

Multiplexed Fluorescence Plate Reader In Situ Protein Expression Assay in Apoptotic HepG2 Cells


 , and
, and
Abstract
1. Introduction
2. Results
2.1. Validation of the In Situ Multiplex Protein Expression Assay
2.1.1. Antibody Dilution Optimization
2.1.2. Cell Number Optimization
2.1.3. Intraassay and Interassay Imprecision Studies
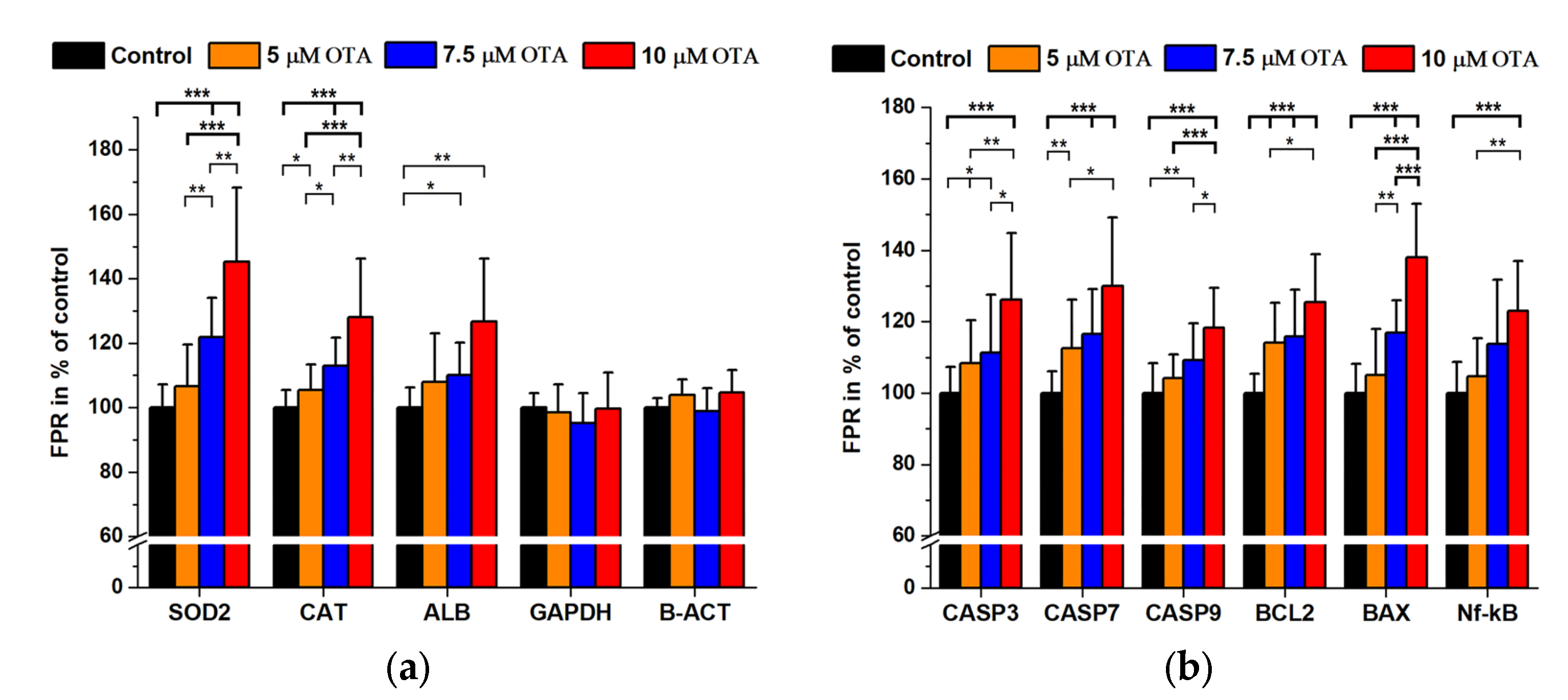
2.2. Ochratoxin A Treatments
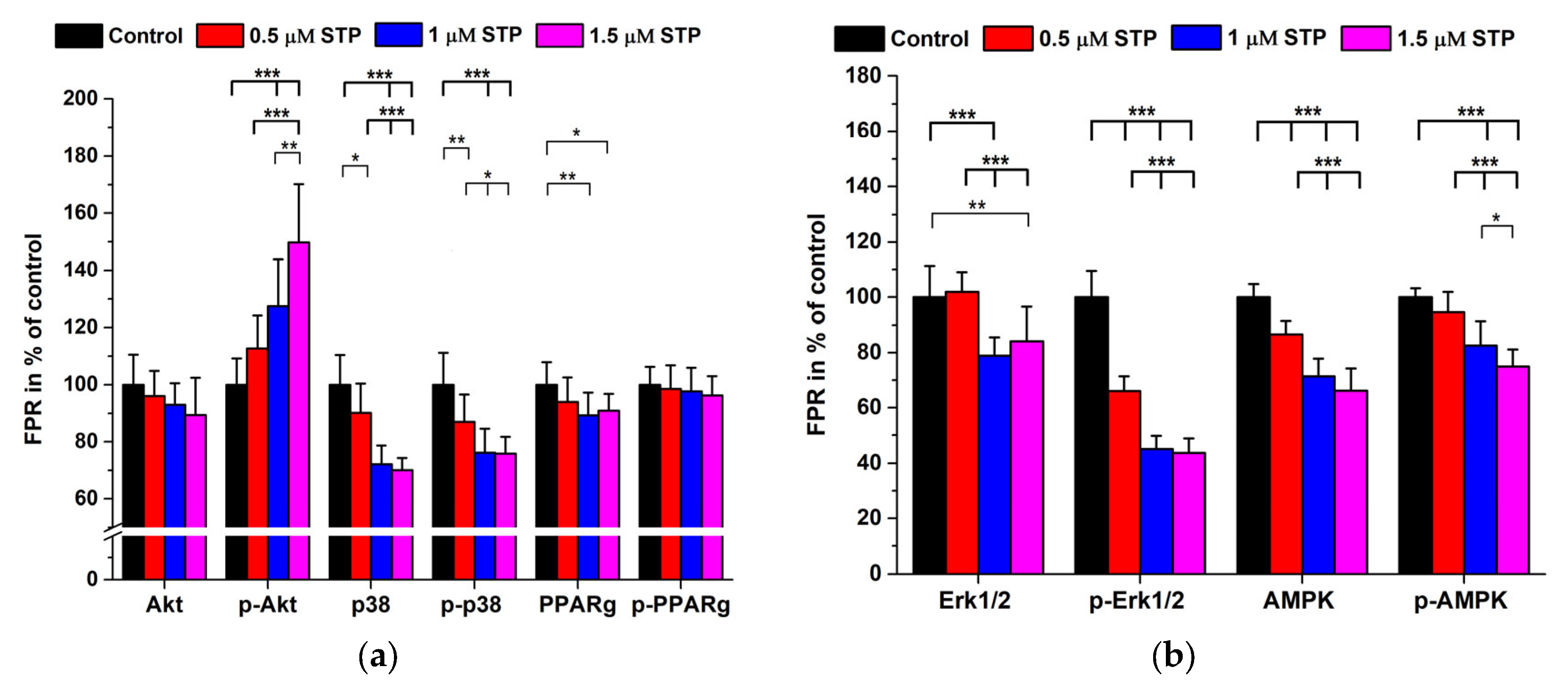
2.3. Staurosporine Treatments
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Chemicals
4.3. Antibodies
4.4. Treatment of HepG2 Cells
4.5. In Situ Antigen Labeling Protocol
- Thorough washing 3 times with Ca, Mg containing PBS.
- Fixing with 150 µL of 3.5% formaldehyde-PBS at RT for 15 min.
- After 3 washing steps permeabilization with 150 µL of 0.1% Triton X 100-PBS at RT for 20 min.
- Blocking with 150 µL of Superblock T20 at 4 °C overnight, tightly capped. Alternatively, blocking could be performed at 37 °C for 1 h with agitation to reduce processing time.
- Removal of blocker by tapping and addition of blocker-diluted antibodies into the wells in 100 µL volume. Incubation for 2 h at 37 °C with 400 rpm in an orbital shaker. The antibody dilutions were 800-fold in most cases except for GAPDH (5000-fold dilution), B-ACT and ALB (2000-fold dilution).
- Washing 5 times with T-TBS (200 µL/well).
- Incubation with secondary antibody 4000-fold diluted with blocker for 1 h at 37 °C with 400 rpm in the orbital shaker (100 µL/well).
- Washing 2 times with T-TBS and once with TBS (200 µL/well).
- Detection of the immune complexes in the emptied wells by incubation for 1 h at RT with 150 µL/well Ampliflu Red/H2O2 reagent (50 mM K-phosphate buffer of pH 7.5 Ampliflu 16 µM, H2O2 10 mM dissolved by 0.1% citric acid, final concentrations). The detection reagent should be prepared before usage and should be kept on ice protected from light.
- Measurement of fluorescence intensities at 540 nm/580 nm excitation/emission settings in a Perkin Elmer EnSpire multimode plate reader (Per-Form Hungaria Ltd. Budapest, Hungary).
4.6. Intracellular Protein Measurement and Data Calculation
4.7. Validation of the Protein Expression Assay
4.7.1. Cell Number Optimization and Imprecision Studies
4.7.2. Statistical Evaluation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Peer, G.; Mestdagh, P.; Vandesompele, J. Accurate RT-qPCR gene expression analysis on cell culture lysates. Sci. Rep. 2012, 2, 222. [Google Scholar] [CrossRef] [PubMed]
- Pillai-Kastoori, L.; Schutz-Geschwender, A.R.; Harford, J.A. A systematic approach to quantitative Western blot analysis. Anal. Biochem. 2020, 593, 113608. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Naguro, I.; Herr, A.E. In Situ Single-Cell Western Blot on Adherent Cell Culture. Angew Chem. Int. Ed. Engl. 2019, 58, 13929–13934. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J.; Spelke, D.P.; Xu, Z.; Kang, C.C.; Schaffer, D.V.; Herr, A.E. Single-cell western blotting. Nat. Methods 2014, 11, 749–755. [Google Scholar] [CrossRef]
- Buccitelli, C.; Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Desire, C.T.; Arrua, R.D.; Strudwick, X.L.; Kopecki, Z.; Cowin, A.J.; Hilder, E.F. The development of microfluidic-based western blotting: Technical advances and future perspectives. J. Chromatogr. A 2023, 1691, 463813. [Google Scholar] [CrossRef]
- Juncker, D.; Bergeron, S.; Laforte, V.; Li, H. Cross-reactivity in antibody microarrays and multiplexed sandwich assays: Shedding light on the dark side of multiplexing. Curr. Opin. Chem. Biol. 2014, 18, 29–37. [Google Scholar] [CrossRef]
- Bolton, J.S.; Chaudhury, S.; Dutta, S.; Gregory, S.; Locke, E.; Pierson, T.; Bergmann-Leitner, E.S. Comparison of ELISA with electro-chemiluminescence technology for the qualitative and quantitative assessment of serological responses to vaccination. Malar. J. 2020, 19, 159. [Google Scholar] [CrossRef]
- Burnette, W.N. "Western blotting": Electrophoretic transfer of proteins from sodium dodecyl sulfate—Polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 1981, 112, 195–203. [Google Scholar] [CrossRef]
- Jungblut, P.; Eckerskorn, C.; Lottspeich, F.; Klose, J. Blotting efficiency investigated by using two-dimensional electrophoresis, hydrophobic membranes and proteins from different sources. Electrophoresis 1990, 11, 581–588. [Google Scholar] [CrossRef]
- He, M.; Herr, A.E. Automated microfluidic protein immunoblotting. Nat. Protoc. 2010, 5, 1844–1856. [Google Scholar] [CrossRef]
- Vallejo-Illarramendi, A.; Marciano, D.K.; Reichardt, L.F. A novel method that improves sensitivity of protein detection in PAGE and Western blot. Electrophoresis 2013, 34, 1148–1150. [Google Scholar] [CrossRef]
- Jin, S.; Furtaw, M.D.; Chen, H.; Lamb, D.T.; Ferguson, S.A.; Arvin, N.E.; Dawod, M.; Kennedy, R.T. Multiplexed Western Blotting Using Microchip Electrophoresis. Anal. Chem. 2016, 88, 6703–6710. [Google Scholar] [CrossRef]
- Sanders, B.J.; Kim, D.C.; Dunn, R.C. Recent Advances in Microscale Western Blotting. Anal. Methods Adv. Methods Appl. 2016, 8, 7002–7013. [Google Scholar] [CrossRef]
- Kielkopf, C.L.; Bauer, W.; Urbatsch, I.L. Analysis of Proteins by Immunoblotting. Cold Spring Harb. Protoc. 2021, 2021, 512–520. [Google Scholar] [CrossRef]
- Edouard, S.; Jaafar, R.; Orain, N.; Parola, P.; Colson, P.; La Scola, B.; Fournier, P.E.; Raoult, D.; Drancourt, M. Automated Western immunoblotting detection of anti-SARS-CoV-2 serum antibodies. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 1309–1317. [Google Scholar] [CrossRef]
- Mahmood, T.; Yang, P.C. Western blot: Technique, theory, and trouble shooting. North Am. J. Med. Sci. 2012, 4, 429–434. [Google Scholar]
- Poulsen, T.B.G.; Karamehmedovic, A.; Aboo, C.; Jorgensen, M.M.; Yu, X.; Fang, X.; Blackburn, J.M.; Nielsen, C.H.; Kragstrup, T.W.; Stensballe, A. Protein array-based companion diagnostics in precision medicine. Expert Rev. Mol. Diagn. 2020, 20, 1183–1198. [Google Scholar] [CrossRef] [PubMed]
- Mountjoy, K.G. ELISA versus LUMINEX assay for measuring mouse metabolic hormones and cytokines: Sharing the lessons I have learned. J. Immunoass. Immunochem. 2021, 42, 154–173. [Google Scholar] [CrossRef]
- Telford, W.G. Multiparametric Analysis of Apoptosis by Flow Cytometry. Methods Mol. Biol. 2018, 1678, 167–202. [Google Scholar] [PubMed]
- Luckert, C.; Schulz, C.; Lehmann, N.; Thomas, M.; Hofmann, U.; Hammad, S.; Hengstler, J.G.; Braeuning, A.; Lampen, A.; Hessel, S. Comparative analysis of 3D culture methods on human HepG2 cells. Arch. Toxicol. 2017, 91, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Koszegi, T.; Poor, M. Ochratoxin A: Molecular Interactions, Mechanisms of Toxicity and Prevention at the Molecular Level. Toxins 2016, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Perez, E.; Ryu, D.; Lee, C.; Lee, H.J. Ochratoxin A Induces Oxidative Stress in HepG2 Cells by Impairing the Gene Expression of Antioxidant Enzymes. Toxins 2021, 13, 271. [Google Scholar] [CrossRef]
- Sali, N.; Nagy, S.; Poor, M.; Koszegi, T. Multiparametric luminescent cell viability assay in toxicology models: A critical evaluation. J. Pharm. Toxicol. Methods 2016, 79, 45–54. [Google Scholar] [CrossRef]
- Kupski, L.; Freitas, M.; Ribeiro, D.; Furlong, E.B.; Fernandes, E. Ochratoxin A activates neutrophils and kills these cells through necrosis, an effect eliminated through its conversion into ochratoxin alpha. Toxicology 2016, 368–369, 91–102. [Google Scholar] [CrossRef]
- Vettorazzi, A.; van Delft, J.; Lopez de Cerain, A. A review on ochratoxin A transcriptomic studies. Food Chem. Toxicol. 2013, 59, 766–783. [Google Scholar] [CrossRef]
- Yang, Q.; He, X.; Li, X.; Xu, W.; Luo, Y.; Yang, X.; Wang, Y.; Li, Y.; Huang, K. DNA damage and S phase arrest induced by Ochratoxin A in human embryonic kidney cells (HEK 293). Mutat. Res. 2014, 765, 22–31. [Google Scholar] [CrossRef]
- Giuliano, M.; Bellavia, G.; Lauricella, M.; D’Anneo, A.; Vassallo, B.; Vento, R.; Tesoriere, G. Staurosporine-induced apoptosis in Chang liver cells is associated with down-regulation of Bcl-2 and Bcl-XL. Int. J. Mol. Med. 2004, 13, 565–571. [Google Scholar] [CrossRef]
- Malsy, M.; Bitzinger, D.; Graf, B.; Bundscherer, A. Staurosporine induces apoptosis in pancreatic carcinoma cells PaTu 8988t and Panc-1 via the intrinsic signaling pathway. Eur. J. Med. Res. 2019, 24, 5. [Google Scholar] [CrossRef]
- Nakano, H.; Omura, S. Chemical biology of natural indolocarbazole products: 30 years since the discovery of staurosporine. J. Antibiot. 2009, 62, 17–26. [Google Scholar] [CrossRef]
- Bilbrough, T.; Piemontese, E.; Seitz, O. Dissecting the role of protein phosphorylation: A chemical biology toolbox. Chem. Soc. Rev. 2022, 51, 5691–5730. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Manns, J.; Daubrawa, M.; Driessen, S.; Paasch, F.; Hoffmann, N.; Loffler, A.; Lauber, K.; Dieterle, A.; Alers, S.; Iftner, T.; et al. Triggering of a novel intrinsic apoptosis pathway by the kinase inhibitor staurosporine: Activation of caspase-9 in the absence of Apaf-1. FASEB J. 2011, 25, 3250–3261. [Google Scholar] [CrossRef]
- Ahlemeyer, B.; Klumpp, S.; Krieglstein, J. Release of cytochrome c into the extracellular space contributes to neuronal apoptosis induced by staurosporine. Brain Res. 2002, 934, 107–116. [Google Scholar] [CrossRef]
- Il’ichev, Y.V.; Perry, J.L.; Ruker, F.; Dockal, M.; Simon, J.D. Interaction of ochratoxin A with human serum albumin. Binding sites localized by competitive interactions with the native protein and its recombinant fragments. Chem. Biol. Interact. 2002, 141, 275–293. [Google Scholar] [CrossRef]
- Faisal, Z.; Derdak, D.; Lemli, B.; Kunsagi-Mate, S.; Balint, M.; Hetenyi, C.; Csepregi, R.; Koszegi, T.; Sueck, F.; Cramer, B.; et al. Interaction of 2’R-ochratoxin A with Serum Albumins: Binding Site, Effects of Site Markers, Thermodynamics, Species Differences of Albumin-binding, and Influence of Albumin on Its Toxicity in MDCK Cells. Toxins 2018, 10, 353. [Google Scholar] [CrossRef]
- Jung, K.Y.; Takeda, M.; Kim, D.K.; Tojo, A.; Narikawa, S.; Yoo, B.S.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of ochratoxin A transport by human organic anion transporters. Life Sci. 2001, 69, 2123–2135. [Google Scholar] [CrossRef]
- Anzai, N.; Jutabha, P.; Endou, H. Molecular mechanism of ochratoxin a transport in the kidney. Toxins 2010, 2, 1381–1398. [Google Scholar] [CrossRef]
- Erceg, S.; Mateo, E.M.; Zipancic, I.; Rodriguez Jimenez, F.J.; Perez Arago, M.A.; Jimenez, M.; Soria, J.M.; Garcia-Esparza, M.A. Assessment of Toxic Effects of Ochratoxin A in Human Embryonic Stem Cells. Toxins 2019, 11, 217. [Google Scholar] [CrossRef]
- Yang, X.; Gao, Y.; Yan, Q.; Bao, X.; Zhao, S.; Wang, J.; Zheng, N. Transcriptome Analysis of Ochratoxin A-Induced Apoptosis in Differentiated Caco-2 Cells. Toxins 2019, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Banjara, S.; Suraweera, C.D.; Hinds, M.G.; Kvansakul, M. The Bcl-2 Family: Ancient Origins, Conserved Structures, and Divergent Mechanisms. Biomolecules 2020, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Hou, L.; Gan, F.; Zhou, X.; Zhou, Y.; Qian, G.; Liu, Z.; Huang, K. Immunotoxicity of ochratoxin A and aflatoxin B1 in combination is associated with the nuclear factor kappa B signaling pathway in 3D4/21 cells. Chemosphere 2018, 199, 718–727. [Google Scholar] [CrossRef]
- Taverna, M.; Marie, A.L.; Mira, J.P.; Guidet, B. Specific antioxidant properties of human serum albumin. Ann. Intensive Care 2013, 3, 4. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Ozcan, Z.; Gul, G.; Yaman, I. Ochratoxin A activates opposing c-MET/PI3K/Akt and MAPK/ERK 1-2 pathways in human proximal tubule HK-2 cells. Arch. Toxicol. 2015, 89, 1313–1327. [Google Scholar] [CrossRef]
- Lawrie, A.M.; Noble, M.E.; Tunnah, P.; Brown, N.R.; Johnson, L.N.; Endicott, J.A. Protein kinase inhibition by staurosporine revealed in details of the molecular interaction with CDK2. Nat. Struct. Biol. 1997, 4, 796–801. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Gani, O.A.; Engh, R.A. Protein kinase inhibition of clinically important staurosporine analogues. Nat. Prod. Rep. 2010, 27, 489–498. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | 2nd AB Only (Signal Mean ± SD) in Cps | Immune Complex (Signal Mean ± SD) in Cps | 1st/2nd AB Dilutions | Background Signal in % |
|---|---|---|---|---|
| SOD2 | 482.0 ± 47.6 | 13,103.4 ± 1082.9 | 800/4000 | 3.7 |
| CAT | 587.0 ± 47.1 | 8403.1 ± 181.3 | 800/4000 | 7.0 |
| Casp3 | 426.0 ± 40.1 | 11,738.1 ± 572.0 | 800/4000 | 3.6 |
| Casp7 | 473.0 ± 45.4 | 11,033.3 ± 608.0 | 800/4000 | 4.3 |
| Casp9 | 288.0 ± 22.7 | 13,338.1 ± 901.0 | 800/4000 | 2.2 |
| BCL2 | 357.7 ± 39.9 | 4031.2 ± 91.4 | 800/4000 | 8.9 |
| BAX | 487.0 ± 62.6 | 7535.6 ± 421.0 | 800/4000 | 6.5 |
| Nf-kB | 258.3 ± 31.2 | 2296.2 ± 219.9 | 800/4000 | 11.2 |
| GAPDH | 443.5 ± 16.1 | 11,578.4 ± 668.4 | 5000/4000 | 3.8 |
| ALB | 321.5 ± 30.7 | 12,096.5 ± 772.1 | 2000/4000 | 2.7 |
| B-ACT | 297.8 ± 13.6 | 26,604.0 ± 1024.3 | 2000/4000 | 1.1 |
| Akt | 358.0 ± 47.6 | 2325.1 ± 52.5 | 800/4000 | 15.4 |
| p-Akt | 154.4 ± 10.7 | 767.4 ± 22.0 | 800/4000 | 20.1 |
| p38 | 299.7 ± 33.6 | 11,710.3 ± 767.7 | 800/4000 | 2.6 |
| p-p38 | 216.1 ± 12.2 | 7477.2 ± 271.0 | 800/4000 | 2.9 |
| PPARg | 194.2 ± 18.7 | 4351.9 ± 225.0 | 800/4000 | 4.5 |
| p-PPARg | 384.0 ± 68.8 | 21,361.2 ± 2108.0 | 800/4000 | 1.8 |
| Erk 1/2 | 237.9 ± 29.4 | 3077.8 ± 37.3 | 800/4000 | 7.7 |
| p-Erk 1/2 | 409.0 ± 50.7 | 9374.8 ± 699.4 | 800/4000 | 4.4 |
| AMPK | 406.5 ± 31.6 | 10,232.1 ± 452.5 | 800/4000 | 4.0 |
| p-AMPK | 289.0 ± 62.8 | 7343.8 ± 436.9 | 800/4000 | 3.9 |
| Intraassay Imprecision | FPR |
|---|---|
| GAPDH (N = 84) | 100 ± 9.08 (mean ± SD) |
| Interassay Imprecision by Independent Measurements | |
| GAPDH control (N = 8 × 4) | 100 ± 5.94 (mean ± SD) |
| GAPDH OTA treated (N = 6 × 4) | 101.1 ± 7.71 (mean ± SD) |
| B-ACT (N = 4 × 4) | 100 ± 4.74 (mean ± SD) |
| B-ACT OTA treated (N = 4 × 4) | 103 ± 4.84 (mean ± SD) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakabfi-Csepregi, R.; Kovács, G.L.; Kaltenecker, P.; Kőszegi, T. Multiplexed Fluorescence Plate Reader In Situ Protein Expression Assay in Apoptotic HepG2 Cells. Int. J. Mol. Sci. 2023, 24, 6564. https://doi.org/10.3390/ijms24076564
Jakabfi-Csepregi R, Kovács GL, Kaltenecker P, Kőszegi T. Multiplexed Fluorescence Plate Reader In Situ Protein Expression Assay in Apoptotic HepG2 Cells. International Journal of Molecular Sciences. 2023; 24(7):6564. https://doi.org/10.3390/ijms24076564
Chicago/Turabian StyleJakabfi-Csepregi, Rita, Gábor L. Kovács, Péter Kaltenecker, and Tamás Kőszegi. 2023. "Multiplexed Fluorescence Plate Reader In Situ Protein Expression Assay in Apoptotic HepG2 Cells" International Journal of Molecular Sciences 24, no. 7: 6564. https://doi.org/10.3390/ijms24076564
APA StyleJakabfi-Csepregi, R., Kovács, G. L., Kaltenecker, P., & Kőszegi, T. (2023). Multiplexed Fluorescence Plate Reader In Situ Protein Expression Assay in Apoptotic HepG2 Cells. International Journal of Molecular Sciences, 24(7), 6564. https://doi.org/10.3390/ijms24076564