
Hematopoietic Stem Cells and the Immune System in Development and Aging


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HSC Juvenescence
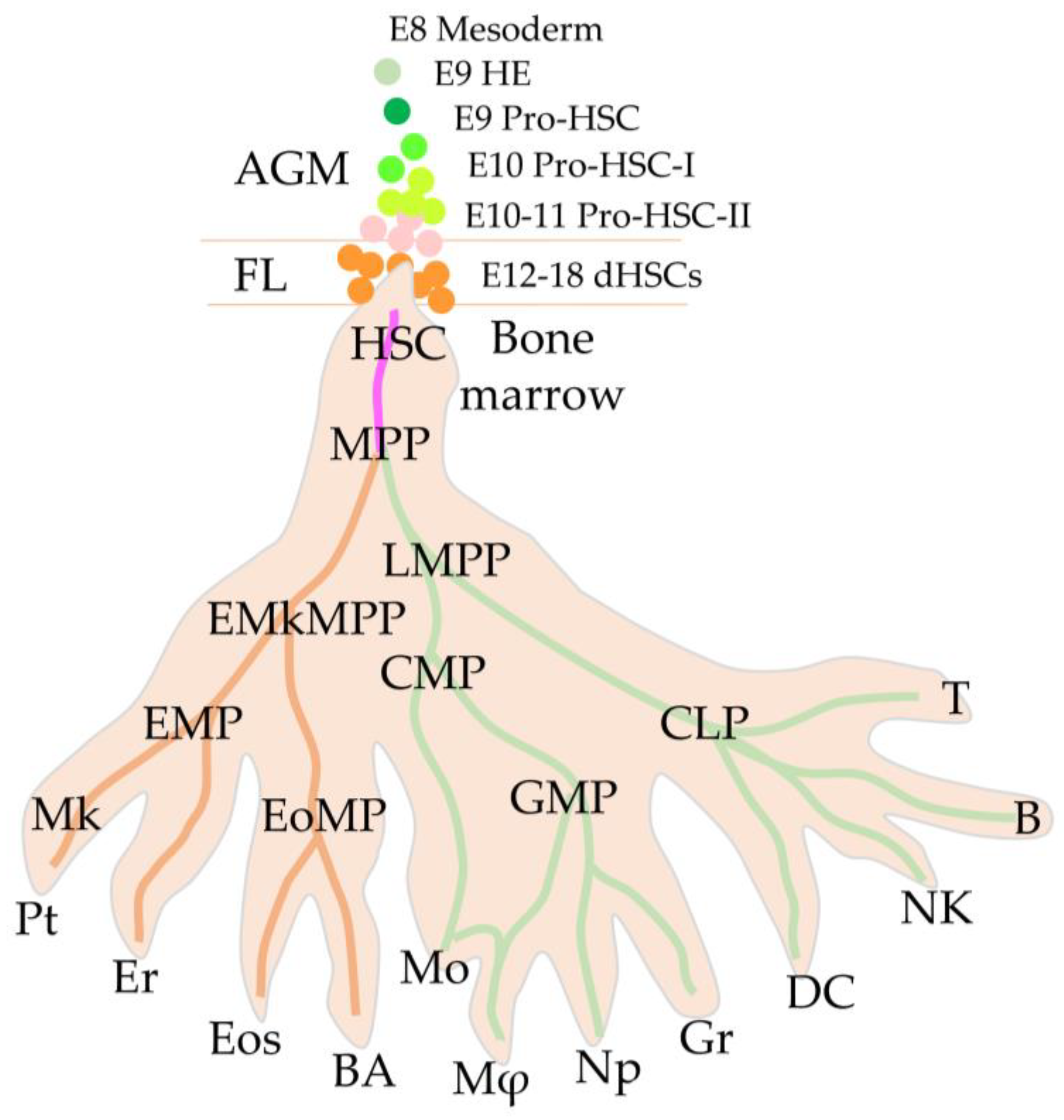
2.1. First Definitive HSCs
2.2. High Regenerative Potential of Embryonic HSCs
2.3. Migration to the Fetal Liver through the Placental Labyrinths
2.4. Embryonic vs. Adult HSCs
3. HSCs Adulthood and Aging
3.1. DNA Damage
3.2. Epigenetic Modifications
3.3. Transcriptomic Alteration
3.4. Self-Renew of Aging HSC and Its Repopulation Ability
3.5. Clonality and CHIP-Mutations
3.6. Differentiation Imbalance, CHIP, and Chromosomal Alterations
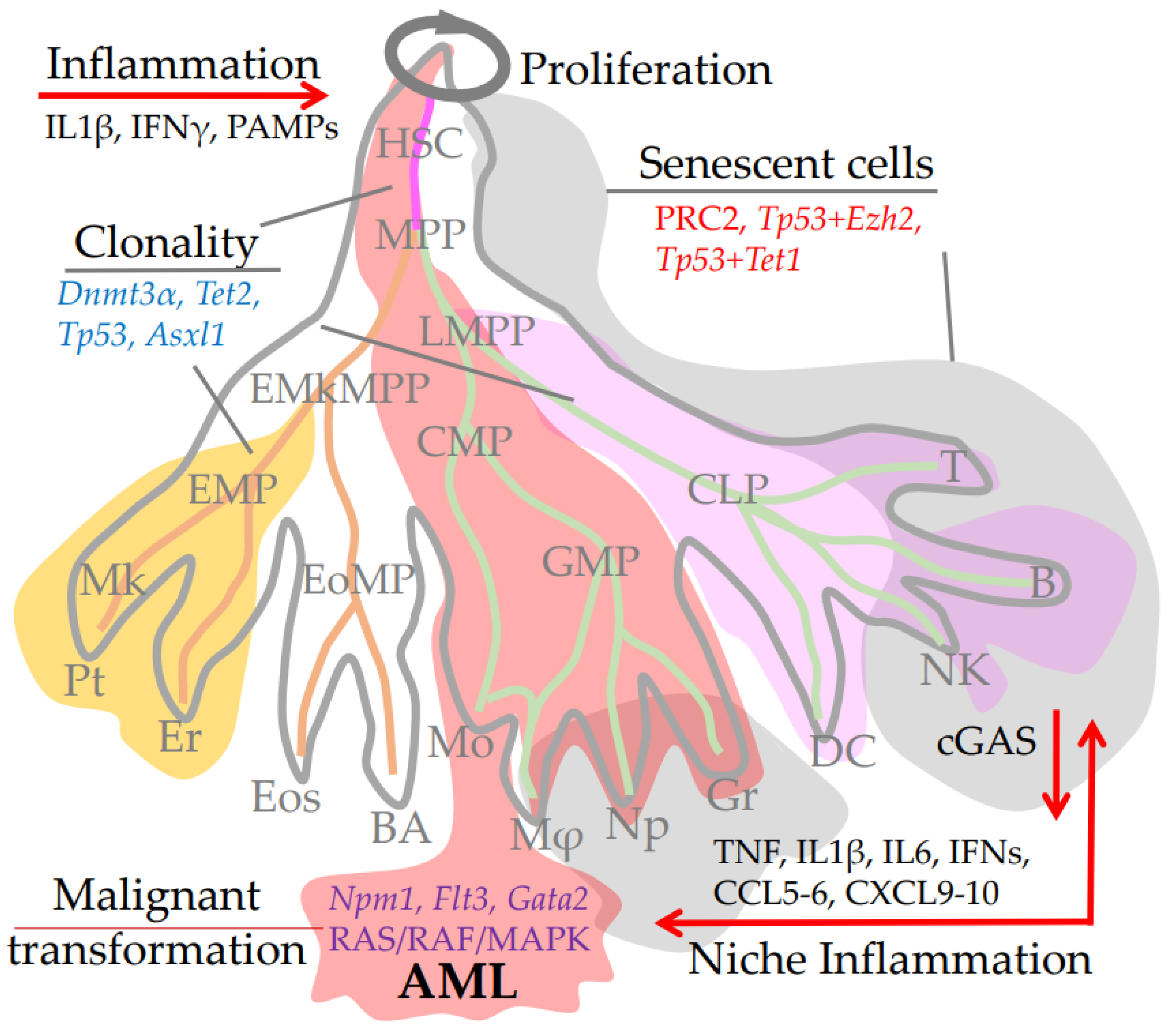
3.7. Inflammation and HSC Niche Aging
4. HSCs Senility and Immune Aging as a Risk Factor for Pathologies
4.1. Lymphopenia, Anaemia, Thrombosis, and Aging
4.2. Loss of Naïve T-Lymphocytes and TCR Repertoire
4.3. Reduced Effectiveness of Antigen Presentation
4.4. Inflammatory Phenotype of Differentiated Immune Cells, Senescence and Aging
5. Conclusion Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Konturek-Ciesla, A.; Bryder, D. Stem Cells, Hematopoiesis and Lineage Tracing: Transplantation-Centric Views and Beyond. Front. Cell Dev. Biol. 2022, 10, 903528. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, T.; Gu, J.; Huang, K.; Zhang, T.; Zhang, Z.; Liu, H.; Tang, J.; Mai, Y.; Zhang, Y.; et al. Characterization and generation of human definitive multipotent hematopoietic stem/progenitor cells. Cell Discov. 2020, 6, 89. [Google Scholar] [CrossRef]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [Green Version]
- Verovskaya, E.V.; Dellorusso, P.V.; Passegué, E. Losing Sense of Self and Surroundings: Hematopoietic Stem Cell Aging and Leukemic Transformation. Trends Mol. Med. 2019, 25, 494–515. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yoon, S.R.; Choi, I.; Jung, H. Causes and Mechanisms of Hematopoietic Stem Cell Aging. Int. J. Mol. Sci. 2019, 20, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Lian, X.; Xu, Y.; Hu, H.; Chang, C.; Zhang, H.; Zhang, L. Hematopoietic stem/progenitor cell senescence is associated with altered expression profiles of cellular memory-involved gene. Biosci. Rep. 2018, 38, BSR20171589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moehrle, B.M.; Geiger, H. Aging of hematopoietic stem cells: DNA damage and mutations? Exp. Hematol. 2016, 44, 895–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, L.; Hu, J.; Li, N.; Gao, J.; Huo, R.; Peng, X.; Zhang, N.; Liu, Y.; Zhao, H.; Liu, R.; et al. The Mechanism of Stem Cell Aging. Stem Cell Rev. Rep. 2022, 18, 1281–1293. [Google Scholar] [CrossRef]
- Latchney, S.E.; Calvi, L.M. The aging hematopoietic stem cell niche: Phenotypic and functional changes and mechanisms that contribute to hematopoietic aging. Semin. Hematol. 2017, 54, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.; Wang, W.; Su, D.M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun. Ageing 2020, 17, 2. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Dong, X.; Zhang, Z.; Zhang, Q.; Zhao, Y. Age-related thymic involution: Mechanisms and functional impact. Aging Cell 2022, 21, e13671. [Google Scholar] [CrossRef] [PubMed]
- Leins, H.; Mulaw, M.; Eiwen, K.; Sakk, V.; Liang, Y.; Denkinger, M.; Geiger, H.; Schirmbeck, R. Aged murine hematopoietic stem cells drive aging-associated immune remodeling. Blood 2018, 132, 565–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerman, I. Accumulation of DNA damage in the aged hematopoietic stem cell compartment. Semin. Hematol. 2017, 54, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Skead, K.; Ang Houle, A.; Abelson, S.; Agbessi, M.; Bruat, V.; Lin, B.; Soave, D.; Shlush, L.; Wright, S.; Dick, J.; et al. Interacting evolutionary pressures drive mutation dynamics and health outcomes in aging blood. Nat. Commun. 2021, 12, 4921. [Google Scholar] [CrossRef]
- Jacobsen, S.E.W.; Nerlov, C. Haematopoiesis in the era of advanced single-cell technologies. Nat. Cell Biol. 2019, 21, 2–8. [Google Scholar] [CrossRef]
- Brown, G. Hematopoietic and Chronic Myeloid Leukemia Stem Cells: Multi-Stability versus Lineage Restriction. Int. J. Mol. Sci. 2022, 23, 13570. [Google Scholar] [CrossRef]
- Yamashita, M.; Iwama, A. Aging and Clonal Behavior of Hematopoietic Stem Cells. Int. J. Mol. Sci. 2022, 23, 1948. [Google Scholar] [CrossRef]
- Watt, S.M.; Hua, P.; Roberts, I. Increasing Complexity of Molecular Landscapes in Human Hematopoietic Stem and Progenitor Cells during Development and Aging. Int. J. Mol. Sci. 2022, 23, 3675. [Google Scholar] [CrossRef]
- Ho, N.P.; Takizawa, H. Inflammation Regulates Haematopoietic Stem Cells and Their Niche. Int. J. Mol. Sci. 2022, 23, 1125. [Google Scholar] [CrossRef]
- Jaiswal, S.; Libby, P. Clonal haematopoiesis: Connecting ageing and inflammation in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 137–144. [Google Scholar] [CrossRef]
- Vink, C.S.; Mariani, S.A.; Dzierzak, E. Embryonic Origins of the Hematopoietic System: Hierarchies and Heterogeneity. Hemasphere 2022, 6, e737. [Google Scholar] [CrossRef] [PubMed]
- Dzierzak, E.; Bigas, A. Blood Development: Hematopoietic Stem Cell Dependence and Independence. Cell Stem Cell 2018, 22, 639–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandarakov, O.; Belyavsky, A.; Semenova, E. Bone Marrow Niches of Hematopoietic Stem and Progenitor Cells. Int. J. Mol. Sci. 2022, 23, 4462. [Google Scholar] [CrossRef]
- Wu, F.; Chen, Z.; Liu, J.; Hou, Y. The Akt-mTOR network at the interface of hematopoietic stem cell homeostasis. Exp. Hematol. 2021, 103, 15–23. [Google Scholar] [CrossRef]
- Belyavsky, A.; Petinati, N.; Drize, N. Hematopoiesis during Ontogenesis, Adult Life, and Aging. Int. J. Mol. Sci. 2021, 22, 9231. [Google Scholar] [CrossRef]
- Morganti, C.; Ito, K. Mitochondrial Contributions to Hematopoietic Stem Cell Aging. Int. J. Mol. Sci. 2021, 22, 11117. [Google Scholar] [CrossRef] [PubMed]
- Liggett, L.A.; Sankaran, V.G. Unraveling Hematopoiesis through the Lens of Genomics. Cell 2020, 182, 1384–1400. [Google Scholar] [CrossRef]
- Laurenti, E.; Göttgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef]
- Ghaffari, S. Haematopoietic stem cell quiescence exposed using mitochondrial membrane potential. Curr. Opin. Hematol. 2023, 30, 1–3. [Google Scholar] [CrossRef]
- Xie, X.; Su, M.; Ren, K.; Ma, X.; Lv, Z.; Li, Z.; Mei, Y.; Ji, P. Clonal hematopoiesis and bone marrow inflammation. Transl. Res. 2022; ahead of print. [Google Scholar] [CrossRef]
- Yamamoto, R.; Wilkinson, A.C.; Nakauchi, H. Changing concepts in hematopoietic stem cells. Science 2018, 362, 895–896. [Google Scholar] [CrossRef] [PubMed]
- Ganuza, M.; Chabot, A.; Tang, X.; Bi, W.; Natarajan, S.; Carter, R.; Gawad, C.; Kang, G.; Cheng, Y.; McKinney-Freeman, S. Murine hematopoietic stem cell activity is derived from pre-circulation embryos but not yolk sacs. Nat. Commun. 2018, 9, 5405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, P.G.; Nakano, H.; Das, P.P.; Chen, M.J.; Rowe, R.G.; Chou, S.S.; Ross, S.J.; Sakamoto, K.M.; Zon, L.I.; Schlaeger, T.M.; et al. Flow-induced protein kinase A-CREB pathway acts via BMP signaling to promote HSC emergence. J. Exp. Med. 2015, 212, 633–648. [Google Scholar] [CrossRef]
- Diaz, M.F.; Li, N.; Lee, H.J.; Adamo, L.; Evans, S.M.; Willey, H.E.; Arora, N.; Torisawa, Y.S.; Vickers, D.A.; Morris, S.A.; et al. Biomechanical forces promote blood development through prostaglandin E2 and the cAMP-PKA signaling axis. J. Exp. Med. 2015, 212, 665–680. [Google Scholar] [CrossRef]
- Chandrakanthan, V.; Rorimpandey, P.; Zanini, F.; Chacon, D.; Olivier, J.; Joshi, S.; Kang, Y.C.; Knezevic, K.; Huang, Y.; Qiao, Q.; et al. Mesoderm-derived PDGFRA(+) cells regulate the emergence of hematopoietic stem cells in the dorsal aorta. Nat. Cell Biol. 2022, 24, 1211–1225. [Google Scholar] [CrossRef]
- Boisset, J.C.; van Cappellen, W.; Andrieu-Soler, C.; Galjart, N.; Dzierzak, E.; Robin, C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature 2010, 464, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Rybtsov, S.A.; Lagarkova, M.A. Development of Hematopoietic Stem Cells in the Early Mammalian Embryo. Biochemistry 2019, 84, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Gao, P.; Tober, J.; Bennett, L.; Chen, C.; Uzun, Y.; Li, Y.; Howell, E.D.; Mumau, M.; Yu, W.; et al. Developmental trajectory of prehematopoietic stem cell formation from endothelium. Blood 2020, 136, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Rybtsov, S.; Batsivari, A.; Bilotkach, K.; Paruzina, D.; Senserrich, J.; Nerushev, O.; Medvinsky, A. Tracing the origin of the HSC hierarchy reveals an SCF-dependent, IL-3-independent CD43(-) embryonic precursor. Stem Cell Rep. 2014, 3, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Ivanovs, A.; Rybtsov, S.; Ng, E.S.; Stanley, E.G.; Elefanty, A.G.; Medvinsky, A. Human haematopoietic stem cell development: From the embryo to the dish. Development 2017, 144, 2323–2337. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, M.; Montecino-Rodriguez, E.; Ferkowicz, M.J.; Porayette, P.; Shelley, W.C.; Conway, S.J.; Dorshkind, K.; Yoder, M.C. Embryonic day 9 yolk sac and intra-embryonic hemogenic endothelium independently generate a B-1 and marginal zone progenitor lacking B-2 potential. Proc. Natl. Acad. Sci. USA 2011, 108, 1468–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simic, M.; Manosalva, I.; Spinelli, L.; Gentek, R.; Shayan, R.R.; Siret, C.; Girard-Madoux, M.; Wang, S.; de Fabritus, L.; Verschoor, J.; et al. Distinct Waves from the Hemogenic Endothelium Give Rise to Layered Lymphoid Tissue Inducer Cell Ontogeny. Cell Rep. 2020, 32, 108004. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Huang, J.; Tavian, M.; Nagai, Y.; Hirose, J.; Zuniga-Pflucker, J.C.; Peault, B.; Kincade, P.W. Tracing the first waves of lymphopoiesis in mice. Development 2006, 133, 2041–2051. [Google Scholar] [CrossRef] [Green Version]
- Taoudi, S.; Gonneau, C.; Moore, K.; Sheridan, J.M.; Blackburn, C.C.; Taylor, E.; Medvinsky, A. Extensive hematopoietic stem cell generation in the AGM region via maturation of VE-cadherin+CD45+ pre-definitive HSCs. Cell Stem Cell 2008, 3, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybtsov, S.; Sobiesiak, M.; Taoudi, S.; Souilhol, C.; Senserrich, J.; Liakhovitskaia, A.; Ivanovs, A.; Frampton, J.; Zhao, S.; Medvinsky, A. Hierarchical organization and early hematopoietic specification of the developing HSC lineage in the AGM region. J. Exp. Med. 2011, 208, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medvinsky, A.; Dzierzak, E. Definitive hematopoiesis is autonomously initiated by the AGM region. Cell 1996, 86, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Arora, N.; Wenzel, P.L.; McKinney-Freeman, S.L.; Ross, S.J.; Kim, P.G.; Chou, S.S.; Yoshimoto, M.; Yoder, M.C.; Daley, G.Q. Effect of developmental stage of HSC and recipient on transplant outcomes. Dev. Cell 2014, 29, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Rybtsov, S.; Ivanovs, A.; Zhao, S.; Medvinsky, A. Concealed expansion of immature precursors underpins acute burst of adult HSC activity in foetal liver. Development 2016, 143, 1284–1289. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Li, X.; Wang, W.; Zhu, P.; Zhou, J.; He, W.; Ding, M.; Xiong, F.; Zheng, X.; Li, Z.; et al. Tracing haematopoietic stem cell formation at single-cell resolution. Nature 2016, 533, 487–492. [Google Scholar] [CrossRef]
- Crosse, E.I.; Gordon-Keylock, S.; Rybtsov, S.; Binagui-Casas, A.; Felchle, H.; Nnadi, N.C.; Kirschner, K.; Chandra, T.; Tamagno, S.; Webb, D.J.; et al. Multi-layered Spatial Transcriptomics Identify Secretory Factors Promoting Human Hematopoietic Stem Cell Development. Cell Stem Cell 2020, 27, 822–839.e8. [Google Scholar] [CrossRef]
- McGarvey, A.C.; Rybtsov, S.; Souilhol, C.; Tamagno, S.; Rice, R.; Hills, D.; Godwin, D.; Rice, D.; Tomlinson, S.R.; Medvinsky, A. A molecular roadmap of the AGM region reveals BMPER as a novel regulator of HSC maturation. J. Exp. Med. 2017, 214, 3731–3751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souilhol, C.; Gonneau, C.; Lendinez, J.G.; Batsivari, A.; Rybtsov, S.; Wilson, H.; Morgado-Palacin, L.; Hills, D.; Taoudi, S.; Antonchuk, J.; et al. Inductive interactions mediated by interplay of asymmetric signalling underlie development of adult haematopoietic stem cells. Nat. Commun. 2016, 7, 10784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souilhol, C.; Lendinez, J.G.; Rybtsov, S.; Murphy, F.; Wilson, H.; Hills, D.; Batsivari, A.; Binagui-Casas, A.; McGarvey, A.C.; MacDonald, H.R.; et al. Developing HSCs become Notch independent by the end of maturation in the AGM region. Blood 2016, 128, 1567–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batsivari, A.; Rybtsov, S.; Souilhol, C.; Binagui-Casas, A.; Hills, D.; Zhao, S.; Travers, P.; Medvinsky, A. Understanding Hematopoietic Stem Cell Development through Functional Correlation of Their Proliferative Status with the Intra-aortic Cluster Architecture. Stem Cell Rep. 2017, 8, 1549–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medvinsky, A.L.; Samoylina, N.L.; Muller, A.M.; Dzierzak, E.A. An early pre-liver intraembryonic source of CFU-S in the developing mouse. Nature 1993, 364, 64–67. [Google Scholar] [CrossRef]
- Muller, A.M.; Medvinsky, A.; Strouboulis, J.; Grosveld, F.; Dzierzak, E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity 1994, 1, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Ivanovs, A.; Rybtsov, S.; Welch, L.; Anderson, R.A.; Turner, M.L.; Medvinsky, A. Highly potent human haemopoietic stem cells first emerge in the intraembryonic aorta-gonad-mesonephros region. Lancet 2013, 381, 12. [Google Scholar] [CrossRef]
- Ivanovs, A.; Rybtsov, S.; Welch, L.; Anderson, R.A.; Turner, M.L.; Medvinsky, A. Highly potent human hematopoietic stem cells first emerge in the intraembryonic aorta-gonad-mesonephros region. J. Exp. Med. 2011, 208, 2417–2427. [Google Scholar] [CrossRef] [Green Version]
- Ivanovs, A.; Rybtsov, S.; Anderson, R.A.; Turner, M.L.; Medvinsky, A. Postmenstrual gestational age should be used with care in studies of early human hematopoietic development. Blood 2013, 121, 3051–3052. [Google Scholar] [CrossRef] [Green Version]
- de Bruijn, M.F.; Ma, X.; Robin, C.; Ottersbach, K.; Sanchez, M.J.; Dzierzak, E. Hematopoietic stem cells localize to the endothelial cell layer in the midgestation mouse aorta. Immunity 2002, 16, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Taoudi, S.; Medvinsky, A. Functional identification of the hematopoietic stem cell niche in the ventral domain of the embryonic dorsal aorta. Proc. Natl. Acad. Sci. USA 2007, 104, 9399–9403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanovs, A.; Rybtsov, S.; Anderson, R.A.; Turner, M.L.; Medvinsky, A. Identification of the niche and phenotype of the first human hematopoietic stem cells. Stem Cell Rep. 2014, 2, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, S.A.; Li, Z.; Rice, S.; Krieg, C.; Fragkogianni, S.; Robinson, M.; Vink, C.S.; Pollard, J.W.; Dzierzak, E. Pro-inflammatory Aorta-Associated Macrophages Are Involved in Embryonic Development of Hematopoietic Stem Cells. Immunity 2019, 50, 1439–1452.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitch, S.R.; Kimber, G.M.; Wilson, N.K.; Parker, A.; Mirshekar-Syahkal, B.; Göttgens, B.; Medvinsky, A.; Dzierzak, E.; Ottersbach, K. Signaling from the sympathetic nervous system regulates hematopoietic stem cell emergence during embryogenesis. Cell Stem Cell 2012, 11, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Hadland, B.; Varnum-Finney, B.; Dozono, S.; Dignum, T.; Nourigat-McKay, C.; Heck, A.M.; Ishida, T.; Jackson, D.L.; Itkin, T.; Butler, J.M.; et al. Engineering a niche supporting hematopoietic stem cell development using integrated single-cell transcriptomics. Nat. Commun. 2022, 13, 1584. [Google Scholar] [CrossRef]
- Kumaravelu, P.; Hook, L.; Morrison, A.M.; Ure, J.; Zhao, S.; Zuyev, S.; Ansell, J.; Medvinsky, A. Quantitative developmental anatomy of definitive haematopoietic stem cells/long-term repopulating units (HSC/RUs): Role of the aorta-gonad-mesonephros (AGM) region and the yolk sac in colonisation of the mouse embryonic liver. Development 2002, 129, 4891–4899. [Google Scholar] [CrossRef]
- Pellin, D.; Loperfido, M.; Baricordi, C.; Wolock, S.L.; Montepeloso, A.; Weinberg, O.K.; Biffi, A.; Klein, A.M.; Biasco, L. A comprehensive single cell transcriptional landscape of human hematopoietic progenitors. Nat. Commun. 2019, 10, 2395. [Google Scholar] [CrossRef] [Green Version]
- Ivanovs, A.; Rybtsov, S.; Anderson, R.A.; Medvinsky, A. Vast Self-Renewal Potential of Human AGM Region HSCs Dramatically Declines in the Umbilical Cord Blood. Stem Cell Rep. 2020, 15, 811–816. [Google Scholar] [CrossRef]
- McKenzie, J.L.; Gan, O.I.; Doedens, M.; Wang, J.C.; Dick, J.E. Individual stem cells with highly variable proliferation and self-renewal properties comprise the human hematopoietic stem cell compartment. Nat. Immunol. 2006, 7, 1225–1233. [Google Scholar] [CrossRef]
- Liu, C.; Chen, B.J.; Deoliveira, D.; Sempowski, G.D.; Chao, N.J.; Storms, R.W. Progenitor cell dose determines the pace and completeness of engraftment in a xenograft model for cord blood transplantation. Blood 2010, 116, 5518–5527. [Google Scholar] [CrossRef] [Green Version]
- Guenechea, G.; Gan, O.I.; Dorrell, C.; Dick, J.E. Distinct classes of human stem cells that differ in proliferative and self-renewal potential. Nat. Immunol. 2001, 2, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Caocci, G.; Greco, M.; La Nasa, G. Bone Marrow Homing and Engraftment Defects of Human Hematopoietic Stem and Progenitor Cells. Mediterr. J. Hematol. Infect. Dis. 2017, 9, e2017032. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lan, Y.; He, W.; Chen, D.; Wang, J.; Zhou, F.; Wang, Y.; Sun, H.; Chen, X.; Xu, C.; et al. Mouse embryonic head as a site for hematopoietic stem cell development. Cell Stem Cell 2012, 11, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Gordon-Keylock, S.; Sobiesiak, M.; Rybtsov, S.; Moore, K.; Medvinsky, A. Mouse extraembryonic arterial vessels harbor precursors capable of maturing into definitive HSCs. Blood 2013, 122, 2338–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gekas, C.; Rhodes, K.E.; Van Handel, B.; Chhabra, A.; Ueno, M.; Mikkola, H.K. Hematopoietic stem cell development in the placenta. Int. J. Dev. Biol. 2010, 54, 1089–1098. [Google Scholar] [CrossRef]
- Ottersbach, K.; Dzierzak, E. The murine placenta contains hematopoietic stem cells within the vascular labyrinth region. Dev. Cell 2005, 8, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Robin, C.; Bollerot, K.; Mendes, S.; Haak, E.; Crisan, M.; Cerisoli, F.; Lauw, I.; Kaimakis, P.; Jorna, R.; Vermeulen, M.; et al. Human placenta is a potent hematopoietic niche containing hematopoietic stem and progenitor cells throughout development. Cell Stem Cell 2009, 5, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Easterbrook, J.; Rybtsov, S.; Gordon-Keylock, S.; Ivanovs, A.; Taoudi, S.; Anderson, R.A.; Medvinsky, A. Analysis of the Spatiotemporal Development of Hematopoietic Stem and Progenitor Cells in the Early Human Embryo. Stem Cell Rep. 2019, 12, 1056–1068. [Google Scholar] [CrossRef] [Green Version]
- Ema, H.; Nakauchi, H. Expansion of hematopoietic stem cells in the developing liver of a mouse embryo. Blood 2000, 95, 2284–2288. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.E.; Wagers, A.J.; Gulati, A.P.; Johnson, F.L.; Weissman, I.L. Physiological migration of hematopoietic stem and progenitor cells. Science 2001, 294, 1933–1936. [Google Scholar] [CrossRef]
- Christensen, J.L.; Wright, D.E.; Wagers, A.J.; Weissman, I.L. Circulation and chemotaxis of fetal hematopoietic stem cells. PLoS Biol. 2004, 2, E75. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.; Yoshimoto, M.; Takebe, T. Fetal liver hematopoiesis: From development to delivery. Stem Cell Res. Ther. 2021, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Holyoake, T.L.; Nicolini, F.E.; Eaves, C.J. Functional differences between transplantable human hematopoietic stem cells from fetal liver, cord blood, and adult marrow. Exp. Hematol. 1999, 27, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, F.E.; Holyoake, T.L.; Cashman, J.D.; Chu, P.P.; Lambie, K.; Eaves, C.J. Unique differentiation programs of human fetal liver stem cells shown both in vitro and in vivo in NOD/SCID mice. Blood 1999, 94, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Manesia, J.K.; Xu, Z.; Broekaert, D.; Boon, R.; van Vliet, A.; Eelen, G.; Vanwelden, T.; Stegen, S.; Van Gastel, N.; Pascual-Montano, A.; et al. Highly proliferative primitive fetal liver hematopoietic stem cells are fueled by oxidative metabolic pathways. Stem Cell Res. 2015, 15, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, R.; Arif, T.; Kalmykova, S.; Kasianov, A.; Lin, M.; Menon, V.; Qiu, J.; Bernitz, J.M.; Moore, K.; Lin, F.; et al. Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 2020, 26, 359–376.e7. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; DiMaulo-Milk, E.; Leslie, J.; Ding, L. Hematopoietic stem cells temporally transition to thrombopoietin dependence in the fetal liver. Sci. Adv. 2022, 8, eabm7688. [Google Scholar] [CrossRef]
- Liu, L.; Inoki, A.; Fan, K.; Mao, F.; Shi, G.; Jin, X.; Zhao, M.; Ney, G.; Jones, M.; Sun, S.; et al. ER-associated degradation preserves hematopoietic stem cell quiescence and self-renewal by restricting mTOR activity. Blood 2020, 136, 2975–2986. [Google Scholar] [CrossRef]
- Vanuytsel, K.; Villacorta-Martin, C.; Lindstrom-Vautrin, J.; Wang, Z.; Garcia-Beltran, W.F.; Vrbanac, V.; Parsons, D.; Lam, E.C.; Matte, T.M.; Dowrey, T.W.; et al. Multi-modal profiling of human fetal liver hematopoietic stem cells reveals the molecular signature of engraftment. Nat. Commun. 2022, 13, 1103. [Google Scholar] [CrossRef]
- Dykstra, B.; Kent, D.; Bowie, M.; McCaffrey, L.; Hamilton, M.; Lyons, K.; Lee, S.J.; Brinkman, R.; Eaves, C. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell Stem Cell 2007, 1, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Hérault, L.; Poplineau, M.; Remy, E.; Duprez, E. Single Cell Transcriptomics to Understand HSC Heterogeneity and Its Evolution upon Aging. Cells 2022, 11, 3125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.C.; Sadek, H.A. Hypoxia and metabolic properties of hematopoietic stem cells. Antioxid. Redox Signal. 2014, 20, 1891–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palis, J. Hematopoietic stem cell-independent hematopoiesis: Emergence of erythroid, megakaryocyte, and myeloid potential in the mammalian embryo. FEBS Lett. 2016, 590, 3965–3974. [Google Scholar] [CrossRef]
- Palis, J. Primitive and definitive erythropoiesis in mammals. Front. Physiol. 2014, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Malik, J.; Kim, A.R.; Tyre, K.A.; Cherukuri, A.R.; Palis, J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013, 98, 1778–1787. [Google Scholar] [CrossRef] [PubMed]
- Tavian, M.; Robin, C.; Coulombel, L.; Péault, B. The human embryo, but not its yolk sac, generates lympho-myeloid stem cells: Mapping multipotent hematopoietic cell fate in intraembryonic mesoderm. Immunity 2001, 15, 487–495. [Google Scholar] [CrossRef] [Green Version]
- McGrath, K.E.; Frame, J.M.; Fegan, K.H.; Bowen, J.R.; Conway, S.J.; Catherman, S.C.; Kingsley, P.D.; Koniski, A.D.; Palis, J. Distinct Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood Cells in the Mammalian Embryo. Cell Rep. 2015, 11, 1892–1904. [Google Scholar] [CrossRef] [Green Version]
- McGrath, K.E.; Koniski, A.D.; Malik, J.; Palis, J. Circulation is established in a stepwise pattern in the mammalian embryo. Blood 2003, 101, 1669–1676. [Google Scholar] [CrossRef] [Green Version]
- Dieterlen-Lievre, F. On the origin of haemopoietic stem cells in the avian embryo: An experimental approach. J. Embryol. Exp. Morphol. 1975, 33, 607–619. [Google Scholar] [CrossRef]
- Cumano, A.; Ferraz, J.C.; Klaine, M.; Di Santo, J.P.; Godin, I. Intraembryonic, but not yolk sac hematopoietic precursors, isolated before circulation, provide long-term multilineage reconstitution. Immunity 2001, 15, 477–485. [Google Scholar] [CrossRef] [Green Version]
- Medvinsky, A.; Rybtsov, S.; Taoudi, S. Embryonic origin of the adult hematopoietic system: Advances and questions. Development 2011, 138, 1017–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, M.; Porayette, P.; Glosson, N.L.; Conway, S.J.; Carlesso, N.; Cardoso, A.A.; Kaplan, M.H.; Yoder, M.C. Autonomous murine T-cell progenitor production in the extra-embryonic yolk sac before HSC emergence. Blood 2012, 119, 5706–5714. [Google Scholar] [CrossRef] [Green Version]
- Samokhvalov, I.M.; Samokhvalova, N.I.; Nishikawa, S. Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 2007, 446, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M.; Kosters, A.; Cornelius, S.; Valiente, N.; Cheng, H.; Latorre, A.; Nishida, C.; Ghosn, E.E.B.; Kobayashi, M. Mast Cell Repopulating Ability Is Lost During the Transition From Pre-HSC to FL HSC. Front. Immunol. 2022, 13, 896396. [Google Scholar] [CrossRef] [PubMed]
- Grimbaldeston, M.A.; Chen, C.C.; Piliponsky, A.M.; Tsai, M.; Tam, S.Y.; Galli, S.J. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 2005, 167, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [Green Version]
- Gentek, R.; Ghigo, C.; Hoeffel, G.; Bulle, M.J.; Msallam, R.; Gautier, G.; Launay, P.; Chen, J.; Ginhoux, F.; Bajenoff, M. Hemogenic Endothelial Fate Mapping Reveals Dual Developmental Origin of Mast Cells. Immunity 2018, 48, 1160–1171.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Liu, S.; Xu, J.; Zhang, X.; Han, D.; Liu, J.; Xia, M.; Yi, L.; Shen, Q.; Xu, S.; et al. Adult Connective Tissue-Resident Mast Cells Originate from Late Erythro-Myeloid Progenitors. Immunity 2018, 49, 640–653.e5. [Google Scholar] [CrossRef] [Green Version]
- Dahlin, J.S.; Hallgren, J. Mast cell progenitors: Origin, development and migration to tissues. Mol. Immunol. 2015, 63, 9–17. [Google Scholar] [CrossRef]
- Silver, A.J.; Bick, A.G.; Savona, M.R. Germline risk of clonal haematopoiesis. Nat. Rev. Genet. 2021, 22, 603–617. [Google Scholar] [CrossRef]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- McRae, H.M.; Voss, A.K.; Thomas, T. Are transplantable stem cells required for adult hematopoiesis? Exp. Hematol. 2019, 75, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Knapp, D.; Hammond, C.A.; Hui, T.; van Loenhout, M.T.J.; Wang, F.; Aghaeepour, N.; Miller, P.H.; Moksa, M.; Rabu, G.M.; Beer, P.A.; et al. Single-cell analysis identifies a CD33(+) subset of human cord blood cells with high regenerative potential. Nat. Cell Biol. 2018, 20, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Benz, C.; Copley, M.R.; Kent, D.G.; Wohrer, S.; Cortes, A.; Aghaeepour, N.; Ma, E.; Mader, H.; Rowe, K.; Day, C.; et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell 2012, 10, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copley, M.R.; Beer, P.A.; Eaves, C.J. Hematopoietic stem cell heterogeneity takes center stage. Cell Stem Cell 2012, 10, 690–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Haan, G.; Lazare, S.S. Aging of hematopoietic stem cells. Blood 2018, 131, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef]
- Akunuru, S.; Geiger, H. Aging, Clonality, and Rejuvenation of Hematopoietic Stem Cells. Trends Mol. Med. 2016, 22, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sun, J.; Ju, Z.; Wang, Z.Q.; Li, T. Nbs1-mediated DNA damage repair pathway regulates haematopoietic stem cell development and embryonic haematopoiesis. Cell Prolif. 2021, 54, e12972. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef]
- Takagi, K.; Yamada, Y.; Gong, J.S.; Sone, T.; Yokota, M.; Tanaka, M. Association of a 5178C-->A (Leu237Met) polymorphism in the mitochondrial DNA with a low prevalence of myocardial infarction in Japanese individuals. Atherosclerosis 2004, 175, 281–286. [Google Scholar] [CrossRef]
- Mansell, E.; Sigurdsson, V.; Deltcheva, E.; Brown, J.; James, C.; Miharada, K.; Soneji, S.; Larsson, J.; Enver, T. Mitochondrial Potentiation Ameliorates Age-Related Heterogeneity in Hematopoietic Stem Cell Function. Cell Stem Cell 2021, 28, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.D.; Ghaffari, S. Mitochondria in the maintenance of hematopoietic stem cells: New perspectives and opportunities. Blood 2019, 133, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Muraguchi, T.; Hoshii, T.; Hirao, A. Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid. Redox Signal. 2008, 10, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sejas, D.P.; Qiu, Y.; Williams, D.A.; Pang, Q. Inflammatory ROS promote and cooperate with the Fanconi anemia mutation for hematopoietic senescence. J. Cell Sci. 2007, 120, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, M.L.; Van Remmen, H.; Drake, J.A.; Yang, H.; Guo, Z.M.; Kewitt, K.; Walter, C.A.; Richardson, A. Does oxidative damage to DNA increase with age? Proc. Natl. Acad. Sci. USA 2001, 98, 10469–10474. [Google Scholar] [CrossRef] [Green Version]
- Caiado, F.; Pietras, E.M.; Manz, M.G. Inflammation as a regulator of hematopoietic stem cell function in disease, aging, and clonal selection. J. Exp. Med. 2021, 218, e20201541. [Google Scholar] [CrossRef]
- Lin, Q.; Wu, L.; Ma, Z.; Chowdhury, F.A.; Mazumder, H.H.; Du, W. Persistent DNA damage-induced NLRP12 improves hematopoietic stem cell function. JCI Insight 2020, 5, e133365. [Google Scholar] [CrossRef]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, E.; Spencer Chapman, M.; Williams, N.; Dawson, K.J.; Mende, N.; Calderbank, E.F.; Jung, H.; Mitchell, T.; Coorens, T.H.H.; Spencer, D.H.; et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 2022, 606, 343–350. [Google Scholar] [CrossRef]
- Cakouros, D.; Gronthos, S. Epigenetic Regulation of Bone Marrow Stem Cell Aging: Revealing Epigenetic Signatures associated with Hematopoietic and Mesenchymal Stem Cell Aging. Aging Dis. 2019, 10, 174–189. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarenkov, A.; Sardina, J.L. Dissecting TET2 Regulatory Networks in Blood Differentiation and Cancer. Cancers 2022, 14, 830. [Google Scholar] [CrossRef] [PubMed]
- Khokhar, E.S.; Borikar, S.; Eudy, E.; Stearns, T.; Young, K.; Trowbridge, J.J. Aging-associated decrease in the histone acetyltransferase KAT6B is linked to altered hematopoietic stem cell differentiation. Exp. Hematol. 2020, 82, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef] [Green Version]
- Sawai, C.M.; Babovic, S.; Upadhaya, S.; Knapp, D.; Lavin, Y.; Lau, C.M.; Goloborodko, A.; Feng, J.; Fujisaki, J.; Ding, L.; et al. Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity 2016, 45, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huang, Y.; Hu, L.; Cheng, T. New insights into Human Hematopoietic Stem and Progenitor Cells via Single-Cell Omics. Stem Cell Rev. Rep. 2022, 18, 1322–1336. [Google Scholar] [CrossRef]
- Hérault, L.; Poplineau, M.; Mazuel, A.; Platet, N.; Remy, É.; Duprez, E. Single-cell RNA-seq reveals a concomitant delay in differentiation and cell cycle of aged hematopoietic stem cells. BMC Biol. 2021, 19, 19. [Google Scholar] [CrossRef]
- Rundberg Nilsson, A.; Soneji, S.; Adolfsson, S.; Bryder, D.; Pronk, C.J. Human and Murine Hematopoietic Stem Cell Aging Is Associated with Functional Impairments and Intrinsic Megakaryocytic/Erythroid Bias. PLoS ONE 2016, 11, e0158369. [Google Scholar] [CrossRef] [Green Version]
- Flohr Svendsen, A.; Yang, D.; Kim, K.; Lazare, S.; Skinder, N.; Zwart, E.; Mura-Meszaros, A.; Ausema, A.; von Eyss, B.; de Haan, G.; et al. A comprehensive transcriptome signature of murine hematopoietic stem cell aging. Blood 2021, 138, 439–451. [Google Scholar] [CrossRef]
- SanMiguel, J.M.; Young, K.; Trowbridge, J.J. Hand in hand: Intrinsic and extrinsic drivers of aging and clonal hematopoiesis. Exp. Hematol. 2020, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Beerman, I. Proliferation: Driver of HSC aging phenotypes? Mech. Ageing Dev. 2020, 191, 111331. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Wandycz, A.M.; Akashi, K.; Globerson, A.; Weissman, I.L. The aging of hematopoietic stem cells. Nat. Med. 1996, 2, 1011–1016. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, Y.J.; Xu, L.P.; Liu, K.Y.; Liu, D.H.; Zhang, X.H.; Chen, H.; Han, W.; Chen, Y.H.; Wang, F.R.; et al. Who is the best donor for a related HLA haplotype-mismatched transplant? Blood 2014, 124, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Pessoa Rodrigues, C.; Akhtar, A. Differential H4K16ac levels ensure a balance between quiescence and activation in hematopoietic stem cells. Sci. Adv. 2021, 7, eabi5987. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cao, B.; Naval-Sanchez, M.; Pham, T.; Sun, Y.B.Y.; Williams, B.; Heazlewood, S.Y.; Deshpande, N.; Li, J.; Kraus, F.; et al. Nicotinamide riboside attenuates age-associated metabolic and functional changes in hematopoietic stem cells. Nat. Commun. 2021, 12, 2665. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fraticelli, A.E.; Weinreb, C.; Wang, S.W.; Migueles, R.P.; Jankovic, M.; Usart, M.; Klein, A.M.; Lowell, S.; Camargo, F.D. Single-cell lineage tracing unveils a role for TCF15 in haematopoiesis. Nature 2020, 583, 585–589. [Google Scholar] [CrossRef]
- Qi, L.; Martin-Sandoval, M.S.; Merchant, S.; Gu, W.; Eckhardt, M.; Mathews, T.P.; Zhao, Z.; Agathocleous, M.; Morrison, S.J. Aspartate availability limits hematopoietic stem cell function during hematopoietic regeneration. Cell Stem Cell 2021, 28, 1982–1999. [Google Scholar] [CrossRef]
- Vasanthakumar, A.; Arnovitz, S.; Marquez, R.; Lepore, J.; Rafidi, G.; Asom, A.; Weatherly, M.; Davis, E.M.; Neistadt, B.; Duszynski, R.; et al. Brca1 deficiency causes bone marrow failure and spontaneous hematologic malignancies in mice. Blood 2016, 127, 310–313. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Wang, Q.; Kao, Y.R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegue, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, T. Lysosomes and Their Role in Regulating the Metabolism of Hematopoietic Stem Cells. Biology 2022, 11, 1410. [Google Scholar] [CrossRef] [PubMed]
- Mogilenko, D.A.; Shchukina, I.; Artyomov, M.N. Immune ageing at single-cell resolution. Nat. Rev. Immunol. 2022, 22, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673. [Google Scholar] [CrossRef]
- Park, S.J.; Bejar, R. Clonal Hematopoiesis in Aging. Curr. Stem Cell Rep. 2018, 4, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.A.; Walsh, K. Clonal hematopoiesis, somatic mosaicism, and age-associated disease. Physiol. Rev. 2023, 103, 649–716. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, Q.; Yu, H.; Capitano, M.L.; Vemula, S.; Nabinger, S.C.; Gao, R.; Yao, C.; Kobayashi, M.; Geng, Z.; et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat. Commun. 2019, 10, 5649. [Google Scholar] [CrossRef] [Green Version]
- Sebert, M.; Gachet, S.; Leblanc, T.; Rousseau, A.; Bluteau, O.; Kim, R.; Ben Abdelali, R.; Sicre de Fontbrune, F.; Maillard, L.; Fedronie, C.; et al. Clonal hematopoiesis driven by chromosome 1q/MDM4 trisomy defines a canonical route toward leukemia in Fanconi anemia. Cell Stem Cell 2023, 30, 153–170. [Google Scholar] [CrossRef]
- Hidalgo, I.; Herrera-Merchan, A.; Ligos, J.M.; Carramolino, L.; Nuñez, J.; Martinez, F.; Dominguez, O.; Torres, M.; Gonzalez, S. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence-like cell cycle arrest. Cell Stem Cell 2012, 11, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Teo, Y.V.; Evans, S.A.; Neretti, N.; Sedivy, J.M. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018, 22, 3480–3492. [Google Scholar] [CrossRef] [Green Version]
- Chu, L.; Qu, Y.; An, Y.; Hou, L.; Li, J.; Li, W.; Fan, G.; Song, B.L.; Li, E.; Zhang, L.; et al. Induction of senescence-associated secretory phenotype underlies the therapeutic efficacy of PRC2 inhibition in cancer. Cell Death Dis. 2022, 13, 155. [Google Scholar] [CrossRef] [PubMed]
- Filipczak, P.T.; Leng, S.; Tellez, C.S.; Do, K.C.; Grimes, M.J.; Thomas, C.L.; Walton-Filipczak, S.R.; Picchi, M.A.; Belinsky, S.A. p53-Suppressed Oncogene TET1 Prevents Cellular Aging in Lung Cancer. Cancer Res. 2019, 79, 1758–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Feusier, J.E.; Arunachalam, S.; Tashi, T.; Baker, M.J.; VanSant-Webb, C.; Ferdig, A.; Welm, B.E.; Rodriguez-Flores, J.L.; Ours, C.; Jorde, L.B.; et al. Large-Scale Identification of Clonal Hematopoiesis and Mutations Recurrent in Blood Cancers. Blood Cancer Discov. 2021, 2, 226–237. [Google Scholar] [CrossRef]
- Loberg, M.A.; Bell, R.K.; Goodwin, L.O.; Eudy, E.; Miles, L.A.; SanMiguel, J.M.; Young, K.; Bergstrom, D.E.; Levine, R.L.; Schneider, R.K.; et al. Sequentially inducible mouse models reveal that Npm1 mutation causes malignant transformation of Dnmt3a-mutant clonal hematopoiesis. Leukemia 2019, 33, 1635–1649. [Google Scholar] [CrossRef] [Green Version]
- McKerrell, T.; Park, N.; Moreno, T.; Grove, C.S.; Ponstingl, H.; Stephens, J.; Crawley, C.; Craig, J.; Scott, M.A.; Hodkinson, C.; et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015, 10, 1239–1245. [Google Scholar] [CrossRef]
- Shih, A.H.; Jiang, Y.; Meydan, C.; Shank, K.; Pandey, S.; Barreyro, L.; Antony-Debre, I.; Viale, A.; Socci, N.; Sun, Y.; et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 2015, 27, 502–515. [Google Scholar] [CrossRef] [Green Version]
- Sportoletti, P.; Celani, L.; Varasano, E.; Rossi, R.; Sorcini, D.; Rompietti, C.; Strozzini, F.; Del Papa, B.; Guarente, V.; Spinozzi, G.; et al. GATA1 epigenetic deregulation contributes to the development of AML with NPM1 and FLT3-ITD cooperating mutations. Leukemia 2019, 33, 1827–1832. [Google Scholar] [CrossRef] [Green Version]
- Challen, G.A.; Goodell, M.A. Clonal hematopoiesis: Mechanisms driving dominance of stem cell clones. Blood 2020, 136, 1590–1598. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Izzo, F.; Lee, S.C.; Poran, A.; Chaligne, R.; Gaiti, F.; Gross, B.; Murali, R.R.; Deochand, S.D.; Ang, C.; Jones, P.W.; et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat. Genet. 2020, 52, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Chen, R.; Yang, Y.; He, H.; Xu, L.; Jiang, Y.; Guo, Z.; He, W.; Jiang, H.; Wang, J. Aging-elevated inflammation promotes DNMT3A R878H-driven clonal hematopoiesis. Acta Pharm. Sin. B 2022, 12, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Guo, X. Decoding and rejuvenating human ageing genomes: Lessons from mosaic chromosomal alterations. Ageing Res. Rev. 2021, 68, 101342. [Google Scholar] [CrossRef]
- Lenaerts, A.; Kucinski, I.; Deboutte, W.; Derecka, M.; Cauchy, P.; Manke, T.; Göttgens, B.; Grosschedl, R. EBF1 primes B-lymphoid enhancers and limits the myeloid bias in murine multipotent progenitors. J. Exp. Med. 2022, 219, e20212437. [Google Scholar] [CrossRef]
- Nechanitzky, R.; Akbas, D.; Scherer, S.; Györy, I.; Hoyler, T.; Ramamoorthy, S.; Diefenbach, A.; Grosschedl, R. Transcription factor EBF1 is essential for the maintenance of B cell identity and prevention of alternative fates in committed cells. Nat. Immunol. 2013, 14, 867–875. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Hughes-Davies, A.; Clayfield, L.; Menendez-Gonzalez, J.B.; Almotiri, A.; Alotaibi, B.; Tonks, A.; Rodrigues, N.P. Gata2 haploinsufficiency promotes proliferation and functional decline of hematopoietic stem cells with myeloid bias during aging. Blood Adv. 2021, 5, 4285–4290. [Google Scholar] [CrossRef]
- Zong, L.; Tanaka-Yano, M.; Park, B.; Yanai, H.; Turhan, F.T.; Croteau, D.L.; Tian, J.; Fang, E.F.; Bohr, V.A.; Beerman, I. NAD(+) augmentation with nicotinamide riboside improves lymphoid potential of Atm(−/−) and old mice HSCs. NPJ Aging Mech. Dis. 2021, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mack, R.; Breslin, P.; Zhang, J. Molecular and cellular mechanisms of aging in hematopoietic stem cells and their niches. J. Hematol. Oncol. 2020, 13, 157. [Google Scholar] [CrossRef]
- Grover, A.; Sanjuan-Pla, A.; Thongjuea, S.; Carrelha, J.; Giustacchini, A.; Gambardella, A.; Macaulay, I.; Mancini, E.; Luis, T.C.; Mead, A.; et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat. Commun. 2016, 7, 11075. [Google Scholar] [CrossRef] [Green Version]
- Nishi, K.; Sakamaki, T.; Kao, K.S.; Sadaoka, K.; Fujii, M.; Takaori-Kondo, A.; Miyanishi, M. Age-Associated Myeloid Biased Hematopoiesis Depends on Relative Decrease of Short-Term Hematopoietic Stem Cell. Blood 2019, 134, 2481. [Google Scholar] [CrossRef]
- Kato, H.; Igarashi, K. To be red or white: Lineage commitment and maintenance of the hematopoietic system by the “inner myeloid”. Haematologica 2019, 104, 1919–1927. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Ramirez, E.; Florian, M.C. Understanding intrinsic hematopoietic stem cell aging. Haematologica 2020, 105, 22–37. [Google Scholar] [CrossRef]
- Zhou, L.; Li, L.W.; Yan, Q.; Petryniak, B.; Man, Y.; Su, C.; Shim, J.; Chervin, S.; Lowe, J.B. Notch-dependent control of myelopoiesis is regulated by fucosylation. Blood 2008, 112, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swann, J.B.; Nusser, A.; Morimoto, R.; Nagakubo, D.; Boehm, T. Retracing the evolutionary emergence of thymopoiesis. Sci. Adv. 2020, 6, eabd9585. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.D.; Wu, D.; Meydani, S.N. Age-associated alterations in immune function and inflammation. Prog. Neuropsychopharmacol. Biol. Psychiatry 2022, 118, 110576. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.A.; Pollina, E.A.; Singh, P.P.; Mahmoudi, S.; Harel, I.; Casey, K.M.; Dulken, B.W.; Kundaje, A.; Brunet, A. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res. 2019, 29, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Matteini, F.; Mulaw, M.A.; Florian, M.C. Aging of the Hematopoietic Stem Cell Niche: New Tools to Answer an Old Question. Front. Immunol. 2021, 12, 738204. [Google Scholar] [CrossRef]
- Matatall, K.A.; Shen, C.C.; Challen, G.A.; King, K.Y. Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cells 2014, 32, 3023–3030. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, M.; Passegué, E. TNF-α Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell 2019, 25, 357–372. [Google Scholar] [CrossRef] [Green Version]
- Bogeska, R.; Mikecin, A.M.; Kaschutnig, P.; Fawaz, M.; Büchler-Schäff, M.; Le, D.; Ganuza, M.; Vollmer, A.; Paffenholz, S.V.; Asada, N.; et al. Inflammatory exposure drives long-lived impairment of hematopoietic stem cell self-renewal activity and accelerated aging. Cell Stem Cell 2022, 29, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Boiko, J.R.; Borghesi, L. Hematopoiesis sculpted by pathogens: Toll-like receptors and inflammatory mediators directly activate stem cells. Cytokine 2012, 57, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Ozato, K. Innate Immune Memory in Hematopoietic Stem/Progenitor Cells: Myeloid-Biased Differentiation and the Role of Interferon. Front. Immunol. 2021, 12, 621333. [Google Scholar] [CrossRef]
- Young, K.; Borikar, S.; Bell, R.; Kuffler, L.; Philip, V.; Trowbridge, J.J. Progressive alterations in multipotent hematopoietic progenitors underlie lymphoid cell loss in aging. J. Exp. Med. 2016, 213, 2259–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montecino-Rodriguez, E.; Dorshkind, K. Use of Busulfan to Condition Mice for Bone Marrow Transplantation. STAR Protoc. 2020, 1, 100159. [Google Scholar] [CrossRef]
- Batey, K.; Kim, J.; Brinster, L.; Gonzalez-Matias, G.; Wu, Z.; Solorzano, S.; Chen, J.; Feng, X.; Young, N.S. Residual effects of busulfan and irradiation on murine hematopoietic stem and progenitor cells. Exp. Hematol. 2022, 105, 22–31. [Google Scholar] [CrossRef]
- DeGregori, J. Aging, inflammation, and HSC. Blood 2020, 136, 153–154. [Google Scholar] [CrossRef]
- Mann, M.; Mehta, A.; de Boer, C.G.; Kowalczyk, M.S.; Lee, K.; Haldeman, P.; Rogel, N.; Knecht, A.R.; Farouq, D.; Regev, A.; et al. Heterogeneous Responses of Hematopoietic Stem Cells to Inflammatory Stimuli Are Altered with Age. Cell Rep. 2018, 25, 2992–3005. [Google Scholar] [CrossRef] [Green Version]
- Kovtonyuk, L.V.; Caiado, F.; Garcia-Martin, S.; Manz, E.M.; Helbling, P.; Takizawa, H.; Boettcher, S.; Al-Shahrour, F.; Nombela-Arrieta, C.; Slack, E.; et al. IL-1 mediates microbiome-induced inflammaging of hematopoietic stem cells in mice. Blood 2022, 139, 44–58. [Google Scholar] [CrossRef]
- Caiado, F.; Kovtonyuk, L.V.; Gonullu, N.G.; Fullin, J.; Boettcher, S.; Manz, M.G. Aging drives Tet2+/− clonal hematopoiesis via IL-1 signaling. Blood 2023, 141, 886–903. [Google Scholar] [CrossRef]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Hormaechea-Agulla, D.; Matatall, K.A.; Le, D.T.; Kain, B.; Long, X.; Kus, P.; Jaksik, R.; Challen, G.A.; Kimmel, M.; King, K.Y. Chronic infection drives Dnmt3a-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell 2021, 28, 1428–1442. [Google Scholar] [CrossRef] [PubMed]
- Hecker, J.S.; Hartmann, L.; Rivière, J.; Buck, M.C.; van der Garde, M.; Rothenberg-Thurley, M.; Fischer, L.; Winter, S.; Ksienzyk, B.; Ziemann, F.; et al. CHIP and hips: Clonal hematopoiesis is common in patients undergoing hip arthroplasty and is associated with autoimmune disease. Blood 2021, 138, 1727–1732. [Google Scholar] [CrossRef]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017, 55, 56–70. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helbling, P.M.; Piñeiro-Yáñez, E.; Gerosa, R.; Boettcher, S.; Al-Shahrour, F.; Manz, M.G.; Nombela-Arrieta, C. Global Transcriptomic Profiling of the Bone Marrow Stromal Microenvironment during Postnatal Development, Aging, and Inflammation. Cell Rep. 2019, 29, 3313–3330. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Xu, P.; Zhang, X.; Liao, M.; Dong, Q.; Cong, T.; Tang, B.; Yang, X.; Ye, M.; Chang, Y.J.; et al. Aging-induced IL27Ra Signaling Impairs Hematopoietic Stem Cells. Blood 2020, 136, 183–198. [Google Scholar] [CrossRef]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Renders, S.; Svendsen, A.F.; Panten, J.; Rama, N.; Maryanovich, M.; Sommerkamp, P.; Ladel, L.; Redavid, A.R.; Gibert, B.; Lazare, S.; et al. Niche derived netrin-1 regulates hematopoietic stem cell dormancy via its receptor neogenin-1. Nat. Commun. 2021, 12, 608. [Google Scholar] [CrossRef]
- Schlegel, M.; Körner, A.; Kaussen, T.; Knausberg, U.; Gerber, C.; Hansmann, G.; Jónasdóttir, H.S.; Giera, M.; Mirakaj, V. Inhibition of neogenin fosters resolution of inflammation and tissue regeneration. J. Clin. Investig. 2018, 128, 4711–4726. [Google Scholar] [CrossRef]
- Frisch, B.J.; Hoffman, C.M.; Latchney, S.E.; LaMere, M.W.; Myers, J.; Ashton, J.; Li, A.J.; Saunders, J., 2nd; Palis, J.; Perkins, A.S.; et al. Aged marrow macrophages expand platelet-biased hematopoietic stem cells via Interleukin1B. JCI Insight 2019, 5, e124213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuribayashi, W.; Oshima, M.; Itokawa, N.; Koide, S.; Nakajima-Takagi, Y.; Yamashita, M.; Yamazaki, S.; Rahmutulla, B.; Miura, F.; Ito, T.; et al. Limited rejuvenation of aged hematopoietic stem cells in young bone marrow niche. J. Exp. Med. 2021, 218, e20192283. [Google Scholar] [CrossRef] [PubMed]
- Spel, L.; Zaffalon, L.; Hou, C.; Nganko, N.; Chapuis, C.; Martinon, F. CDC42 regulates PYRIN inflammasome assembly. Cell Rep. 2022, 41, 111636. [Google Scholar] [CrossRef]
- Guidi, N.; Marka, G.; Sakk, V.; Zheng, Y.; Florian, M.C.; Geiger, H. An aged bone marrow niche restrains rejuvenated hematopoietic stem cells. Stem Cells 2021, 39, 1101–1106. [Google Scholar] [CrossRef]
- Shevyrev, D.; Tereshchenko, V.; Blinova, E.; Knauer, N.; Pashkina, E.; Sizikov, A.; Kozlov, V. Regulatory T Cells Fail to Suppress Fast Homeostatic Proliferation In Vitro. Life 2021, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- Min, B. Spontaneous T Cell Proliferation: A Physiologic Process to Create and Maintain Homeostatic Balance and Diversity of the Immune System. Front. Immunol. 2018, 9, 547. [Google Scholar] [CrossRef]
- Shevyrev, D.; Blinova, E.A.; Kozlov, V.A. The influence of humoral factors of homeostatistic proliferation on t-regulatory cells in vitro. Bull. Sib. Med. 2019, 18, 286–293. [Google Scholar] [CrossRef]
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.Q.; Miao, H. Lymphopenia predicts disease severity of COVID-19: A descriptive and predictive study. Signal. Transduct. Target. Ther. 2020, 5, 33. [Google Scholar] [CrossRef]
- Moxham, V.F.; Karegli, J.; Phillips, R.E.; Brown, K.L.; Tapmeier, T.T.; Hangartner, R.; Sacks, S.H.; Wong, W. Homeostatic proliferation of lymphocytes results in augmented memory-like function and accelerated allograft rejection. J. Immunol. 2008, 180, 3910–3918. [Google Scholar] [CrossRef] [Green Version]
- Milman, N.; Pedersen, A.N.; Ovesen, L.; Schroll, M. Hemoglobin concentrations in 358 apparently healthy 80-year-old Danish men and women. Should the reference interval be adjusted for age? Aging Clin. Exp. Res. 2008, 20, 8–14. [Google Scholar] [CrossRef]
- Vlasschaert, C.; McNaughton, A.J.M.; Chong, M.; Cook, E.K.; Hopman, W.; Kestenbaum, B.; Robinson-Cohen, C.; Garland, J.; Moran, S.M.; Paré, G.; et al. Association of Clonal Hematopoiesis of Indeterminate Potential with Worse Kidney Function and Anemia in Two Cohorts of Patients with Advanced Chronic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 985–995. [Google Scholar] [CrossRef]
- Stauder, R.; Valent, P.; Theurl, I. Anemia at older age: Etiologies, clinical implications, and management. Blood 2018, 131, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Bruserud, Ø.; Vo, A.K.; Rekvam, H. Hematopoiesis, Inflammation and Aging—The Biological Background and Clinical Impact of Anemia and Increased C-Reactive Protein Levels on Elderly Individuals. J. Clin. Med. 2022, 11, 706. [Google Scholar] [CrossRef] [PubMed]
- Girelli, D.; Busti, F. Anemia and adverse outcomes in the elderly: A detrimental inflammatory loop? Haematologica 2019, 104, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P. Does clonal hematopoiesis explain unexplained anemia? Blood 2020, 135, 1080–1082. [Google Scholar] [CrossRef] [Green Version]
- Montenont, E.; Rondina, M.T.; Campbell, R.A. Altered functions of platelets during aging. Curr. Opin. Hematol. 2019, 26, 336–342. [Google Scholar] [CrossRef]
- Karal-Ogly, D.D.; Shumeev, A.N.; Keburiya, V.V.; Mintel, M.V.; Rybtsov, S.A. Age-Related Changes in the Clustering of Blood Populations in Cynomolgus Monkeys Depend on Sex and Immune Status. Life 2023, 13, 316. [Google Scholar] [CrossRef]
- da Silva, P.F.L.; Ogrodnik, M.; Kucheryavenko, O.; Glibert, J.; Miwa, S.; Cameron, K.; Ishaq, A.; Saretzki, G.; Nagaraja-Grellscheid, S.; Nelson, G.; et al. The bystander effect contributes to the accumulation of senescent cells in vivo. Aging Cell 2019, 18, e12848. [Google Scholar] [CrossRef]
- Poscablo, D.M.; Worthington, A.K.; Smith-Berdan, S.; Forsberg, E.C. Megakaryocyte progenitor cell function is enhanced upon aging despite the functional decline of aged hematopoietic stem cells. Stem Cell Rep. 2021, 16, 1598–1613. [Google Scholar] [CrossRef]
- Le Blanc, J.; Lordkipanidzé, M. Platelet Function in Aging. Front. Cardiovasc. Med. 2019, 6, 109. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heringer-Walther, S.; Eckert, K.; Schumacher, S.M.; Uharek, L.; Wulf-Goldenberg, A.; Gembardt, F.; Fichtner, I.; Schultheiss, H.P.; Rodgers, K.; Walther, T. Angiotensin-(1–7) stimulates hematopoietic progenitor cells in vitro and in vivo. Haematologica 2009, 94, 857–860. [Google Scholar] [CrossRef] [Green Version]
- Jokubaitis, V.J.; Sinka, L.; Driessen, R.; Whitty, G.; Haylock, D.N.; Bertoncello, I.; Smith, I.; Péault, B.; Tavian, M.; Simmons, P.J. Angiotensin-converting enzyme (CD143) marks hematopoietic stem cells in human embryonic, fetal, and adult hematopoietic tissues. Blood 2008, 111, 4055–4063. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Kim, J.; Metter, E.J.; Nguyen, H.; Truong, T.; Lustig, A.; Ferrucci, L.; Weng, N.P. Changes in blood lymphocyte numbers with age in vivo and their association with the levels of cytokines/cytokine receptors. Immun. Ageing 2016, 13, 24. [Google Scholar] [CrossRef] [Green Version]
- Britanova, O.V.; Putintseva, E.V.; Shugay, M.; Merzlyak, E.M.; Turchaninova, M.A.; Staroverov, D.B.; Bolotin, D.A.; Lukyanov, S.; Bogdanova, E.A.; Mamedov, I.Z.; et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol. 2014, 192, 2689–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Nguyen, T.; Achour, A.; Ko, A.; Cifello, J.; Ling, C.; Sharma, J.; Hiroi, T.; Zhang, Y.; Chia, C.W.; et al. Longitudinal analysis reveals age-related changes in the T cell receptor repertoire of human T cell subsets. J. Clin. Investig. 2022, 132, e158122. [Google Scholar] [CrossRef]
- Zlotoff, D.A.; Zhang, S.L.; De Obaldia, M.E.; Hess, P.R.; Todd, S.P.; Logan, T.D.; Bhandoola, A. Delivery of progenitors to the thymus limits T-lineage reconstitution after bone marrow transplantation. Blood 2011, 118, 1962–1970. [Google Scholar] [CrossRef]
- Shevyrev, D.; Tereshchenko, V.; Kozlov, V.; Sennikov, S. Phylogeny, Structure, Functions, and Role of AIRE in the Formation of T-Cell Subsets. Cells 2022, 11, 194. [Google Scholar] [CrossRef]
- Lee, Y.N.; Frugoni, F.; Dobbs, K.; Tirosh, I.; Du, L.; Ververs, F.A.; Ru, H.; Ott de Bruin, L.; Adeli, M.; Bleesing, J.H.; et al. Characterization of T and B cell repertoire diversity in patients with RAG deficiency. Sci. Immunol. 2016, 1, eaah6109. [Google Scholar] [CrossRef] [Green Version]
- Shevyrev, D.; Tereshchenko, V.; Kozlov, V. Immune Equilibrium Depends on the Interaction Between Recognition and Presentation Landscapes. Front. Immunol. 2021, 12, 706136. [Google Scholar] [CrossRef] [PubMed]
- Jagger, A.; Shimojima, Y.; Goronzy, J.J.; Weyand, C.M. Regulatory T cells and the immune aging process: A mini-review. Gerontology 2014, 60, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A. Clinical perspectives on the age-related increase of immunosuppressive activity. J. Mol. Med. 2022, 100, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Flores, R.R.; Jang, I.H.; Saathoff, A.; Robbins, P.D. Immune Senescence, Immunosenescence and Aging. Front. Aging 2022, 3, 900028. [Google Scholar] [CrossRef] [PubMed]
- Niroula, A.; Sekar, A.; Murakami, M.A.; Trinder, M.; Agrawal, M.; Wong, W.J.; Bick, A.G.; Uddin, M.M.; Gibson, C.J.; Griffin, G.K.; et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat. Med. 2021, 27, 1921–1927. [Google Scholar] [CrossRef] [PubMed]
- Beer, J.; Crotta, S.; Breithaupt, A.; Ohnemus, A.; Becker, J.; Sachs, B.; Kern, L.; Llorian, M.; Ebert, N.; Labroussaa, F.; et al. Impaired immune response drives age-dependent severity of COVID-19. J. Exp. Med. 2022, 219, e20220621. [Google Scholar] [CrossRef]
- Wong, C.; Goldstein, D.R. Impact of aging on antigen presentation cell function of dendritic cells. Curr. Opin. Immunol. 2013, 25, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Zacca, E.R.; Crespo, M.I.; Acland, R.P.; Roselli, E.; Núñez, N.G.; Maccioni, M.; Maletto, B.A.; Pistoresi-Palencia, M.C.; Morón, G. Aging Impairs the Ability of Conventional Dendritic Cells to Cross-Prime CD8+ T Cells upon Stimulation with a TLR7 Ligand. PLoS ONE 2015, 10, e0140672. [Google Scholar] [CrossRef]
- Goudsmit, J.; van den Biggelaar, A.H.J.; Koudstaal, W.; Hofman, A.; Koff, W.C.; Schenkelberg, T.; Alter, G.; Mina, M.J.; Wu, J.W. Immune age and biological age as determinants of vaccine responsiveness among elderly populations: The Human Immunomics Initiative research program. Eur. J. Epidemiol. 2021, 36, 753–762. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.A.; McGowan, S.J.; et al. An aged immune system drives senescence and ageing of solid organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Li, W. Phagocyte dysfunction, tissue aging and degeneration. Ageing Res. Rev. 2013, 12, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Uhl, B.; Vadlau, Y.; Zuchtriegel, G.; Nekolla, K.; Sharaf, K.; Gaertner, F.; Massberg, S.; Krombach, F.; Reichel, C.A. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood 2016, 128, 2327–2337. [Google Scholar] [CrossRef] [Green Version]
- Pluvinage, J.V.; Haney, M.S.; Smith, B.A.H.; Sun, J.; Iram, T.; Bonanno, L.; Li, L.; Lee, D.P.; Morgens, D.W.; Yang, A.C.; et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 2019, 568, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; Pothof, J.; Vijg, J.; Hoeijmakers, J.H.J. The central role of DNA damage in the ageing process. Nature 2021, 592, 695–703. [Google Scholar] [CrossRef]
- Ubaida-Mohien, C.; Moaddel, R.; Moore, A.Z.; Kuo, P.L.; Faghri, F.; Tharakan, R.; Tanaka, T.; Nalls, M.A.; Ferrucci, L. Proteomics and Epidemiological Models of Human Aging. Front. Physiol. 2021, 12, 674013. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal. Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef]
- Minton, K. Inflammation: Inflammasome-related ageing. Nat. Rev. Immunol. 2017, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Sayed, N.; Huang, Y.; Nguyen, K.; Krejciova-Rajaniemi, Z.; Grawe, A.P.; Gao, T.; Tibshirani, R.; Hastie, T.; Alpert, A.; Cui, L.; et al. An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nat. Aging 2021, 1, 598–615. [Google Scholar] [CrossRef] [PubMed]
- Saul, D.; Kosinsky, R.L.; Atkinson, E.J.; Doolittle, M.L.; Zhang, X.; LeBrasseur, N.K.; Pignolo, R.J.; Robbins, P.D.; Niedernhofer, L.J.; Ikeno, Y.; et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat. Commun. 2022, 13, 4827. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chen, Z.; Shen, W.; Huang, G.; Sedivy, J.M.; Wang, H.; Ju, Z. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: The regulation and intervention. Signal. Transduct. Target. Ther. 2021, 6, 245. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, A.K.; Damgaard, A.; Mackey, A.L.; Schjerling, P.; Magnusson, P.; Olesen, A.T.; Kjaer, M.; Scheele, C. An anti-inflammatory phenotype in visceral adipose tissue of old lean mice, augmented by exercise. Sci. Rep. 2019, 9, 12069. [Google Scholar] [CrossRef] [Green Version]
- Ou, M.Y.; Zhang, H.; Tan, P.C.; Zhou, S.B.; Li, Q.F. Adipose tissue aging: Mechanisms and therapeutic implications. Cell Death Dis. 2022, 13, 300. [Google Scholar] [CrossRef]
- Suzuki, H.; Mikami, T.; Iwahara, N.; Akiyama, Y.; Wanibuchi, M.; Komatsu, K.; Yokoyama, R.; Hirano, T.; Hosoda, R.; Horio, Y.; et al. Aging-associated inflammation and fibrosis in arachnoid membrane. BMC Neurol. 2021, 21, 169. [Google Scholar] [CrossRef]
- Nishiura, H.; Imasaka, M.; Yamanegi, K.; Fujimoto, J.; Ohmuraya, M. Immune Aging and How It Works for Inflammation and Fibrosis. Front. Physiol. 2021, 12, 795508. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shevyrev, D.; Tereshchenko, V.; Berezina, T.N.; Rybtsov, S. Hematopoietic Stem Cells and the Immune System in Development and Aging. Int. J. Mol. Sci. 2023, 24, 5862. https://doi.org/10.3390/ijms24065862
Shevyrev D, Tereshchenko V, Berezina TN, Rybtsov S. Hematopoietic Stem Cells and the Immune System in Development and Aging. International Journal of Molecular Sciences. 2023; 24(6):5862. https://doi.org/10.3390/ijms24065862
Chicago/Turabian StyleShevyrev, Daniil, Valeriy Tereshchenko, Tatiana N. Berezina, and Stanislav Rybtsov. 2023. "Hematopoietic Stem Cells and the Immune System in Development and Aging" International Journal of Molecular Sciences 24, no. 6: 5862. https://doi.org/10.3390/ijms24065862
APA StyleShevyrev, D., Tereshchenko, V., Berezina, T. N., & Rybtsov, S. (2023). Hematopoietic Stem Cells and the Immune System in Development and Aging. International Journal of Molecular Sciences, 24(6), 5862. https://doi.org/10.3390/ijms24065862