193]. Moreover, genetic knockdown of TRPC1 also reduced angiotensin II-induced increase in [Ca2+]i and permeability in HUVECs [206]. Likewise, thrombin-induced extracellular Ca2+ entry and increase in endothelial permeability was dramatically enhanced by TRPC1 up-regulation in hPAECs (Table 6) [201]. In agreement with these findings, genetic knockdown or deletion of, respectively, TRPC1 and TRPC4 inhibited the nuclear translocation of the pro-inflammatory transcription factor, NF-κB, in hPAECs (Table 6). The ISOC was instrumental to activate AMPK and PKCδ, which are both required to activate NF-κB and increase endothelial permeability [207]. Similarly, tumor necrosis factor-α (TNF-α) induced ISOC activation and thereby promoted endothelial hyperpermeability in EA.hy926 monolayers by causing the up-regulation of TRPC1, but not TRPC4, protein [208]. TNF-α is a primary inflammatory mediator that increases vascular permeability during the early stages of sepsis, and its stimulatory effect on endothelial cells can be counteracted by inhibiting the ISOC with the chromogranin A (CGA)-derived fragment, CGA47–66 [208]. Finally, lead/malathion poisoning also increases the endothelial expression of TRPC1 and TRPC4 proteins and thereby causes an elevation in SOCE and permeability at the rat blood–brain barrier [209]. Therefore, these findings indicate that the ISOC supports the endothelial Ca2+ signals that reorganize the cytoskeletal architecture to induce gaps between adjoining cells, disrupt the endothelial barrier and increase vascular permeability [210].
An additional process that is seemingly mediated by the endothelial ISOC through NF-κB activation is cell survival during thrombin stimulation. In accord, HUVECs challenged with a prolonged exposure to thrombin activate the transcriptional pro-inflammatory program, but do not undergo apoptosis, due to concomitant NF-κB-dependent A20 expression [194]. More recent work suggested that TRPC1, as well as TRPC4, play an anti-apoptotic role also in rPAECs. Acute hypoxia resulted in the up-regulation of Galectin-3 (Gal-3), a member of the family of β-galactoside-binding lectins, which induced apoptosis by down-regulating both TRPC1 and TRPC4 proteins [211]. Therefore, the ISOC could be effectively targeted to treat PAH.
Recent evidence suggested that endothelial TRPC1-containing channels could also control angiogenesis. For instance, endothelial TRPC1 supported VEGF-induced angiogenic sprouting of intersegmental vessels in zebrafish larvae [212], maintained the plateau phase of VEGF-induced extracellular Ca2+ entry in MAECs [213], induced wound healing in HMECs [214], and promoted vascular regrowth and recovery of myocardial function in a mouse model of acute myocardial infarction [215]. The glycosidase protein, Klotho, promotes TRPC1 association to VEGF receptor 2, thereby favouring TRPC1-mediated extracellular Ca2+ entry in response to InsP3-dependent reduction in [Ca2+]ER [213]. In addition, VEGF could exploit TRPC4 to stimulate migration and tube formation in retinal microvascular endothelial cells [216]. Finally, the genetic silencing of TRPC1 and TRPC4 impaired proliferation and bidimensional tube formation in HUVECs [38,217].
Table 6.
Endothelial functions modulated by the ISOC.
Table 6.
Endothelial functions modulated by the ISOC.
| Endothelial Cell Type | Evidence of ISOC Involvement | Function | Reference |
|---|---|---|---|
| HUVECs, HMECs | TRPC1over, anti-TRPC1 antibody, 1 µM La3+ | VEGF-induced increase in endothelial permeability | [193] |
| HUVECs | siTRPC1 | Angiotensin II-induced increase in endothelial permeability | [206] |
| HUVECs | siTRPC1 | Thrombin-induced apoptosis | [194] |
| HUVECs | siTRPC1, TRPC1over | Ca2+- and TNFα-induced VCAM-1 upregulation and monocyte adhesion | [219] |
| hPAECs | TRPC1over | Thrombin-induced increase in endothelial permeability | [201] |
| hPAECs, mPAECs | siTRPC1, TRPC4-/- mice | Nuclear translocation of NF-κB | [207] |
| Zebrafish | TRPC1-/- | Vascular development | [212] |
| HUVECs | siTRPC1, siTRPC4 | Proliferation and tube formation | [38,217] |
| HRMECs | siTRPC4 | VEGF-induced migration and tube formation | [216] |
| MAECs | siTRPC1 | VEGF-induced Ca2+ entry | [213] |
| MCAECs | TRPC1-/- mouse | Migration and tube formation in vitro and neovessel formation in vivo | [215] |
Abbreviations: HMECs: human dermal microvascular endothelial cells; hPAECs: human pulmonary artery endothelial cells; HRMECs: human retinal microvascular endothelial cells; MAECs: mouse aortic endothelial cells; MCAECs: mouse coronary artery endothelial cells; mPAECs: mouse pulmonary artery endothelial cells; HUVECs: human umbilical vein endothelial cells; siTRPC1: small interfering RNA selectively targeting TRPC1; TRPC1over: TRPC1 overexpression.
Conversely, endothelium-dependent NO release and vasodilation were not affected in a transgenic mouse model devoid of TRPC1 [218]. This finding strongly suggests that TRPC1 is not tightly coupled to eNOS in vascular endothelial cells [218].
6. The ICRAC-Like Current: The Third Store-Operated Current in Endothelial Cells
Some of the earlier reports of the endothelial ISOC described a membrane current that actually displayed intermediate electrophysiological features between ISOC and ICRAC (Table 7) [49]. Vaca and Kunze showed that, in BAECs, tBHQ elicited a non-selective cation current that displayed an inwardly-rectifying IV relationship, presented a single-channel conductance of 5 pS between −40 and −80 mV, reversed at ≈+6 mV, and could also be activated in response to physiological depletion of the InsP3-sensitive ER Ca2+ pool with ATP or bradykinin (Table 7) [44,189]. Subsequently, the Stevens group showed that thapsigargin activated an inwardly-rectifying cation current, which reversed between +30 mV and +40 mV and presented Ca2+-dependent inactivation, in rPAECs (Table 7) [69,140,220,221]. Intriguingly, the Na+ permeability of this store-dependent current was progressively reduced by a gradual increase in extracellular Ca2+ concentration [86]. This behaviour, which is known as anomalous mole fraction [52], is typical of Ca2+-selective channels, but the Erev (+40 mV, more negative than the Erev of a pure inward Ca2+ current) and the occurrence of a small outward current (which reflects cytosolic K+ efflux) indicate that this current is also contributed to by monovalent cations and is therefore non-selective. The Nilius group also showed that depleting the ER by dialyzing the cytosol with InsP3 through the patch-pipette and adding tBHQ to the bath elicited a store-operated current in MAECs (Table 7) [87]. This current reversed at ≈+40 mV and displayed a less robust inward rectification and lower Ca2+ permeability (PCa/PNa ≈ 160 vs. PCa/PNa ≈ 1000) as compared to the archetypal ICRAC [52,222]. Finally, Frieden and colleagues found that, in EA.hy926 cells, thapsigargin evoked a store-dependent current that presented intermediate electrophysiological features between Ca2+-selective and non-selective cation channels (Table 7): (1) inwardly-rectifying IV relationship; (2) sensitivity to low micromolar doses of La3+; (3) anomalous mole fraction; and (4) rather negative Erev, i.e., ≈−7 mV [46]. In our opinion, these pieces of evidence strongly suggest that this endothelial store-operated current should not be coined either as ISOC or as ICRAC, but rather as ICRAC-like [46], as suggested for the analogous store-dependent currents described in rat left ventricular adult cardiomyocytes [90,118], human gingival keratinocytes [128], and bovine adrenal cells [129].
Table 7.
Electrophysiological features of the endothelial ICRAC-like current.
6.1. STIM1, TRPC1, TRPC4, and Orai1 Mediate the Endothelial ICRAC-Like Current
The gating mechanisms and the electrophysiological features of the ICRAC-like current hint at a model according to which the channel pore is contributed to by both TRPC and Orai1 subunits, which assemble into a heteromeric complex that is informed about ER Ca2+ content by STIM1 [90,119,123]. In accord, thapsigargin-evoked ICRAC-like currents in rPAECs were significantly inhibited by the genetic knockdown of TRPC1 through a specific siRNA [86]. Subsequently, it was found that this ICRAC-like current requires spectrin-protein 4.1 interaction (Figure 4) [220] and protein 4.1 binding to TRPC4 (Figure 4) [69]. In parallel, the Nilius group discovered that the ICRAC-like current was suppressed upon genetic knockdown of TRPC4 with a selective siRNA and in MAECs isolated from TRPC4-deficient mice [87]. A landmark study to understand how the ISOC can be turned into an ICRAC-like current was published in 2012, when the Stevens group exploited a novel Förster resonance energy transfer (FRET) approach to demonstrate that Orai1 could be incorporated into a protein 4.1-bound TRPC1/TRPC4 channel in rPAECs (Figure 4) [51]. They found that Orai1 is constitutively coupled to TRPC4 and can associate to TRPC1 in response to ER Ca2+ depletion [51]. This supermolecular channel complex includes 1 TRPC1 subunit and 2 TRPC4 subunits (Figure 4) [51,123], while the exact Orai1 stoichiometry remains to be investigated. Electrophysiological recordings revealed that genetic knockdown of Orai1 reduced the amplitude of the inward current at −80 mV, shifted the Erev from +40 mV to +20 mV, reduced the Ca2+-dependent inactivation, and abolished the anomalous mole fraction behaviour [51]. A subsequent report from the same group confirmed that, although Orai1 is not strictly required to mediate Ca2+ entry in rPAECs, it favours the coupling between ER and PM channels during low ER Ca2+ depletion events and that it restricts the Na+ permeability of the ICRAC-like current [50]. Thus, as observed with the ISOC, Orai1 does not contribute to SOCE, but it finely regulates the cation permeability of CRAC-like channels: from non-selective (no Orai1 present) to Ca2+ (semi)-selective (Orai1 present in combination with TRPC channels). Finally, Cioffi and coworkers showed that genetic deletion of STIM1 reduced SOCE and that STIM1 phosphorylation upon ER Ca2+ release contributed to inactivate the ICRAC-like current in rPAECs [223]. The evidence that Orai1 can be incorporated into and confer CRAC-like features to TRPC1/TRPC4 heteromers can be crucial to unveiling the molecular architecture of the wide range of store-dependent currents, i.e., from truly to less Ca2+-selective, also recorded in other types of mammalian cells.
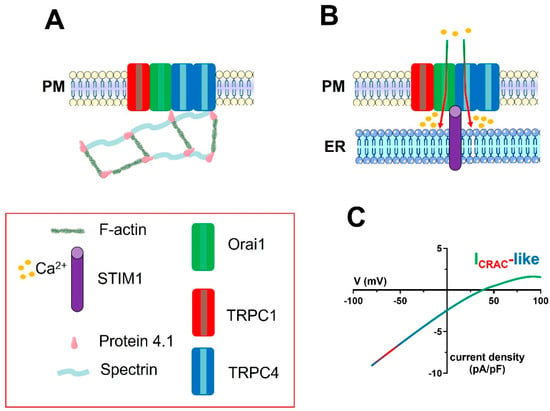
Figure 4.
The molecular architecture of the ICRAC-like channel in vascular endothelial cells. A series of studies carried out on rPAECs demonstrated that the ion channel signalplex mediating the endothelial ICRAC−like currents is contributed to by one TRPC1 subunit and two TRPC4 subunits, as well as by Orai1, and is located within caveolae. TRPC4 is physically associated with both Orai1 and the actin-binding protein, protein 4.1. The latter, in turn, must be associated with the spectrin membrane skeleton (A). A reduction in [Ca2+]ER (not shown) causes the STIM1-dependent activation of the ion channel signalplex on the PM (B). STIM1 is likely to physically interact with Orai1, TRPC1, and TRPC4, but this hypothesis remains to be experimentally probed. Orai1 incorporation into the STIM1/TRPC1/TRPC4 complex determines the Ca2+−selectivity of the store-operated current, which can therefore be defined as ICRAC-like (C). For sake of clarity, the spectrin−F-actin network beneath the plasma membrane has not been shown in Panel B.
6.2. The Role of the ICRAC-Like Current in Endothelial Function
The role of the endothelial ICRAC-like current has been mainly investigated in rat pulmonary vasculature [51], in which it stimulates the development of intercellular gaps in extra-alveolar, but not intra-alveolar, endothelial cells [140,224]. This observation is consistent with the finding that, in rat PMECs (rPMECs), SOCE is lower as compared to rPAECs [140,225] and the ICRAC-like current is not detectable [140]. Intriguingly, blocking type 4 phosphodiesterase with rolipram increased intracellular levels of cAMP, abolished endothelial ICRAC-like currents in pulmonary artery, and promoted their occurrence in lung capillaries [140]. These findings concur with the notion that the cAMP-producing agonists, epinephrine, and isoproterenol, interfere with endothelial gap formation, and further reveal that the ICRAC-like current controls endothelial cell permeability [210]. Therefore, aberrant activation of the ICRAC-like current could sensitize alveolar endothelium to Ca2+-dependent openings that result in pulmonary edema [140,226]. It turns out that, at least in rodent pulmonary vasculature, the endothelial cell barrier could be finely tuned by endothelial Ca2+ signals supported by both the ISOC (Section 4.3) and the ICRAC-like current. Conversely, the ICRAC has been recorded in hPAECs (Table 2) and controls permeability in hPMECs (Table 4).
Endothelium-dependent NO production and vasodilation were impaired in the TRPC4 knockout mouse model lacking endothelial ICRAC-like currents [87]. In this view, it is worth of recalling that: (1) TRPC1 deletion does not generally affect endothelium-dependent NO production [218]; (2) eNOS is preferentially coupled with Orai1 [28,159]; and (3) Orai1 can be constitutively associated to TRPC4 [51]. Therefore, it is reasonable to speculate that, within the STIM1/TRPC1/TRPC4/Orai1 signalplex, the eNOS is selectively coupled to Orai1 via TRPC4.
7. The Endothelial SOCE Machinery: Open Questions
The evidence discussed so far, which derives from almost three decades of intense research, indicates that the endothelial SOCE can be mediated by at least three different ionic currents: (1) ICRAC, which is a Ca2+-selective current; (2) ISOC, which is a non-selective cation current; and (3) ICRAC-like, which bears intermediate electrophysiological features between ICRAC and ISOC. Orai1 incorporation into the channel signalplex is likely to make the difference between a Ca2+-selective and a non-selective cation channel. It is, however, not surprising that many compelling issues regarding the molecular architecture and the role of endothelial SOCE in cardiovascular physiology and pathology remain to be elucidated.
First, may these three currents coexist in the same endothelial cell type? Pioneering work from the Ambudkar group showed that, following depletion of the ER Ca2+ pool, ICRAC and ISOC coexist in HSG cells, and that the ICRAC is activated first to stimulate the Ca2+-dependent insertion of TRPC1 on the PM [85]. However, the Orai1-mediated ICRAC is masked by the larger TRPC1-mediated ISOC and can be easily detected only after genetic silencing of the latter conductance [85]. Intriguingly, patch-clamp whole-cell recordings revealed that store depletion first activated a tiny ICRAC-like current and then a larger ISOC in human gingival keratinocytes [128,227]. This study showed that genetic deletion of TRPC4, a core component of the ICRAC-like current, also suppressed the subsequent ISOC [128]. These findings could help explaining how Orai1 and TRPC1/TRPC4 interact in endothelial cells and some discrepancies between different studies. For instance, both ICRAC [38] and ISOC [37,43,188,190] were recorded in HUVECs. However, Abdullaev et al. did not report about a large non-selective cation current, although they were able to record the nearly unmeasurable endothelial ICRAC [38]. This investigation exploited a Cs+-based intracellular solution, which is routinely employed to measure the ICRAC, while TRPC1 may display a poor permeability to Cs+ [228]. Conversely, the pipette solution employed to record the ISOC was based upon K+ [37] or Na+ [43,193], which readily permeate TRPC1 and can generate large outward currents. In addition, Abdullaev et al. dialyzed the cytosol with 20 mM BAPTA to record the ICRAC [38], which might have inadvertently prevented the local Ca2+ entry through Orai1 from inducing TRPC1 exocytosis on the PM. Therefore, these authors might have found the ideal conditions to record the ICRAC long before the role of Orai1 in stimulating the ISOC was identified [85]. In line with this hypothesis, VEGF-induced ISOC in HUVECs was inhibited by 1 µM La3+ [193], which selectively targets Orai1 rather than TRPC1 [52,152,229]. In our opinion, these data collectively support the notion that ICRAC and ISOC coexist in HUVECs. Another popular model to investigate the role of SOCE in endothelial physiopathology is represented by pulmonary endothelium. Two independent studies showed that both the ICRAC [38] and the ISOC [192] are present in hPAECs: while the former is mediated by STIM1/Orai1 [38], studies conducted on both hPAECs [192] and hPMECs [41] suggest that the latter is mediated by STIM1, TRPC1, and TRPC4. Like in HUVECs, it is yet to assess whether the ISOC recorded in hPAECs requires previous Orai1-mediated Ca2+ entry. Intriguingly, this ISOC is again sensitive to 1 µM La3+ [192]. Intriguingly, while SOCE in mPMECs is sensitive to the genetic blockade of STIM1, TRPC1, and TRPC4, but not of Orai1 (see Section 5.2), SOCE in rPAECs is due to an ICRAC-like current that requires the assembly of a STIM1/TRPC1/TRPC4/Orai1 supermolecular complex (see Section 6.1). Therefore, the molecular machinery responsible for SOCE activation in human and rodent lung endothelia could be different, and a further divergence can be observed between mouse and rat PAECs. Nevertheless, the evidence that the small molecule inhibitor BTP2, which is commonly employed to inhibit Orai1, halts sepsis- and ventilation-induced ALI suggests that Orai1 can somehow interact with TRPC1 and TRPC4 also in mice pulmonary endothelial cells [178,183]. In our opinion, this finding raises the question as to whether Orai1 can be incorporated into the STIM1/TRPC1/TRPC4 signalplex also in mPMECs. This hypothesis is supported by two pieces of evidence: (1) genetic deletion of Orai1 does not remarkably affect SOCE amplitude upon activation of endothelial ICRAC-like currents (this feature would explain why SOCE is unaffected in mPMECs transfected with a siOrai1) [50]; and (2) extracellular Na+ influx through CRAC-like channels can also regulate endothelial function (this feature would explain the beneficial effect of the systemic infusion of BTP-2 against LPS-induced pulmonary damage in mice) [178,230]. Electrophysiological measurements of the store-operated currents in mPMECs, which are still lacking, will help definitively in solving this issue.
Second, why have only a few studies identified a clear role of TRPC4 in either the ISOC or the ICRAC-like current? Is the endothelial ISOC strictly dependent on TRPC4 or can it be contributed to only by TRPC1? Ahmed et al. reported that the application of a specific antibody selectively targeting an extracellular epitope of TRPC1 significantly reduced, but not fully suppressed, InsP3-evoked ISOC in HUVECs [43]. The remaining current could, therefore, be due to an incomplete block of TRPC1 because of its physical association with TRPC4 within the channel signalplex. It should, however, be pointed out that TRPC4 expression on the PM may depend on endothelial cell confluence [231]. The Groschner group has convincingly shown that TRPC4 is recruited into the PM and mediates extracellular Ca2+ entry only in sub-confluent, proliferating vascular endothelial cells. Conversely, TRPC4 did not support Ca2+ influx in either isolated or fully confluent endothelial cells [232]. Under such conditions, therefore, TRPC-based store-operated currents are likely to be contributed to only by STIM1 and TRPC1.
Third, what are reasons for such molecular heterogeneity in SOCE machinery in vascular endothelial cells? It has been suggested that Orai1 and TRPC1/TRPC4 channels generate spatially separated Ca2+ microdomains that engage distinct Ca2+-dependent effectors, such as NFAT and NF-κB (Figure 5) [85,121]. In vascular endothelial cells, the ICRAC primarily regulates ER Ca2+ refilling, angiogenesis, and NO release, and platelet aggregation (see Section 4.3), but may also support platelet aggregation and thrombi formation as well as systemic inflammation and pulmonary vascular hyperpermeability (see Section 4.4). Conversely, the ISOC and the ICRAC-like current have long been associated with the fine regulation of endothelial permeability (see Section 5.3 and Section 6.2). Intriguingly, ICRAC and ISOC engage two different transcription factors, i.e., NFAT and NF-κB, respectively, to stimulate the gene expression pro-inflammatory program (Figure 5) [178,194,207]. Recent findings, however, showed that the ISOC could also support the angiogenic activity (Figure 5 and Table 6) and that the ICRAC-like can induce vasorelaxation via NO release [87]. These findings suggest that Orai1 and TRPC1/TRPC4 channels could converge on the same endothelial processes (e.g., permeability and vasorelaxation) either via distinct (permeability, NFAT vs. NF-κB) or similar (vasorelaxation, eNOS) Ca2+-dependent effectors. The pure Ca2+-selective ICRAC could then regulate additional Ca2+-dependent processes, such as ER Ca2+ refilling and van Willebrand factor release.
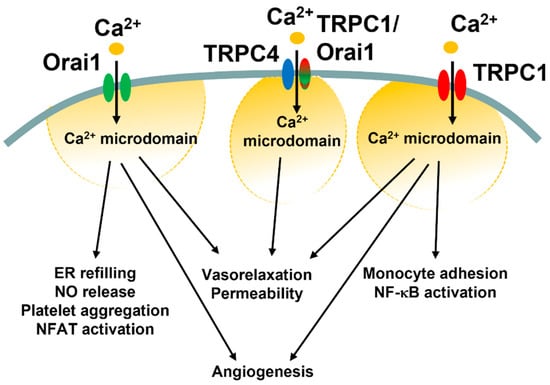
Figure 5.
Endothelial functions regulated by Ca2+ entry through the diverse SOCE mechanisms. This illustration summarizes the different endothelial functions regulated by the ICRAC (Orai1 in green), ICRAC-like currents (TRPC4 in blue plus TRPC1/Orai1 in red/green), and ISOC (TRPC1 in red).
Fourth, does endothelial SOCE cooperate with perinuclear InsP3Rs via the CICR mechanism to stimulate an increase in perinuclear or nuclear Ca2+? Early studies demonstrated that growth factors induced oscillations in nuclear Ca2+ concentration in both micro- [233] and macro-vascular [234] endothelial cells and that these oscillations were abrogated by the pharmacological blockade of SOCE. Future work could assess whether the endothelial SOCE stimulates perinuclear Ca2+ release through InsP3Rs, thereby inducing the nuclear translocation of NFAT and/or NF-κB, and whether the nuclear Ca2+ oscillations sustained by SOCE recruit nuclear localized transcriptional regulators, such as DREAM and myocyte enhancer factor 2C.
Fifth, do other Ca2+-entry pathways play a role in endothelial SOCE? This question does not simply arise from the evidence that other TRPC isoforms may support SOCE in heterologous expression studies, but from landmark studies conducted in native endothelial cells. It has been shown that, in EA.hy926 cells, depleting the ER Ca2+ store with thapsigargin activated both the STIM1/Orai1 machinery and TRPC3-mediated Ca2+ entry [40]. In accord, depletion of the ER Ca2+ content was found to stimulate DAG production via PLC activation, which could be explained by the known Ca2+-dependence of PLC activity; DAG, in turn, stimulated the novel PKCη to activate Src kinase and phosphorylate TRPC3, thereby promoting further Ca2+ entry [40]. In addition, TRPC1 can assemble into a heteromeric channel with TRPV4 in rat mesenteric artery endothelial cells [235], and a reduction in [Ca2+]ER can favour the translocation of TRPV4/TRPC1 complexes into the PM, where they are activated by STIM1 and mediate a large ISOC [236]. These findings were confirmed in MAECs and HUVECs [42], and it was further shown that this novel mode of endothelial SOCE could induce vasorelaxation via NO release in resistance arterioles [237]. It would be tempting to speculate that TRPC1 associates to TRPV4 when TRPC4 is not (or only barely) expressed on the PM, but this hypothesis remains to be experimentally probed.
Sixth, which store-dependent current(s) mediate SOCE in ECFCs? SOCE supports ECFC proliferation, migration, and tube formation both in vitro [25,61,238,239,240,241,242] and in vivo [26,27]. Electrophysiological recordings revealed that a store-operated current is not detectable in ECFCs, whereas Ca2+ imaging revealed that their SOCE is impaired by the genetic knockdown of STIM1, Orai1, and TRPC1 [30,39,239]. If SOCE is supported by TRPC1, but the depletion of the ER Ca2+ content does not lead to the activation of a measurable membrane current, the latter could be mediated by a channel pore that is contributed to by both Orai1 and TRPC1. Future work will have to assess whether STIM1, Orai1, TRPC1, and, possibly, TRPC4 assemble within the same ion channel signalplex and mediate an ICRAC-like current in ECFCs. The evidence that, in ECFCs, SOCE activation results in NF-κB activation [25,238] strongly suggests that the tight coupling between the Ca2+-sensitive transcription factor and the SOCE machinery is provided by TRPC1.
8. Conclusions
SOCE maintains the endothelial Ca2+ response to extracellular stimulation and thereby regulates a plethora of endothelial functions, including angiogenesis, permeability, platelet aggregation, and NO release. Three distinct store-operated currents can mediate SOCE in vascular endothelial cells: (1) the Ca2+-selective ICRAC, which is mediated by STIM1 and Orai1; (2) the non-selective ISOC, which is mediated by STIM1, TRPC1, and TRPC4; and (3) the moderately Ca2+-selective, ICRAC-like current, which is mediated by STIM1, TRPC1, TRPC4, and Orai1. The incorporation of Orai1 into the STIM1/TRPC1/TRPC4 complex, therefore, turns the non-selective ISOC into a Ca2+ (semi)-selective current. As highlighted in Section 7, many questions remain open. In our opinion, the most challenging issues that need to be urgently addressed to move a significant step forward are the following: (1) may these three Ca2+-permeable currents coexist in the same endothelial cell type or does their endothelial distribution change along the vascular network and/or from one species to another? (2) are the ICRAC, ISOC, and ICRAC-like currents coupled to distinct Ca2+-dependent decoders, and do they modulate different endothelial functions? (3) does Orai1-mediated extracellular Ca2+ entry result in TRPC1 translocation to the plasma membrane also in endothelial cells? Exploiting the novel technologies offered by both molecular biology (e.g., CRISPR/Cas9, single-cell RNA sequencing, endothelial-specific GCaMP3 mice) and imaging techniques (e.g., multiphoton microscopy to detect submembrane Ca2+ entry events) will help in solving these issues. Understanding the molecular architecture of this mechanism is indispensable in efficiently targeting endothelial SOCE in patients affected by life-threatening cardiovascular disorders, such as atherosclerosis, cerebrovascular dysfunction, sepsis-induced systemic inflammation, and PAH.
Author Contributions
Conceptualization, F.M.; writing—original draft preparation, F.M.; writing—review and editing, F.M., V.B., A.P., G.G., R.B.-R. and T.S.; visualization, V.B., A.P., G.G., R.B.-R. and T.S.; supervision, F.M.; project administration, F.M.; funding acquisition, F.M. and G.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by: the Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022)-Dept. of Biology and Biotechnology “L. Spallanzani”, the University of Pavia (F.M.), Fondo Ricerca Giovani from the University of Pavia (F.M.), and the EU Horizon 2020 FETOPEN-2018-2020 Program under Grant Agreement N. 828984 (LION-HEARTED). R.B.-R is supported by the National Council of Science and Technology (CONACYT), Identification Number: CVU121216.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McCarron, J.G.; Wilson, C.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Lee, M.D. Heterogeneity and emergent behaviour in the vascular endothelium. Curr. Opin. Pharmacol. 2019, 45, 23–32. [Google Scholar] [CrossRef] [PubMed]
- McCarron, J.G.; Lee, M.D.; Wilson, C. The Endothelium Solves Problems that Endothelial Cells Do Not Know Exist. Trends Pharmacol. Sci. 2017, 38, 322–338. [Google Scholar] [CrossRef]
- Kruger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K(+) (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca(2+) Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef]
- Moccia, F.; Gerbino, A.; Lionetti, V.; Miragoli, M.; Munaron, L.M.; Pagliaro, P.; Pasqua, T.; Penna, C.; Rocca, C.; Samaja, M.; et al. COVID-19-associated cardiovascular morbidity in older adults: A position paper from the Italian Society of Cardiovascular Researches. GeroScience 2020, 42, 1021–1049. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Di Nezza, F.; Pellavio, G.; Faris, P.S.; Negri, S.; De Luca, A.; Laforenza, U.; Ambrosone, L.; Rosti, V.; et al. Nicotinic acid adenine dinucleotide phosphate activates two-pore channel TPC1 to mediate lysosomal Ca(2+) release in endothelial colony-forming cells. J. Cell. Physiol. 2021, 236, 688–705. [Google Scholar] [CrossRef]
- Faris, P.; Negri, S.; Perna, A.; Rosti, V.; Guerra, G.; Moccia, F. Therapeutic Potential of Endothelial Colony-Forming Cells in Ischemic Disease: Strategies to Improve their Regenerative Efficacy. Int. J. Mol. Sci. 2020, 21, 7406. [Google Scholar] [CrossRef]
- Poletto, V.; Rosti, V.; Biggiogera, M.; Guerra, G.; Moccia, F.; Porta, C. The role of endothelial colony forming cells in kidney cancer’s pathogenesis, and in resistance to anti-VEGFR agents and mTOR inhibitors: A speculative review. Crit. Rev. Oncol. Hematol. 2018, 132, 89–99. [Google Scholar] [CrossRef]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Wilson, C.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Clusters of specialized detector cells provide sensitive and high fidelity receptor signaling in the intact endothelium. FASEB J. 2016, 30, 2000–2013. [Google Scholar] [CrossRef]
- Wilson, C.; Zhang, X.; Lee, M.D.; MacDonald, M.; Heathcote, H.R.; Alorfi, N.M.N.; Buckley, C.; Dolan, S.; McCarron, J.G. Disrupted Endothelial Cell Heterogeneity and Network Organization Impair Vascular Function in Prediabetic Obesity. Metabolism 2020, 111, 154340. [Google Scholar] [CrossRef]
- Wilson, C.; Lee, M.D.; McCarron, J.G. Acetylcholine released by endothelial cells facilitates flow-mediated dilatation. J. Physiol. 2016, 594, 7267–7307. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca(2+) Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Zhao, L.; Jing, R.; Trexler, C.; Wang, H.; Li, Y.; Tang, H.; Huang, F.; Zhang, F.; Fang, X.; et al. Inositol 1,4,5-Trisphosphate Receptors in Endothelial Cells Play an Essential Role in Vasodilation and Blood Pressure Regulation. J. Am. Heart Assoc. 2019, 8, e011704. [Google Scholar] [CrossRef]
- Francis, M.; Waldrup, J.R.; Qian, X.; Solodushko, V.; Meriwether, J.; Taylor, M.S. Functional Tuning of Intrinsic Endothelial Ca2+ Dynamics in Swine Coronary Arteries. Circ. Res. 2016, 118, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Genova, T.; Gaglioti, D.; Munaron, L. Regulation of Vessel Permeability by TRP Channels. Front. Physiol. 2020, 11, 421. [Google Scholar] [CrossRef]
- Smani, T.; Gomez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.M.; Rosado, J.A. TRP Channels in Angiogenesis and Other Endothelial Functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Raqeeb, A.; Torres-Jácome, J.; Guzman-Silva, A.; Guerra, G.; Tanzi, F.; Moccia, F. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J. Vasc. Res. 2012, 49, 65–76. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309. [Google Scholar] [CrossRef]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca(2+) Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef] [PubMed]
- Noy, P.J.; Gavin, R.L.; Colombo, D.; Haining, E.J.; Reyat, J.S.; Payne, H.; Thielmann, I.; Lokman, A.B.; Neag, G.; Yang, J.; et al. Tspan18 is a novel regulator of the Ca2+ channel Orai1 and von Willebrand factor release in endothelial cells. Haematologica 2018, 104, 1892–1905. [Google Scholar] [CrossRef]
- Weber, E.W.; Han, F.; Tauseef, M.; Birnbaumer, L.; Mehta, D.; Muller, W.A. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J. Exp. Med. 2015, 212, 1883–1899. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca(2+) Signaling, Angiogenesis and Vasculogenesis: Just What It Takes to Make a Blood Vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef]
- Balducci, V.; Faris, P.; Balbi, C.; Costa, A.; Negri, S.; Rosti, V.; Bollini, S.; Moccia, F. The human amniotic fluid stem cell secretome triggers intracellular Ca(2+) oscillations, NF-kappaB nuclear translocation and tube formation in human endothelial colony-forming cells. J. Cell. Mol. Med. 2021, 25, 8074–8086. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal Cell-Derived Factor-1alpha Promotes Endothelial Colony-Forming Cell Migration Through the Ca(2+)-Dependent Activation of the Extracellular Signal-Regulated Kinase 1/2 and Phosphoinositide 3-Kinase/AKT Pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef]
- Balbi, C.; Lodder, K.; Costa, A.; Moimas, S.; Moccia, F.; Van Herwaarden, T.; Rosti, V.; Campagnoli, F.; Palmeri, A.; De Biasio, P.; et al. Reactivating endogenous mechanisms of cardiac regeneration via paracrine boosting using the human amniotic fluid stem cell secretome. Int. J. Cardiol. 2019, 287, 87–95. [Google Scholar] [CrossRef]
- Blatter, L.A. Tissue Specificity: SOCE: Implications for Ca2+ Handling in Endothelial Cells. Adv. Exp. Med. Biol. 2017, 993, 343–361. [Google Scholar] [CrossRef] [PubMed]
- Groschner, K.; Shrestha, N.; Fameli, N. Cardiovascular and Hemostatic Disorders: SOCE in Cardiovascular Cells: Emerging Targets for Therapeutic Intervention. Adv. Exp. Med. Biol. 2017, 993, 473–503. [Google Scholar] [CrossRef]
- Moccia, F.; Dragoni, S.; Lodola, F.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Store-dependent Ca(2+) entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr. Med. Chem. 2012, 19, 5802–5818. [Google Scholar] [CrossRef]
- Moccia, F. Calcium Signaling in Endothelial Colony Forming Cells in Health and Disease. Adv. Exp. Med. Biol. 2020, 1131, 1013–1030. [Google Scholar] [CrossRef] [PubMed]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Fierro, L.; Lund, P.E.; Parekh, A.B. Comparison of the activation of the Ca2+ release-activated Ca2+ current ICRAC to InsP3 in Jurkat T-lymphocytes, pulmonary artery endothelia and RBL-1 cells. Pflug. Arch. 2000, 440, 580–587. [Google Scholar] [CrossRef]
- Oike, M.; Gericke, M.; Droogmans, G.; Nilius, B. Calcium entry activated by store depletion in human umbilical vein endothelial cells. Cell Calcium 1994, 16, 367–376. [Google Scholar] [CrossRef]
- Fasolato, C.; Nilius, B. Store depletion triggers the calcium release-activated calcium current (ICRAC) in macrovascular endothelial cells: A comparison with Jurkat and embryonic kidney cell lines. Pflug. Arch. 1998, 436, 69–74. [Google Scholar] [CrossRef]
- Kohler, R.; Brakemeier, S.; Kuhn, M.; Degenhardt, C.; Buhr, H.; Pries, A.; Hoyer, J. Expression of ryanodine receptor type 3 and TRP channels in endothelial cells: Comparison of in situ and cultured human endothelial cells. Cardiovasc. Res. 2001, 51, 160–168. [Google Scholar] [CrossRef]
- Groschner, K.; Hingel, S.; Lintschinger, B.; Balzer, M.; Romanin, C.; Zhu, X.; Schreibmayer, W. Trp proteins form store-operated cation channels in human vascular endothelial cells. FEBS Lett. 1998, 437, 101–106. [Google Scholar] [CrossRef]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef]
- Antigny, F.; Jousset, H.; Konig, S.; Frieden, M. Thapsigargin activates Ca(2)+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium 2011, 49, 115–127. [Google Scholar] [CrossRef]
- Sundivakkam, P.C.; Freichel, M.; Singh, V.; Yuan, J.P.; Vogel, S.M.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. The Ca(2+) sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca(2+) entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol. Pharmacol. 2012, 81, 510–526. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cheng, K.T.; Wong, C.O.; O’Neil, R.G.; Birnbaumer, L.; Ambudkar, I.S.; Yao, X. Heteromeric TRPV4-C1 channels contribute to store-operated Ca(2+) entry in vascular endothelial cells. Cell Calcium 2011, 50, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Ahmmed, G.U.; Mehta, D.; Vogel, S.; Holinstat, M.; Paria, B.C.; Tiruppathi, C.; Malik, A.B. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J. Biol. Chem. 2004, 279, 20941–20949. [Google Scholar] [CrossRef]
- Vaca, L.; Kunze, D.L. Depletion of intracellular Ca2+ stores activates a Ca(2+)-selective channel in vascular endothelium. Am. J. Physiol. 1994, 267, C920–C925. [Google Scholar] [CrossRef]
- Mendelowitz, D.; Bacal, K.; Kunze, D.L. Bradykinin-activated calcium influx pathway in bovine aortic endothelial cells. Am. J. Physiol. 1992, 262, H942–H948. [Google Scholar] [CrossRef]
- Girardin, N.C.; Antigny, F.; Frieden, M. Electrophysiological characterization of store-operated and agonist-induced Ca2+ entry pathways in endothelial cells. Pflug. Arch. 2010, 460, 109–120. [Google Scholar] [CrossRef][Green Version]
- Gericke, M.; Droogmans, G.; Nilius, B. Thapsigargin discharges intracellular calcium stores and induces transmembrane currents in human endothelial cells. Pflug. Arch. 1993, 422, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, D.L.; Wu, S.; Stevens, T. On the endothelial cell I(SOC). Cell Calcium 2003, 33, 323–336. [Google Scholar] [CrossRef]
- Nilius, B.; Droogmans, G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001, 81, 1415–1459. [Google Scholar] [CrossRef]
- Xu, N.; Cioffi, D.L.; Alexeyev, M.; Rich, T.C.; Stevens, T. Sodium entry through endothelial store-operated calcium entry channels: Regulation by Orai1. Am. J. Physiol. Cell Physiol. 2015, 308, C277–C288. [Google Scholar] [CrossRef]
- Cioffi, D.L.; Wu, S.; Chen, H.; Alexeyev, M.; St Croix, C.M.; Pitt, B.R.; Uhlig, S.; Stevens, T. Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ. Res. 2012, 110, 1435–1444. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef]
- Moccia, F.; Guerra, G. Ca(2+) Signalling in Endothelial Progenitor Cells: Friend or Foe? J. Cell. Physiol. 2016, 231, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front. Physiol. 2021, 12, 629119. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Endolysosomal Ca(2+) signaling in cardiovascular health and disease. Int. Rev. Cell Mol. Biol. 2021, 363, 203–269. [Google Scholar] [CrossRef] [PubMed]
- Aromolaran, A.S.; Zima, A.V.; Blatter, L.A. Role of glycolytically generated ATP for CaMKII-mediated regulation of intracellular Ca2+ signaling in bovine vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2007, 293, C106–C118. [Google Scholar] [CrossRef]
- Huser, J.; Blatter, L.A. Elementary events of agonist-induced Ca2+ release in vascular endothelial cells. Am. J. Physiol. 1997, 273, C1775–C1782. [Google Scholar] [CrossRef]
- Huser, J.; Holda, J.R.; Kockskamper, J.; Blatter, L.A. Focal agonist stimulation results in spatially restricted Ca2+ release and capacitative Ca2+ entry in bovine vascular endothelial cells. J. Physiol. 1999, 514, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Sedova, M.; Klishin, A.; Huser, J.; Blatter, L.A. Capacitative Ca2+ entry is graded with degree of intracellular Ca2+ store depletion in bovine vascular endothelial cells. J. Physiol. 2000, 523, 549–559. [Google Scholar] [CrossRef]
- Tsai, F.C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjo, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef]
- Sanchez-Hernandez, Y.; Laforenza, U.; Bonetti, E.; Fontana, J.; Dragoni, S.; Russo, M.; Avelino-Cruz, J.E.; Schinelli, S.; Testa, D.; Guerra, G.; et al. Store-operated Ca(2+) entry is expressed in human endothelial progenitor cells. Stem Cells Dev. 2010, 19, 1967–1981. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca(2+) signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef]
- Scarpellino, G.; Genova, T.; Avanzato, D.; Bernardini, M.; Bianco, S.; Petrillo, S.; Tolosano, E.; de Almeida Vieira, J.R.; Bussolati, B.; Fiorio Pla, A.; et al. Purinergic Calcium Signals in Tumor-Derived Endothelium. Cancers 2019, 11, 766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Chen, X.Y.; Dong, H.; Xu, F. Cyclopiazonic Acid-Induced Ca(2+) Store Depletion Initiates Endothelium-Dependent Hyperpolarization-Mediated Vasorelaxation of Mesenteric Arteries in Healthy and Colitis Mice. Front. Physiol. 2021, 12, 639857. [Google Scholar] [CrossRef]
- Malli, R.; Frieden, M.; Hunkova, M.; Trenker, M.; Graier, W.F. Ca2+ refilling of the endoplasmic reticulum is largely preserved albeit reduced Ca2+ entry in endothelial cells. Cell Calcium 2007, 41, 63–76. [Google Scholar] [CrossRef]
- Malli, R.; Frieden, M.; Trenker, M.; Graier, W.F. The role of mitochondria for Ca2+ refilling of the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 12114–12122. [Google Scholar] [CrossRef]
- Elliott, S.J.; Doan, T.N. Oxidant stress inhibits the store-dependent Ca(2+)-influx pathway of vascular endothelial cells. Biochem. J. 1993, 292, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Lemos, F.O.; Bultynck, G.; Parys, J.B. A comprehensive overview of the complex world of the endo- and sarcoplasmic reticulum Ca(2+)-leak channels. Biochim. Biophys. Acta. Mol. Cell Res. 2021, 1868, 119020. [Google Scholar] [CrossRef]
- Cioffi, D.L.; Wu, S.; Alexeyev, M.; Goodman, S.R.; Zhu, M.X.; Stevens, T. Activation of the endothelial store-operated ISOC Ca2+ channel requires interaction of protein 4.1 with TRPC4. Circ. Res. 2005, 97, 1164–1172. [Google Scholar] [CrossRef]
- Naser, N.; Januszewski, A.S.; Brown, B.E.; Jenkins, A.J.; Hill, M.A.; Murphy, T.V. Advanced glycation end products acutely impair ca(2+) signaling in bovine aortic endothelial cells. Front. Physiol. 2013, 4, 38. [Google Scholar] [CrossRef]
- Bishara, N.B.; Ding, H. Glucose enhances expression of TRPC1 and calcium entry in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H171–H178. [Google Scholar] [CrossRef]
- Morgan, A.J.; Jacob, R. Ionomycin enhances Ca2+ influx by stimulating store-regulated cation entry and not by a direct action at the plasma membrane. Biochem. J. 1994, 300, 665–672. [Google Scholar] [CrossRef]
- Wilson, C.; Lee, M.D.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Girkin, J.M.; Saunter, C.D.; McCarron, J.G. Mitochondrial ATP production provides long-range control of endothelial inositol trisphosphate-evoked calcium signaling. J. Biol. Chem. 2019, 294, 737–758. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca(2+) signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef]
- Ong, H.L.; Cheng, K.T.; Liu, X.; Bandyopadhyay, B.C.; Paria, B.C.; Soboloff, J.; Pani, B.; Gwack, Y.; Srikanth, S.; Singh, B.B.; et al. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J. Biol. Chem. 2007, 282, 9105–9116. [Google Scholar] [CrossRef]
- Huang, G.N.; Zeng, W.; Kim, J.Y.; Yuan, J.P.; Han, L.; Muallem, S.; Worley, P.F. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 2006, 8, 1003–1010. [Google Scholar] [CrossRef]
- Lopez, J.J.; Salido, G.M.; Pariente, J.A.; Rosado, J.A. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J. Biol. Chem. 2006, 281, 28254–28264. [Google Scholar] [CrossRef]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J. Biol. Chem. 2008, 283, 25296–25304. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Ambudkar, I.S. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J. Biol. Chem. 2008, 283, 12935–12940. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca(2)+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca(2)+ signals required for specific cell functions. PLoS Biol. 2011, 9, e1001025. [Google Scholar] [CrossRef]
- Brough, G.H.; Wu, S.; Cioffi, D.; Moore, T.M.; Li, M.; Dean, N.; Stevens, T. Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway. FASEB J. 2001, 15, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Freichel, M.; Suh, S.H.; Pfeifer, A.; Schweig, U.; Trost, C.; Weissgerber, P.; Biel, M.; Philipp, S.; Freise, D.; Droogmans, G.; et al. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4-/- mice. Nat. Cell Biol. 2001, 3, 121–127. [Google Scholar] [CrossRef]
- Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC Channels in the SOCE Scenario. Cells 2020, 9, 126. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; De Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 2017, 63, 33–39. [Google Scholar] [CrossRef]
- Sabourin, J.; Boet, A.; Rucker-Martin, C.; Lambert, M.; Gomez, A.M.; Benitah, J.P.; Perros, F.; Humbert, M.; Antigny, F. Ca(2+) handling remodeling and STIM1L/Orai1/TRPC1/TRPC4 upregulation in monocrotaline-induced right ventricular hypertrophy. J. Mol. Cell. Cardiol. 2018, 118, 208–224. [Google Scholar] [CrossRef]
- Nascimento Da Conceicao, V.; Sun, Y.; Zboril, E.K.; De la Chapa, J.J.; Singh, B.B. Loss of Ca(2+) entry via Orai-TRPC1 induces ER stress, initiating immune activation in macrophages. J. Cell Sci. 2019, 133, jcs237610. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S.; Cahalan, M.D. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1989, 1, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Zweifach, A.; Lewis, R.S. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci. USA 1993, 90, 6295–6299. [Google Scholar] [CrossRef]
- Lewis, R.S. Store-Operated Calcium Channels: From Function to Structure and Back Again. Cold Spring Harb. Perspect. Biol. 2020, 12, a035055. [Google Scholar] [CrossRef]
- Emrich, S.M.; Yoast, R.E.; Trebak, M. Physiological Functions of CRAC Channels. Annu. Rev. Physiol. 2021, 84, 355–379. [Google Scholar] [CrossRef]
- Emrich, S.M.; Yoast, R.E.; Xin, P.; Arige, V.; Wagner, L.E.; Hempel, N.; Gill, D.L.; Sneyd, J.; Yule, D.I.; Trebak, M. Omnitemporal choreographies of all five STIM/Orai and IP3Rs underlie the complexity of mammalian Ca(2+) signaling. Cell Rep. 2021, 34, 108760. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.N.; Zhang, X.; Wu, S.; Janoshazi, A.; Bolimuntha, S.; Putney, J.W.; Trebak, M. Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message. Sci. Signal. 2015, 8, ra74. [Google Scholar] [CrossRef]
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J.W. Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities. J. Cell Sci. 2012, 125, 4354–4361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xin, P.; Yoast, R.E.; Emrich, S.M.; Johnson, M.T.; Pathak, T.; Benson, J.C.; Azimi, I.; Gill, D.L.; Monteith, G.R.; et al. Distinct pharmacological profiles of ORAI1, ORAI2, and ORAI3 channels. Cell Calcium 2020, 91, 102281. [Google Scholar] [CrossRef]
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol. 2007, 17, 794–800. [Google Scholar] [CrossRef]
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The native ORAI channel trio underlies the diversity of Ca(2+) signaling events. Nat. Commun. 2020, 11, 2444. [Google Scholar] [CrossRef]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Ong, H.L.; Saadi, H.; Son, G.Y.; Shokatian, Z.; Terry, L.E.; Trebak, M.; Yule, D.I.; Ambudkar, I. Functional communication between IP3R and STIM2 at subthreshold stimuli is a critical checkpoint for initiation of SOCE. Proc. Natl. Acad. Sci. USA 2022, 119, e2114928118. [Google Scholar] [CrossRef]
- Ong, H.L.; De Souza, L.B.; Zheng, C.; Cheng, K.T.; Liu, X.; Goldsmith, C.M.; Feske, S.; Ambudkar, I.S. STIM2 enhances receptor-stimulated Ca(2)(+) signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions. Sci. Signal. 2015, 8, ra3. [Google Scholar] [CrossRef] [PubMed]
- Miederer, A.M.; Alansary, D.; Schwar, G.; Lee, P.H.; Jung, M.; Helms, V.; Niemeyer, B.A. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun. 2015, 6, 6899. [Google Scholar] [CrossRef] [PubMed]
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 2011, 194, 335–346. [Google Scholar] [CrossRef]
- Ramesh, G.; Jarzembowski, L.; Schwarz, Y.; Poth, V.; Konrad, M.; Knapp, M.L.; Schwar, G.; Lauer, A.A.; Grimm, M.O.W.; Alansary, D.; et al. A short isoform of STIM1 confers frequency-dependent synaptic enhancement. Cell Rep. 2021, 34, 108844. [Google Scholar] [CrossRef]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution and regulation of TRPC channels in store-operated Ca2+ entry. Curr. Top. Membr. 2013, 71, 149–179. [Google Scholar] [CrossRef]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca(2+) signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell. Physiol. 2018, 233, 3901–3917. [Google Scholar] [CrossRef]
- Chen, X.; Sooch, G.; Demaree, I.S.; White, F.A.; Obukhov, A.G. Transient Receptor Potential Canonical (TRPC) Channels: Then and Now. Cells 2020, 9, 1983. [Google Scholar] [CrossRef]
- Ong, H.L.; Ambudkar, I.S. The Endoplasmic Reticulum-Plasma Membrane Junction: A Hub for Agonist Regulation of Ca(2+) Entry. Cold Spring Harb. Perspect. Biol. 2020, 12, a035253. [Google Scholar] [CrossRef]
- Ong, H.L.; Jang, S.I.; Ambudkar, I.S. Distinct contributions of Orai1 and TRPC1 to agonist-induced [Ca(2+)](i) signals determine specificity of Ca(2+)-dependent gene expression. PLoS ONE 2012, 7, e47146. [Google Scholar] [CrossRef]
- Di Buduo, C.A.; Abbonante, V.; Marty, C.; Moccia, F.; Rumi, E.; Pietra, D.; Soprano, P.M.; Lim, D.; Cattaneo, D.; Iurlo, A.; et al. Defective interaction of mutant calreticulin and SOCE in megakaryocytes from patients with myeloproliferative neoplasms. Blood 2020, 135, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Shalygin, A.; Kolesnikov, D.; Glushankova, L.; Gusev, K.; Skopin, A.; Skobeleva, K.; Kaznacheyeva, E.V. Role of STIM2 and Orai proteins in regulating TRPC1 channel activity upon calcium store depletion. Cell Calcium 2021, 97, 102432. [Google Scholar] [CrossRef]
- Dyrda, A.; Koenig, S.; Frieden, M. STIM1 long and STIM1 gate differently TRPC1 during store-operated calcium entry. Cell Calcium 2020, 86, 102134. [Google Scholar] [CrossRef]
- Sanchez-Collado, J.; Lopez, J.J.; Jardin, I.; Berna-Erro, A.; Camello, P.J.; Cantonero, C.; Smani, T.; Salido, G.M.; Rosado, J.A. Orai1alpha, but not Orai1beta, co-localizes with TRPC1 and is required for its plasma membrane location and activation in HeLa cells. Cell. Mol. Life Sci. 2022, 79, 33. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Yuan, J.P.; Kim, M.S.; Choi, Y.J.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol. Cell 2008, 32, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Sabourin, J.; Sauc, S.; Bernheim, L.; Koenig, S.; Frieden, M. TRPC1 and TRPC4 channels functionally interact with STIM1L to promote myogenesis and maintain fast repetitive Ca(2+) release in human myotubes. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 806–813. [Google Scholar] [CrossRef]
- Sabourin, J.; Bartoli, F.; Antigny, F.; Gomez, A.M.; Benitah, J.P. Transient Receptor Potential Canonical (TRPC)/Orai1-dependent Store-operated Ca2+ Channels: New Targets of Aldosterone in Cardiomyocytes. J. Biol. Chem. 2016, 291, 13394–13409. [Google Scholar] [CrossRef]
- Broker-Lai, J.; Kollewe, A.; Schindeldecker, B.; Pohle, J.; Nguyen Chi, V.; Mathar, I.; Guzman, R.; Schwarz, Y.; Lai, A.; Weissgerber, P.; et al. Heteromeric channels formed by TRPC1, TRPC4 and TRPC5 define hippocampal synaptic transmission and working memory. EMBO J. 2017, 36, 2770–2789. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; Subedi, K.P.; Son, G.Y.; Liu, X.; Ambudkar, I.S. Tuning store-operated calcium entry to modulate Ca(2+)-dependent physiological processes. Biochim. Biophys. Acta. Mol. Cell Res. 2019, 1866, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Abramowitz, J.; Birnbaumer, L. The TRPC family of TRP channels: Roles inferred (mostly) from knockout mice and relationship to ORAI proteins. Handb. Exp. Pharmacol. 2014, 223, 1055–1075. [Google Scholar] [CrossRef]
- Cioffi, D.L.; Barry, C.; Stevens, T. Store-operated calcium entry channels in pulmonary endothelium: The emerging story of TRPCS and Orai1. Adv. Exp. Med. Biol. 2010, 661, 137–154. [Google Scholar] [CrossRef]
- Liao, Y.; Plummer, N.W.; George, M.D.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl. Acad. Sci. USA 2009, 106, 3202–3206. [Google Scholar] [CrossRef]
- Liao, Y.; Erxleben, C.; Abramowitz, J.; Flockerzi, V.; Zhu, M.X.; Armstrong, D.L.; Birnbaumer, L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc. Natl. Acad. Sci. USA 2008, 105, 2895–2900. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Erxleben, C.; Yildirim, E.; Abramowitz, J.; Armstrong, D.L.; Birnbaumer, L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 4682–4687. [Google Scholar] [CrossRef]
- Molnar, T.; Yarishkin, O.; Iuso, A.; Barabas, P.; Jones, B.; Marc, R.E.; Phuong, T.T.; Krizaj, D. Store-Operated Calcium Entry in Muller Glia Is Controlled by Synergistic Activation of TRPC and Orai Channels. J. Neurosci. 2016, 36, 3184–3198. [Google Scholar] [CrossRef]
- Fatherazi, S.; Presland, R.B.; Belton, C.M.; Goodwin, P.; Al-Qutub, M.; Trbic, Z.; Macdonald, G.; Schubert, M.M.; Izutsu, K.T. Evidence that TRPC4 supports the calcium selective I(CRAC)-like current in human gingival keratinocytes. Pflug. Arch. 2007, 453, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Philipp, S.; Trost, C.; Warnat, J.; Rautmann, J.; Himmerkus, N.; Schroth, G.; Kretz, O.; Nastainczyk, W.; Cavalie, A.; Hoth, M.; et al. TRP4 (CCE1) protein is part of native calcium release-activated Ca2+-like channels in adrenal cells. J. Biol. Chem. 2000, 275, 23965–23972. [Google Scholar] [CrossRef]
- Gees, M.; Owsianik, G.; Nilius, B.; Voets, T. TRP channels. Compr. Physiol. 2012, 2, 563–608. [Google Scholar] [CrossRef]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 2001, 29, 645–655. [Google Scholar] [CrossRef]
- Philipp, S.; Cavalie, A.; Freichel, M.; Wissenbach, U.; Zimmer, S.; Trost, C.; Marquart, A.; Murakami, M.; Flockerzi, V. A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. EMBO J. 1996, 15, 6166–6171. [Google Scholar] [CrossRef] [PubMed]
- Jacob, R. Agonist-stimulated divalent cation entry into single cultured human umbilical vein endothelial cells. J. Physiol. 1990, 421, 55–77. [Google Scholar] [CrossRef] [PubMed]
- Hallam, T.J.; Jacob, R.; Merritt, J.E. Influx of bivalent cations can be independent of receptor stimulation in human endothelial cells. Biochem. J. 1989, 259, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Gericke, M.; Oike, M.; Droogmans, G.; Nilius, B. Inhibition of capacitative Ca2+ entry by a Cl- channel blocker in human endothelial cells. Eur. J. Pharmacol. 1994, 269, 381–384. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef]
- Malli, R.; Naghdi, S.; Romanin, C.; Graier, W.F. Cytosolic Ca2+ prevents the subplasmalemmal clustering of STIM1: An intrinsic mechanism to avoid Ca2+ overload. J. Cell Sci. 2008, 121, 3133–3139. [Google Scholar] [CrossRef]
- Naghdi, S.; Waldeck-Weiermair, M.; Fertschai, I.; Poteser, M.; Graier, W.F.; Malli, R. Mitochondrial Ca2+ uptake and not mitochondrial motility is required for STIM1-Orai1-dependent store-operated Ca2+ entry. J. Cell Sci. 2010, 123, 2553–2564. [Google Scholar] [CrossRef]
- King, J.; Hamil, T.; Creighton, J.; Wu, S.; Bhat, P.; McDonald, F.; Stevens, T. Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvasc. Res. 2004, 67, 139–151. [Google Scholar] [CrossRef]
- Wu, S.; Cioffi, E.A.; Alvarez, D.; Sayner, S.L.; Chen, H.; Cioffi, D.L.; King, J.; Creighton, J.R.; Townsley, M.; Goodman, S.R.; et al. Essential role of a Ca2+-selective, store-operated current (ISOC) in endothelial cell permeability: Determinants of the vascular leak site. Circ. Res. 2005, 96, 856–863. [Google Scholar] [CrossRef]
- Moccia, F.; Baruffi, S.; Spaggiari, S.; Coltrini, D.; Berra-Romani, R.; Signorelli, S.; Castelli, L.; Taglietti, V.; Tanzi, F. P2y1 and P2y2 receptor-operated Ca2+ signals in primary cultures of cardiac microvascular endothelial cells. Microvasc. Res. 2001, 61, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Kito, H.; Yamamura, H.; Suzuki, Y.; Yamamura, H.; Ohya, S.; Asai, K.; Imaizumi, Y. Regulation of store-operated Ca2+ entry activity by cell cycle dependent up-regulation of Orai2 in brain capillary endothelial cells. Biochem. Biophys. Res. Commun. 2015, 459, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Galeano-Otero, I.; Del Toro, R.; Khatib, A.M.; Rosado, J.A.; Ordonez-Fernandez, A.; Smani, T. SARAF and Orai1 Contribute to Endothelial Cell Activation and Angiogenesis. Front. Cell Dev. Biol. 2021, 9, 639952. [Google Scholar] [CrossRef]
- Sachdeva, R.; Fleming, T.; Schumacher, D.; Homberg, S.; Stilz, K.; Mohr, F.; Wagner, A.H.; Tsvilovskyy, V.; Mathar, I.; Freichel, M. Methylglyoxal evokes acute Ca(2+) transients in distinct cell types and increases agonist-evoked Ca(2+) entry in endothelial cells via CRAC channels. Cell Calcium 2019, 78, 66–75. [Google Scholar] [CrossRef]
- Albarran, L.; Lopez, J.J.; Amor, N.B.; Martin-Cano, F.E.; Berna-Erro, A.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Naik, J.S.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Reduced membrane cholesterol after chronic hypoxia limits Orai1-mediated pulmonary endothelial Ca(2+) entry. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H359–H369. [Google Scholar] [CrossRef]
- Yamamura, H.; Suzuki, Y.; Asai, K.; Imaizumi, Y.; Yamamura, H. Oxidative stress facilitates cell death by inhibiting Orai1-mediated Ca(2+) entry in brain capillary endothelial cells. Biochem. Biophys. Res. Commun. 2020, 523, 153–158. [Google Scholar] [CrossRef]
- Deak, A.T.; Groschner, L.N.; Alam, M.R.; Seles, E.; Bondarenko, A.I.; Graier, W.F.; Malli, R. The endocannabinoid N-arachidonoyl glycine (NAGly) inhibits store-operated Ca2+ entry by preventing STIM1-Orai1 interaction. J. Cell Sci. 2013, 126, 879–888. [Google Scholar] [CrossRef]
- Terry, L.E.; VerMeer, M.; Giles, J.; Tran, Q.K. Suppression of store-operated Ca(2+) entry by activation of GPER: Contribution to a clamping effect on endothelial Ca(2+) signaling. Biochem. J. 2017, 474, 3627–3642. [Google Scholar] [CrossRef]
- Yazbeck, P.; Tauseef, M.; Kruse, K.; Amin, M.R.; Sheikh, R.; Feske, S.; Komarova, Y.; Mehta, D. STIM1 Phosphorylation at Y361 Recruits Orai1 to STIM1 Puncta and Induces Ca(2+) Entry. Sci. Rep. 2017, 7, 42758. [Google Scholar] [CrossRef]
- Sundivakkam, P.C.; Natarajan, V.; Malik, A.B.; Tiruppathi, C. Store-operated Ca2+ entry (SOCE) induced by protease-activated receptor-1 mediates STIM1 protein phosphorylation to inhibit SOCE in endothelial cells through AMP-activated protein kinase and p38beta mitogen-activated protein kinase. J. Biol. Chem. 2013, 288, 17030–17041. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting Stim and Orai Proteins as an Alternative Approach in Anticancer Therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef]
- Moccia, F.; Dragoni, S.; Poletto, V.; Rosti, V.; Tanzi, F.; Ganini, C.; Porta, C. Orai1 and Transient Receptor Potential Channels as novel molecular targets to impair tumor neovascularisation in renal cell carcinoma and other malignancies. Anticancer. Agents Med. Chem. 2014, 14, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Di Giuro, C.M.L.; Shrestha, N.; Malli, R.; Groschner, K.; Van Breemen, C.; Fameli, N. Na (+)/Ca(2+) exchangers and Orai channels jointly refill endoplasmic reticulum (ER) Ca(2+) via ER nanojunctions in vascular endothelial cells. Pflug. Arch. 2017, 469, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Bonetti, E.; Dragoni, S.; Fontana, J.; Lodola, F.; Romani, R.B.; Laforenza, U.; Rosti, V.; Tanzi, F. Hematopoietic progenitor and stem cells circulate by surfing on intracellular Ca2+ waves: A novel target for cell-based therapy and anti-cancer treatment? Curr. Signal Transd. Therapy 2012, 7, 161–176. [Google Scholar] [CrossRef]
- Wang, S.; Xu, L.; Wu, Y.; Shen, H.; Lin, Z.; Fang, Y.; Zhang, L.; Shen, B.; Liu, Y.; Wu, K. Parathyroid Hormone Promotes Human Umbilical Vein Endothelial Cell Migration and Proliferation Through Orai1-Mediated Calcium Signaling. Front. Cardiovasc. Med. 2022, 9, 844671. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Weisbrod, R.M.; Pimental, D.R.; Tong, X.; Bolotina, V.M.; Cohen, R.A. Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid. Redox. Signal. 2012, 17, 1099–1108. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Berra-Romani, R. Targeting the endothelial Ca2+ tool kit to rescue endothelial dysfunction in obesity associated-hypertension. Curr. Med. Chem. 2019, 27, 240–257. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Blatter, L.A. Nitric oxide inhibits capacitative Ca2+ entry and enhances endoplasmic reticulum Ca2+ uptake in bovine vascular endothelial cells. J. Physiol. 2002, 539, 77–91. [Google Scholar] [CrossRef]
- Hirano, K.; Hirano, M.; Hanada, A. Involvement of STIM1 in the proteinase-activated receptor 1-mediated Ca2+ influx in vascular endothelial cells. J. Cell. Biochem. 2009, 108, 499–507. [Google Scholar] [CrossRef]
- Kassan, M.; Zhang, W.; Aissa, K.A.; Stolwijk, J.; Trebak, M.; Matrougui, K. Differential role for stromal interacting molecule 1 in the regulation of vascular function. Pflug. Arch. 2015, 467, 1195–1202. [Google Scholar] [CrossRef]
- Nishimoto, M.; Mizuno, R.; Fujita, T.; Isshiki, M. Stromal interaction molecule 1 modulates blood pressure via NO production in vascular endothelial cells. Hypertens. Res. 2018, 41, 506–514. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Angelone, T. Targeting endothelial ion signalling to rescue cerebral blood flow in cerebral disorders. Vascul. Pharmacol. 2022, 145, 106997. [Google Scholar] [CrossRef]
- Mapelli, L.; Gagliano, G.; Soda, T.; Laforenza, U.; Moccia, F.; D’Angelo, E.U. Granular Layer Neurons Control Cerebellar Neurovascular Coupling Through an NMDA Receptor/NO-Dependent System. J. Neurosci. 2017, 37, 1340–1351. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca(2+) signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium 2021, 99, 102454. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca(2+) signals and nitric oxide release in human brain microvascular endothelial cells. Cell. Mol. Life Sci. 2020, 77, 2235–2253. [Google Scholar] [CrossRef] [PubMed]
- Agrud, A.; Subburaju, S.; Goel, P.; Ren, J.; Kumar, A.S.; Caldarone, B.J.; Dai, W.; Chavez, J.; Fukumura, D.; Jain, R.K.; et al. Gabrb3 endothelial cell-specific knockout mice display abnormal blood flow, hypertension, and behavioral dysfunction. Sci. Rep. 2022, 12, 4922. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Scolari, F.; Vismara, M.; Brunetti, V.; Faris, P.; Terribile, G.; Sancini, G.; Berra-Romani, R.; Moccia, F. GABA(A) and GABA(B) Receptors Mediate GABA-Induced Intracellular Ca(2+) Signals in Human Brain Microvascular Endothelial Cells. Cells 2022, 11, 3860. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Pellavio, G.; Orgiu, M.; Negri, S.; Forcaia, G.; Var-Gaz-Guadarrama, V.; Garcia-Carrasco, M.; Botta, L.; Sancini, G.; et al. Histamine induces intracellular Ca(2+) oscillations and nitric oxide release in endothelial cells from brain microvascular circulation. J. Cell. Physiol. 2020, 235, 1515–1530. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic Acid Evokes an Increase in Intracellular Ca(2+) Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca(2+) oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca(2+) release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef]
- Li, J.; Bruns, A.F.; Hou, B.; Rode, B.; Webster, P.J.; Bailey, M.A.; Appleby, H.L.; Moss, N.K.; Ritchie, J.E.; Yuldasheva, N.Y.; et al. Orai3 Surface Accumulation and Calcium Entry Evoked by Vascular Endothelial Growth Factor. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Stolwijk, J.A.; Zhang, X.; Gueguinou, M.; Zhang, W.; Matrougui, K.; Renken, C.; Trebak, M. Calcium Signaling Is Dispensable for Receptor Regulation of Endothelial Barrier Function. J. Biol. Chem. 2016, 291, 22894–22912. [Google Scholar] [CrossRef]
- Chen, F.; Zhu, L.; Cai, L.; Zhang, J.; Zeng, X.; Li, J.; Su, Y.; Hu, Q. A stromal interaction molecule 1 variant up-regulates matrix metalloproteinase-2 expression by strengthening nucleoplasmic Ca2+ signaling. Biochim. Biophys. Acta 2016, 1863, 617–629. [Google Scholar] [CrossRef]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.H.; Zheng, H.; Si, H.; Jin, Y.; Peng, J.M.; He, L.; Zhou, Y.; Munoz-Garay, C.; Zawieja, D.C.; Kuo, L.; et al. Stromal interaction molecule 1 (STIM1) and Orai1 mediate histamine-evoked calcium entry and nuclear factor of activated T-cells (NFAT) signaling in human umbilical vein endothelial cells. J. Biol. Chem. 2014, 289, 29446–29456. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.X.; Yuan, J.N.; Zhang, F.R.; Liu, Y.Y.; Zhang, T.T.; Li, K.; Lv, X.F.; Zhou, J.G.; Huang, L.Y.; Shang, J.Y.; et al. Inhibition of Orai1-mediated Ca(2+) entry limits endothelial cell inflammation by suppressing calcineurin-NFATc4 signaling pathway. Biochem. Biophys. Res. Commun. 2018, 495, 1864–1870. [Google Scholar] [CrossRef] [PubMed]
- Gandhirajan, R.K.; Meng, S.; Chandramoorthy, H.C.; Mallilankaraman, K.; Mancarella, S.; Gao, H.; Razmpour, R.; Yang, X.F.; Houser, S.R.; Chen, J.; et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Investig. 2013, 123, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Daskoulidou, N.; Zeng, B.; Berglund, L.M.; Jiang, H.; Chen, G.L.; Kotova, O.; Bhandari, S.; Ayoola, J.; Griffin, S.; Atkin, S.L.; et al. High glucose enhances store-operated calcium entry by upregulating ORAI/STIM via calcineurin-NFAT signalling. J. Mol. Med. 2015, 93, 511–521. [Google Scholar] [CrossRef]
- Bai, S.; Wei, Y.; Hou, W.; Yao, Y.; Zhu, J.; Hu, X.; Chen, W.; Du, Y.; He, W.; Shen, B.; et al. Orai-IGFBP3 signaling complex regulates high-glucose exposure-induced increased proliferation, permeability, and migration of human coronary artery endothelial cells. BMJ Open Diabetes Res. Care 2020, 8, e001400. [Google Scholar] [CrossRef]
- Zou, M.; Dong, H.; Meng, X.; Cai, C.; Li, C.; Cai, S.; Xue, Y. Store-operated Ca2+ entry plays a role in HMGB1-induced vascular endothelial cell hyperpermeability. PLoS ONE 2015, 10, e0123432. [Google Scholar] [CrossRef]
- Yang, H.; Xue, Y.; Kuang, S.; Zhang, M.; Chen, J.; Liu, L.; Shan, Z.; Lin, Q.; Li, X.; Yang, M.; et al. Involvement of Orai1 in tunicamycin-induced endothelial dysfunction. Korean J. Physiol. Pharmacol. 2019, 23, 95–102. [Google Scholar] [CrossRef]
- Song, X.; Liu, Y.; Dong, L.; Wang, Y. Stromal-Interacting Molecule 1 (Stim1)/Orai1 Modulates Endothelial Permeability in Ventilator-Induced Lung Injury. Med. Sci. Monit. 2018, 24, 9413–9423. [Google Scholar] [CrossRef] [PubMed]
- Berlansky, S.; Sallinger, M.; Grabmayr, H.; Humer, C.; Bernhard, A.; Fahrner, M.; Frischauf, I. Calcium Signals during SARS-CoV-2 Infection: Assessing the Potential of Emerging Therapies. Cells 2022, 11, 253. [Google Scholar] [CrossRef]
- Qiu, X.; Liang, X.; Li, H.; Sun, R. LPS-induced vein endothelial cell injury and acute lung injury have Btk and Orai 1 to regulate SOC-mediated calcium influx. Int. Immunopharmacol. 2021, 90, 107039. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Zhu, Z.; Su, Q.; Li, T.; Song, Q. Toll-like receptor 4 is involved in bacterial endotoxin-induced endothelial cell injury and SOC-mediated calcium regulation. Cell Biol. Int. 2012, 36, 475–481. [Google Scholar] [CrossRef]
- Pasyk, E.; Inazu, M.; Daniel, E.E. CPA enhances Ca2+ entry in cultured bovine pulmonary arterial endothelial cells in an IP3-independent manner. Am. J. Physiol. 1995, 268, H138–H146. [Google Scholar] [CrossRef]
- Encabo, A.; Romanin, C.; Birke, F.W.; Kukovetz, W.R.; Groschner, K. Inhibition of a store-operated Ca2+ entry pathway in human endothelial cells by the isoquinoline derivative LOE 908. Br. J. Pharmacol. 1996, 119, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Vaca, L.; Kunze, D.L. Depletion and refilling of intracellular Ca2+ stores induce oscillations of Ca2+ current. Am. J. Physiol. 1993, 264, H1319–H1322. [Google Scholar] [CrossRef]
- Zhang, H.; Inazu, M.; Weir, B.; Buchanan, M.; Daniel, E. Cyclopiazonic acid stimulates Ca2+ influx through non-specific cation channels in endothelial cells. Eur. J. Pharmacol. 1994, 251, 119–125. [Google Scholar] [CrossRef]
- Paffett, M.L.; Naik, J.S.; Resta, T.C.; Walker, B.R. Reduced store-operated Ca2+ entry in pulmonary endothelial cells from chronically hypoxic rats. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1135–L1142. [Google Scholar] [CrossRef]
- Fantozzi, I.; Zhang, S.; Platoshyn, O.; Remillard, C.V.; Cowling, R.T.; Yuan, J.X. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L1233–L1245. [Google Scholar] [CrossRef] [PubMed]
- Jho, D.; Mehta, D.; Ahmmed, G.; Gao, X.P.; Tiruppathi, C.; Broman, M.; Malik, A.B. Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2 influx. Circ. Res. 2005, 96, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Thippegowda, P.B.; Singh, V.; Sundivakkam, P.C.; Xue, J.; Malik, A.B.; Tiruppathi, C. Ca2+ influx via TRPC channels induces NF-kappaB-dependent A20 expression to prevent thrombin-induced apoptosis in endothelial cells. Am. J. Physiol. Cell Physiol. 2010, 298, C656–C664. [Google Scholar] [CrossRef]
- Sundivakkam, P.C.; Kwiatek, A.M.; Sharma, T.T.; Minshall, R.D.; Malik, A.B.; Tiruppathi, C. Caveolin-1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am. J. Physiol. Cell Physiol. 2009, 296, C403–C413. [Google Scholar] [CrossRef]
- Paria, B.C.; Bair, A.M.; Xue, J.; Yu, Y.; Malik, A.B.; Tiruppathi, C. Ca2+ influx induced by protease-activated receptor-1 activates a feed-forward mechanism of TRPC1 expression via nuclear factor-kappaB activation in endothelial cells. J. Biol. Chem. 2006, 281, 20715–20727. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Freichel, M.; Vogel, S.M.; Paria, B.C.; Mehta, D.; Flockerzi, V.; Malik, A.B. Impairment of store-operated Ca2+ entry in TRPC4(-/-) mice interferes with increase in lung microvascular permeability. Circ. Res. 2002, 91, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Antoniotti, S.; Fiorio Pla, A.; Barral, S.; Scalabrino, O.; Munaron, L.; Lovisolo, D. Interaction between TRPC channel subunits in endothelial cells. J. Recept. Signal Transduct. Res. 2006, 26, 225–240. [Google Scholar] [CrossRef]
- Murata, T.; Lin, M.I.; Stan, R.V.; Bauer, P.M.; Yu, J.; Sessa, W.C. Genetic evidence supporting caveolae microdomain regulation of calcium entry in endothelial cells. J. Biol. Chem. 2007, 282, 16631–16643. [Google Scholar] [CrossRef]
- Pani, B.; Ong, H.L.; Brazer, S.C.; Liu, X.; Rauser, K.; Singh, B.B.; Ambudkar, I.S. Activation of TRPC1 by STIM1 in ER-PM microdomains involves release of the channel from its scaffold caveolin-1. Proc. Natl. Acad. Sci. USA 2009, 106, 20087–20092. [Google Scholar] [CrossRef]
- Mehta, D.; Ahmmed, G.U.; Paria, B.C.; Holinstat, M.; Voyno-Yasenetskaya, T.; Tiruppathi, C.; Minshall, R.D.; Malik, A.B. RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. Role in signaling increased endothelial permeability. J. Biol. Chem. 2003, 278, 33492–33500. [Google Scholar] [CrossRef] [PubMed]
- Beliveau, E.; Lessard, V.; Guillemette, G. STIM1 positively regulates the Ca2+ release activity of the inositol 1,4,5-trisphosphate receptor in bovine aortic endothelial cells. PLoS ONE 2014, 9, e114718. [Google Scholar] [CrossRef]
- Thakore, P.; Earley, S. Transient Receptor Potential Channels and Endothelial Cell Calcium Signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef]
- Mace, K.E.; Lussier, M.P.; Boulay, G.; Terry-Powers, J.L.; Parfrey, H.; Perraud, A.L.; Riches, D.W. TRUSS, TNF-R1, and TRPC ion channels synergistically reverse endoplasmic reticulum Ca2+ storage reduction in response to m1 muscarinic acetylcholine receptor signaling. J. Cell. Physiol. 2010, 225, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.X.; Guo, R.W.; Liu, B.; Wang, X.M.; Qi, F.; Guo, C.M.; Shi, Y.K.; Wang, H. Role of TRPC1 and NF-kappaB in mediating angiotensin II-induced Ca2+ entry and endothelial hyperpermeability. Peptides 2009, 30, 1368–1373. [Google Scholar] [CrossRef]
- Bair, A.M.; Thippegowda, P.B.; Freichel, M.; Cheng, N.; Ye, R.D.; Vogel, S.M.; Yu, Y.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. Ca2+ entry via TRPC channels is necessary for thrombin-induced NF-kappaB activation in endothelial cells through AMP-activated protein kinase and protein kinase Cdelta. J. Biol. Chem. 2009, 284, 563–574. [Google Scholar] [CrossRef]
- Luo, L.; Liu, S.; Zhang, D.; Wei, F.; Gu, N.; Zeng, Y.; Chen, X.; Xu, S.; Liu, S.; Xiang, T. Chromogranin A (CGA)-derived polypeptide (CGA(47–66)) inhibits TNF-alpha-induced vascular endothelial hyper-permeability through SOC-related Ca(2+) signaling. Peptides 2020, 131, 170297. [Google Scholar] [CrossRef]
- Balbuena, P.; Li, W.; Rzigalinski, B.A.; Ehrich, M. Malathion/oxon and lead acetate increase gene expression and protein levels of transient receptor potential canonical channel subunits TRPC1 and TRPC4 in rat endothelial cells of the blood-brain barrier. Int. J. Toxicol. 2012, 31, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, D.L.; Lowe, K.; Alvarez, D.F.; Barry, C.; Stevens, T. TRPing on the lung endothelium: Calcium channels that regulate barrier function. Antioxid. Redox Signal. 2009, 11, 765–776. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Zeng, X.; Chen, S.; Yang, X.; Zhang, L. Galectin-3 mediates pulmonary vascular endothelial cell dynamics via TRPC1/4 under acute hypoxia. J. Biochem. Mol. Toxicol. 2020, 34, e22463. [Google Scholar] [CrossRef]
- Yu, P.C.; Gu, S.Y.; Bu, J.W.; Du, J.L. TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ. Res. 2010, 106, 1221–1232. [Google Scholar] [CrossRef]
- Kusaba, T.; Okigaki, M.; Matui, A.; Murakami, M.; Ishikawa, K.; Kimura, T.; Sonomura, K.; Adachi, Y.; Shibuya, M.; Shirayama, T.; et al. Klotho is associated with VEGF receptor-2 and the transient receptor potential canonical-1 Ca2+ channel to maintain endothelial integrity. Proc. Natl. Acad. Sci. USA 2010, 107, 19308–19313. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, C.; Manetti, M.; Rosa, I.; Romano, E.; Blagojevic, J.; Bellando-Randone, S.; Bruni, C.; Lepri, G.; Guiducci, S.; Ibba-Manneschi, L.; et al. Proangiogenic effects of soluble alpha-Klotho on systemic sclerosis dermal microvascular endothelial cells. Arthritis Res. Ther. 2017, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Peng, Y.; Gao, M.; Zhu, Y.; Zhu, Y.; Yu, F.; Zhou, T.; Shao, J.; Feng, L.; Ma, X. Endothelial Transient Receptor Potential Canonical Channel Regulates Angiogenesis and Promotes Recovery After Myocardial Infarction. J. Am. Heart Assoc. 2022, 11, e023678. [Google Scholar] [CrossRef] [PubMed]
- Song, H.B.; Jun, H.O.; Kim, J.H.; Fruttiger, M.; Kim, J.H. Suppression of transient receptor potential canonical channel 4 inhibits vascular endothelial growth factor-induced retinal neovascularization. Cell Calcium 2015, 57, 101–108. [Google Scholar] [CrossRef]
- Antigny, F.; Girardin, N.; Frieden, M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J. Biol. Chem. 2012, 287, 5917–5927. [Google Scholar] [CrossRef]
- Schmidt, K.; Dubrovska, G.; Nielsen, G.; Fesus, G.; Uhrenholt, T.R.; Hansen, P.B.; Gudermann, T.; Dietrich, A.; Gollasch, M.; De Wit, C.; et al. Amplification of EDHF-type vasodilatations in TRPC1-deficient mice. Br. J. Pharmacol. 2010, 161, 1722–1733. [Google Scholar] [CrossRef]
- Li, S.; Ning, H.; Ye, Y.; Wei, W.; Guo, R.; Song, Q.; Liu, L.; Liu, Y.; Na, L.; Niu, Y.; et al. Increasing extracellular Ca(2+) sensitizes TNF-alpha-induced vascular cell adhesion molecule-1 (VCAM-1) via a TRPC1/ERK1/2/NFkappaB-dependent pathway in human vascular endothelial cells. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 1566–1577. [Google Scholar] [CrossRef]
- Wu, S.; Sangerman, J.; Li, M.; Brough, G.H.; Goodman, S.R.; Stevens, T. Essential control of an endothelial cell ISOC by the spectrin membrane skeleton. J. Cell Biol. 2001, 154, 1225–1233. [Google Scholar] [CrossRef]
- Moore, T.M.; Brough, G.H.; Babal, P.; Kelly, J.J.; Li, M.; Stevens, T. Store-operated calcium entry promotes shape change in pulmonary endothelial cells expressing Trp1. Am. J. Physiol. 1998, 275, L574–L582. [Google Scholar] [CrossRef]
- Bavencoffe, A.; Zhu, M.X.; Tian, J.B. New Aspects of the Contribution of ER to SOCE Regulation: TRPC Proteins as a Link Between Plasma Membrane Ion Transport and Intracellular Ca(2+) Stores. Adv. Exp. Med. Biol. 2017, 993, 239–255. [Google Scholar] [CrossRef]
- Vasauskas, A.A.; Chen, H.; Wu, S.; Cioffi, D.L. The serine-threonine phosphatase calcineurin is a regulator of endothelial store-operated calcium entry. Pulm. Circ. 2014, 4, 116–127. [Google Scholar] [CrossRef]
- Chetham, P.M.; Babal, P.; Bridges, J.P.; Moore, T.M.; Stevens, T. Segmental regulation of pulmonary vascular permeability by store-operated Ca2+ entry. Am. J. Physiol. 1999, 276, L41–L50. [Google Scholar] [CrossRef]
- Kelly, J.J.; Moore, T.M.; Babal, P.; Diwan, A.H.; Stevens, T.; Thompson, W.J. Pulmonary microvascular and macrovascular endothelial cells: Differential regulation of Ca2+ and permeability. Am. J. Physiol. 1998, 274, L810–L819. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, J.; Xu, C.; Han, X.; Gao, Y.; Chen, H. Inhibition of SOCs Attenuates Acute Lung Injury Induced by Severe Acute Pancreatitis in Rats and PMVECs Injury Induced by Lipopolysaccharide. Inflammation 2016, 39, 1049–1058. [Google Scholar] [CrossRef]
- Fatherazi, S.; Belton, C.M.; Izutsu, K.T. Sequential activation of store-operated currents in human gingival keratinocytes. J. Investig. Dermatol. 2003, 121, 120–131. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zarayskiy, V.; Monje, F.; Peter, K.; Csutora, P.; Khodorov, B.I.; Bolotina, V.M. Store-operated Orai1 and IP3 receptor-operated TRPC1 channel. Channels 2007, 1, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Turin, I.; Laforenza, U.; Potenza, D.M.; Bottino, C.; Glasnov, T.N.; Prestia, M.; Ferulli, F.; Saitta, A.; Mosca, A.; et al. Store-operated Ca2+ entry does not control proliferation in primary cultures of human metastatic renal cellular carcinoma. BioMed Res. Int. 2014, 2014, 739494. [Google Scholar] [CrossRef]
- Xu, N.; Ayers, L.; Pastukh, V.; Alexeyev, M.; Stevens, T.; Tambe, D.T. Impact of Na+ permeation on collective migration of pulmonary arterial endothelial cells. PLoS ONE 2021, 16, e0250095. [Google Scholar] [CrossRef] [PubMed]
- Groschner, K. Polymodal TRPC signaling: Emerging role in phenotype switching and tissue remodeling. Commun. Integr. Biol. 2010, 3, 393–395. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Graziani, A.; Poteser, M.; Heupel, W.M.; Schleifer, H.; Krenn, M.; Drenckhahn, D.; Romanin, C.; Baumgartner, W.; Groschner, K. Cell-cell contact formation governs Ca2+ signaling by TRPC4 in the vascular endothelium: Evidence for a regulatory TRPC4-beta-catenin interaction. J. Biol. Chem. 2010, 285, 4213–4223. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Tritto, S.; Signorelli, S.; Taglietti, V.; Tanzi, F. Epidermal growth factor induces intracellular Ca2+ oscillations in microvascular endothelial cells. J. Cell. Physiol. 2003, 194, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Ariyoshi, H.; Kambayashi, J.; Fujitani, K.; Shinoki, N.; Sakon, M.; Kawasaki, T.; Monden, M. Separate analysis of nuclear and cytosolic Ca2+ concentrations in human umbilical vein endothelial cells. J. Cell. Biochem. 1996, 63, 23–36. [Google Scholar] [CrossRef]
- Ma, X.; Qiu, S.; Luo, J.; Ma, Y.; Ngai, C.Y.; Shen, B.; Wong, C.O.; Huang, Y.; Yao, X. Functional role of vanilloid transient receptor potential 4-canonical transient receptor potential 1 complex in flow-induced Ca2+ influx. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cao, J.; Luo, J.; Nilius, B.; Huang, Y.; Ambudkar, I.S.; Yao, X. Depletion of intracellular Ca2+ stores stimulates the translocation of vanilloid transient receptor potential 4-c1 heteromeric channels to the plasma membrane. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2249–2255. [Google Scholar] [CrossRef]
- Greenberg, H.Z.E.; Carlton-Carew, S.R.E.; Zargaran, A.K.; Jahan, K.S.; Birnbaumer, L.; Albert, A.P. Heteromeric TRPV4/TRPC1 channels mediate calcium-sensing receptor-induced relaxations and nitric oxide production in mesenteric arteries: Comparative study using wild-type and TRPC1(-/-) mice. Channels 2019, 13, 410–423. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef]
- Lodola, F.; Laforenza, U.; Bonetti, E.; Lim, D.; Dragoni, S.; Bottino, C.; Ong, H.L.; Guerra, G.; Ganini, C.; Massa, M.; et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS ONE 2012, 7, e42541. [Google Scholar] [CrossRef]
- Dragoni, S.; Reforgiato, M.; Zuccolo, E.; Poletto, V.; Lodola, F.; Ruffinatti, F.A.; Bonetti, E.; Guerra, G.; Barosi, G.; Rosti, V.; et al. Dysregulation of VEGF-induced proangiogenic Ca2+ oscillations in primary myelofibrosis-derived endothelial colony-forming cells. Exp. Hematol. 2015, 43, 1019–1030. [Google Scholar] [CrossRef]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, A.D.; Riccardi, A.; et al. VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Dragoni, S.; Cinelli, M.; Montagnani, S.; Amato, B.; Rosti, V.; Guerra, G.; Tanzi, F. How to utilize Ca2+ signals to rejuvenate the repairative phenotype of senescent endothelial progenitor cells in elderly patients affected by cardiovascular diseases: A useful therapeutic support of surgical approach? BMC Surg. 2013, 13 (Suppl. 2), S46. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).