
A Lipidated Single-B-Chain Derivative of Relaxin Exhibits Improved In Vitro Serum Stability without Altering Activity

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
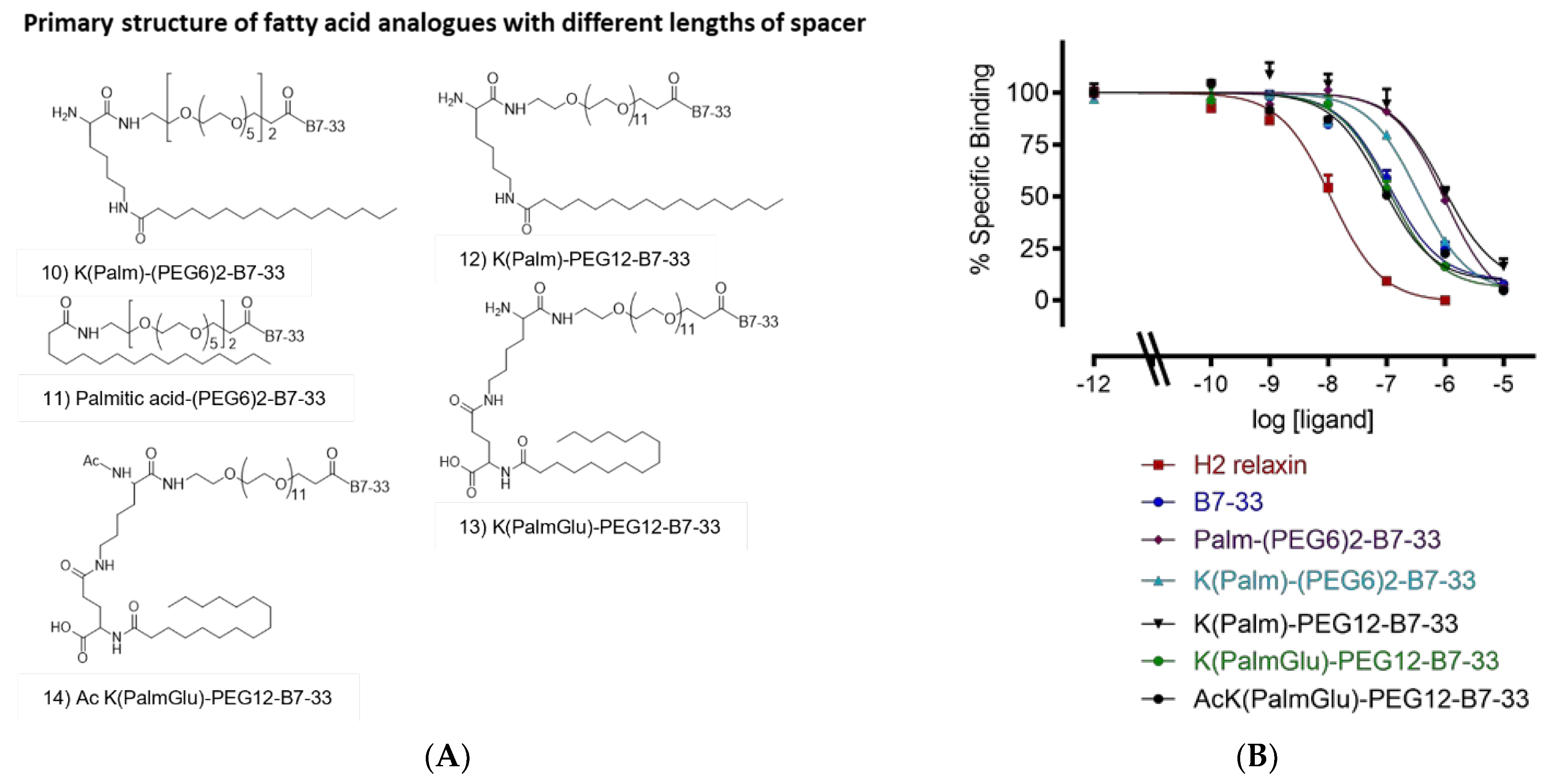
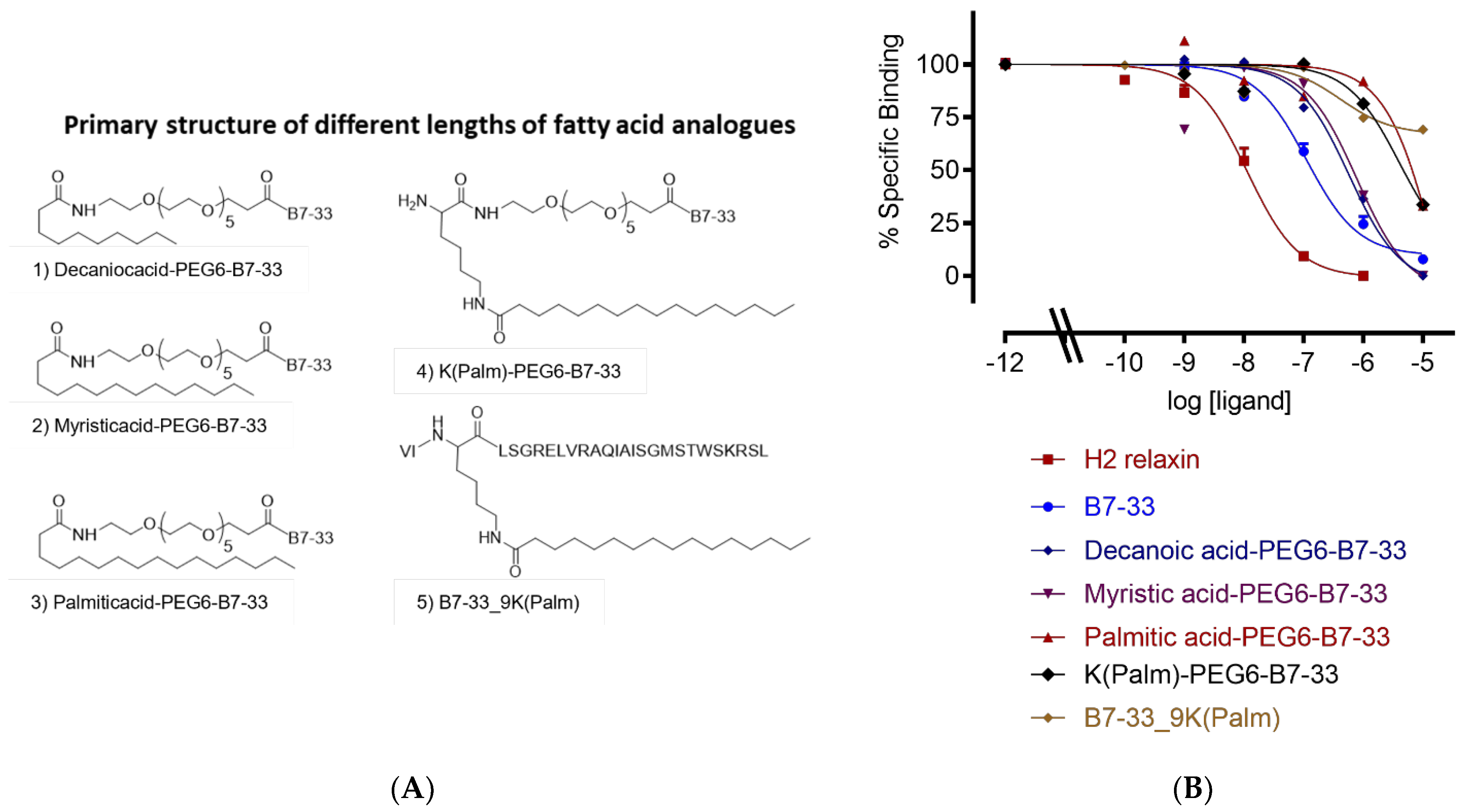
2.1. Different Lengths of Fatty Acids
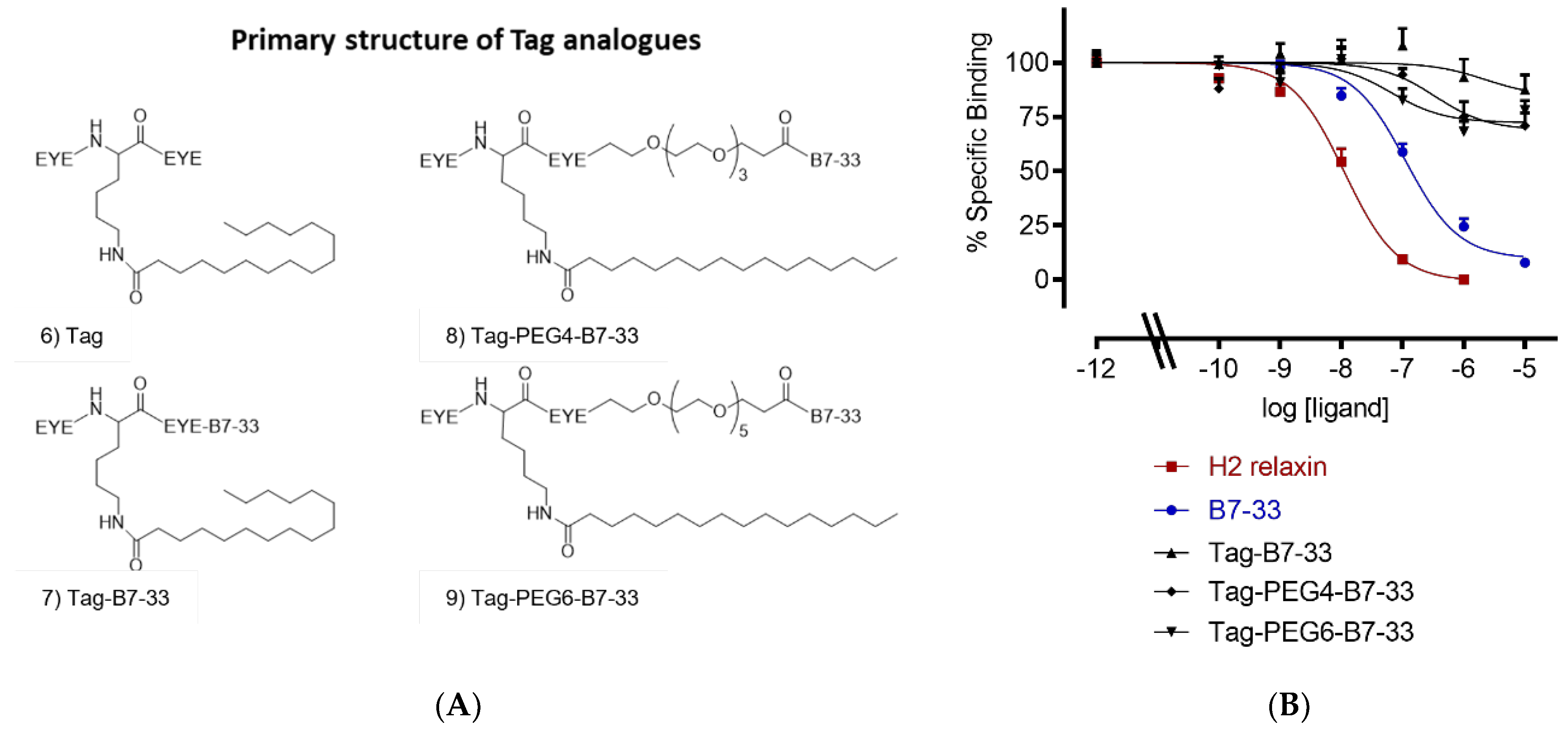
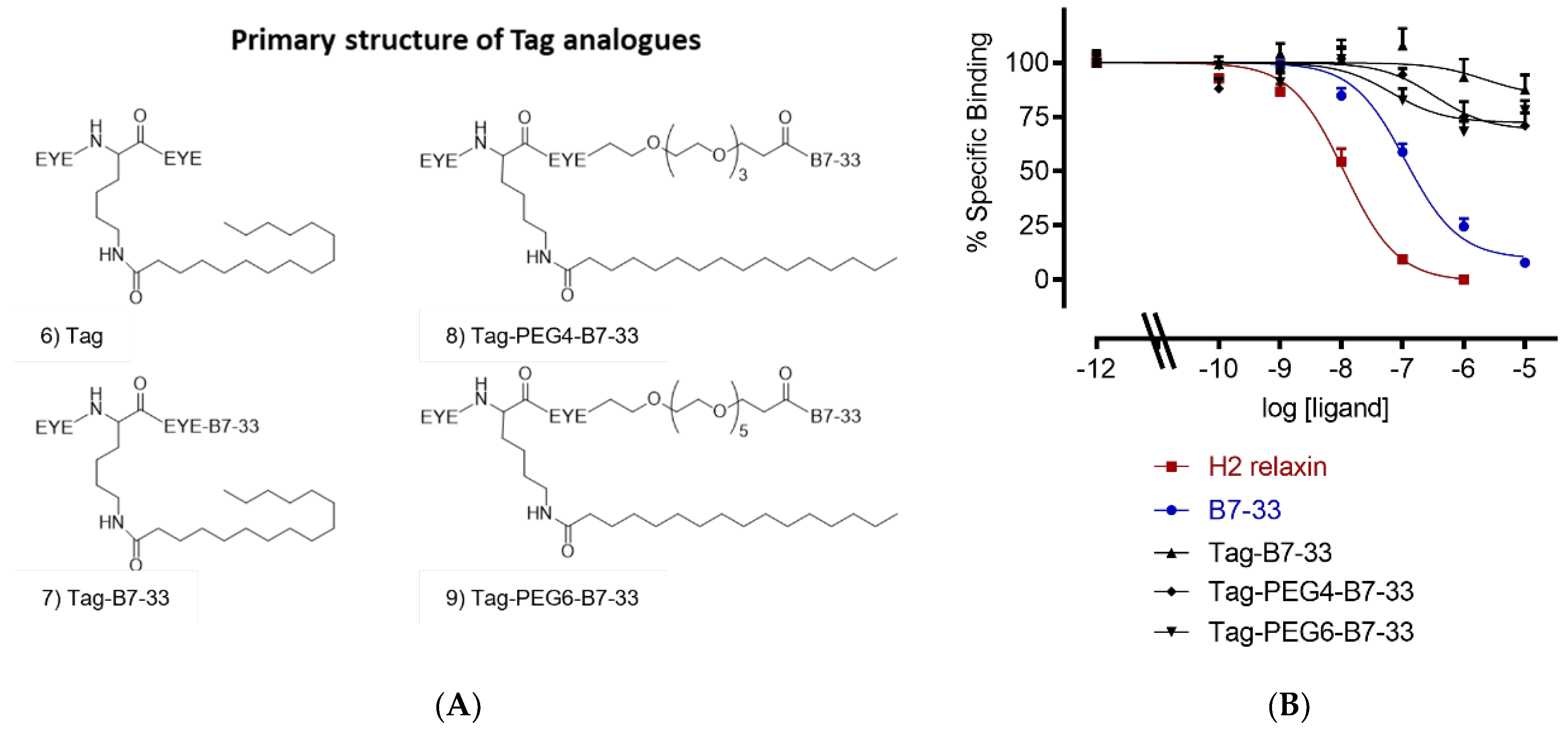
2.2. Solubilizing Tag
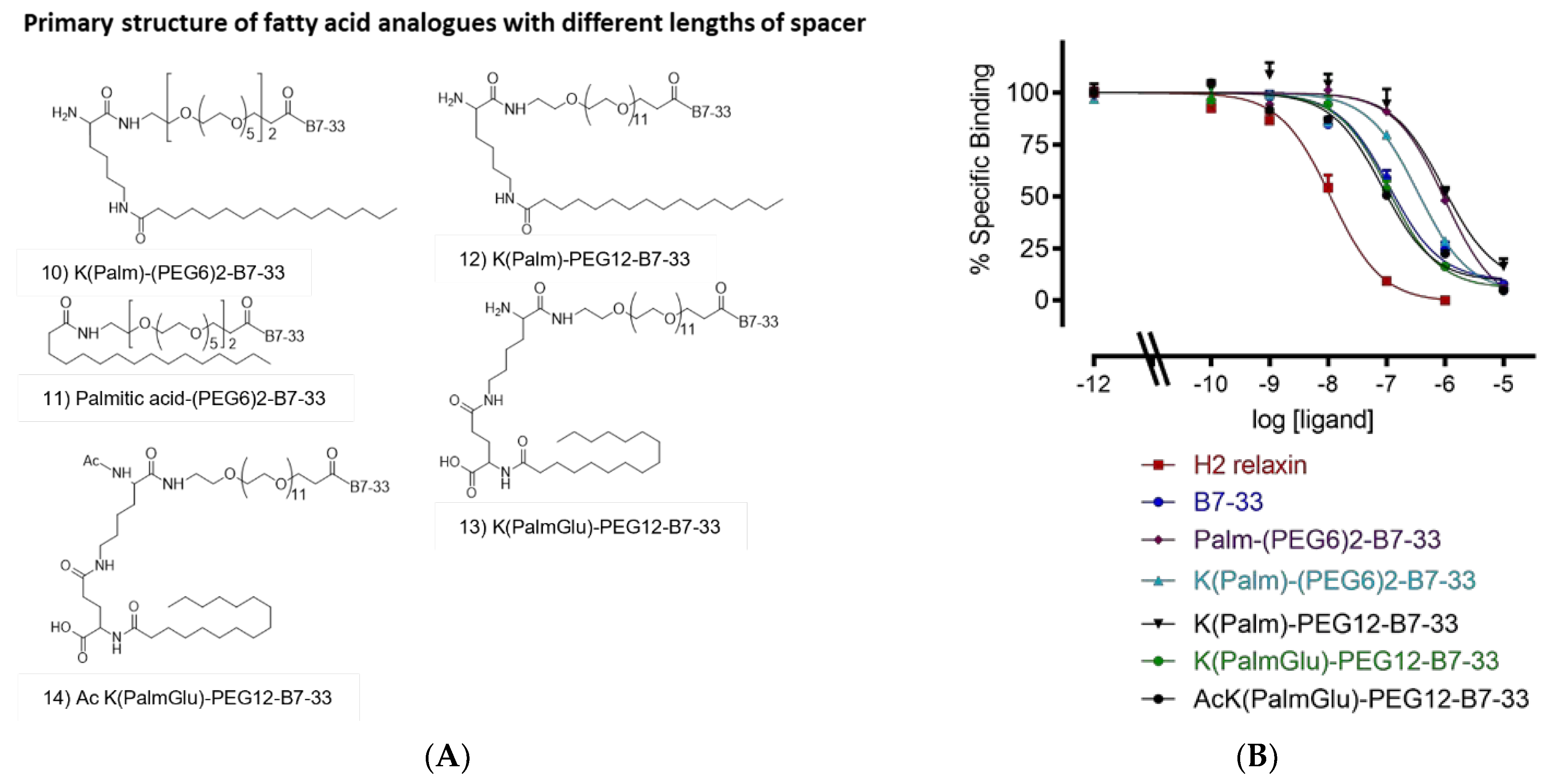
2.3. Different Lengths of Spacer
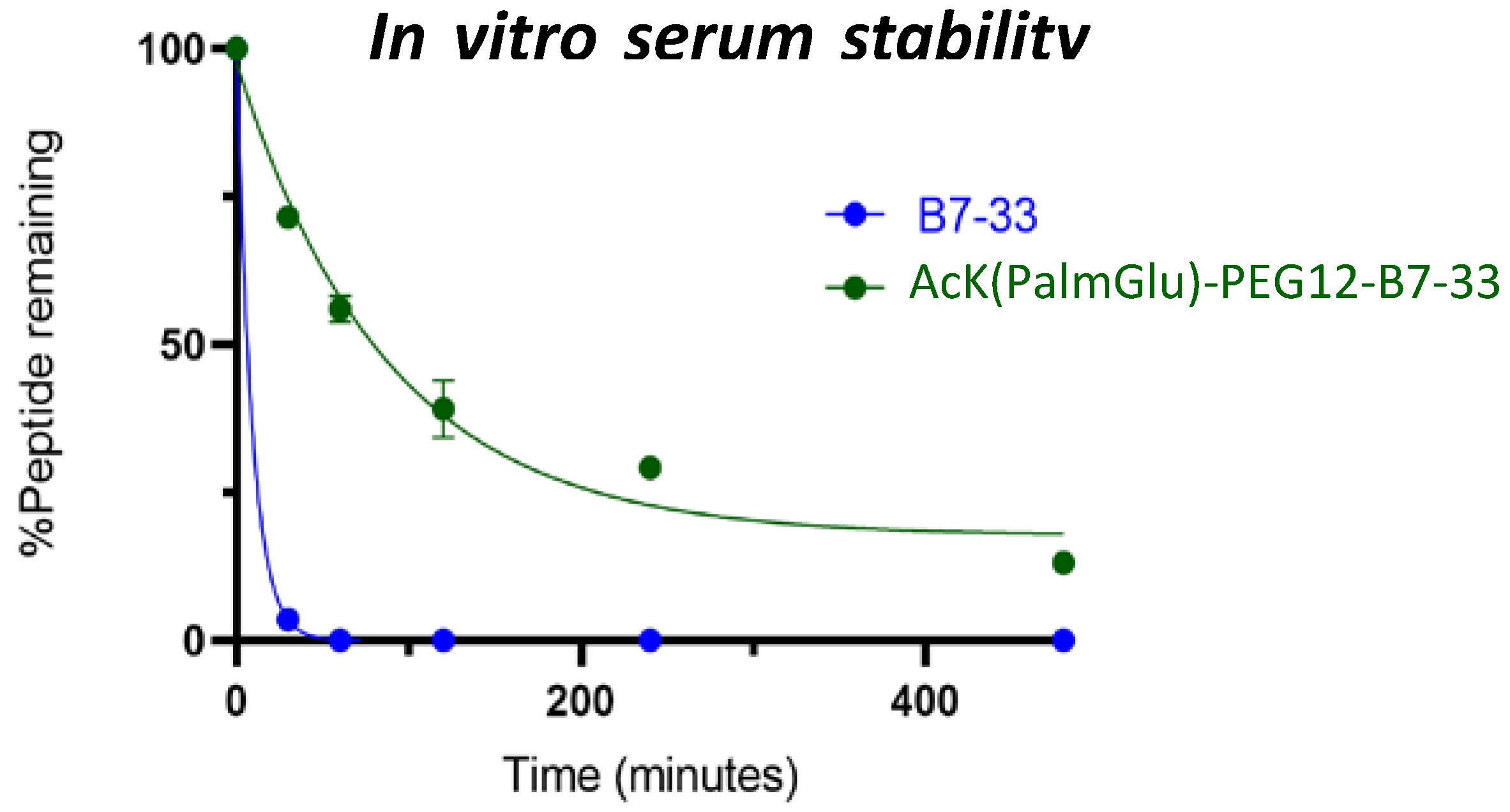
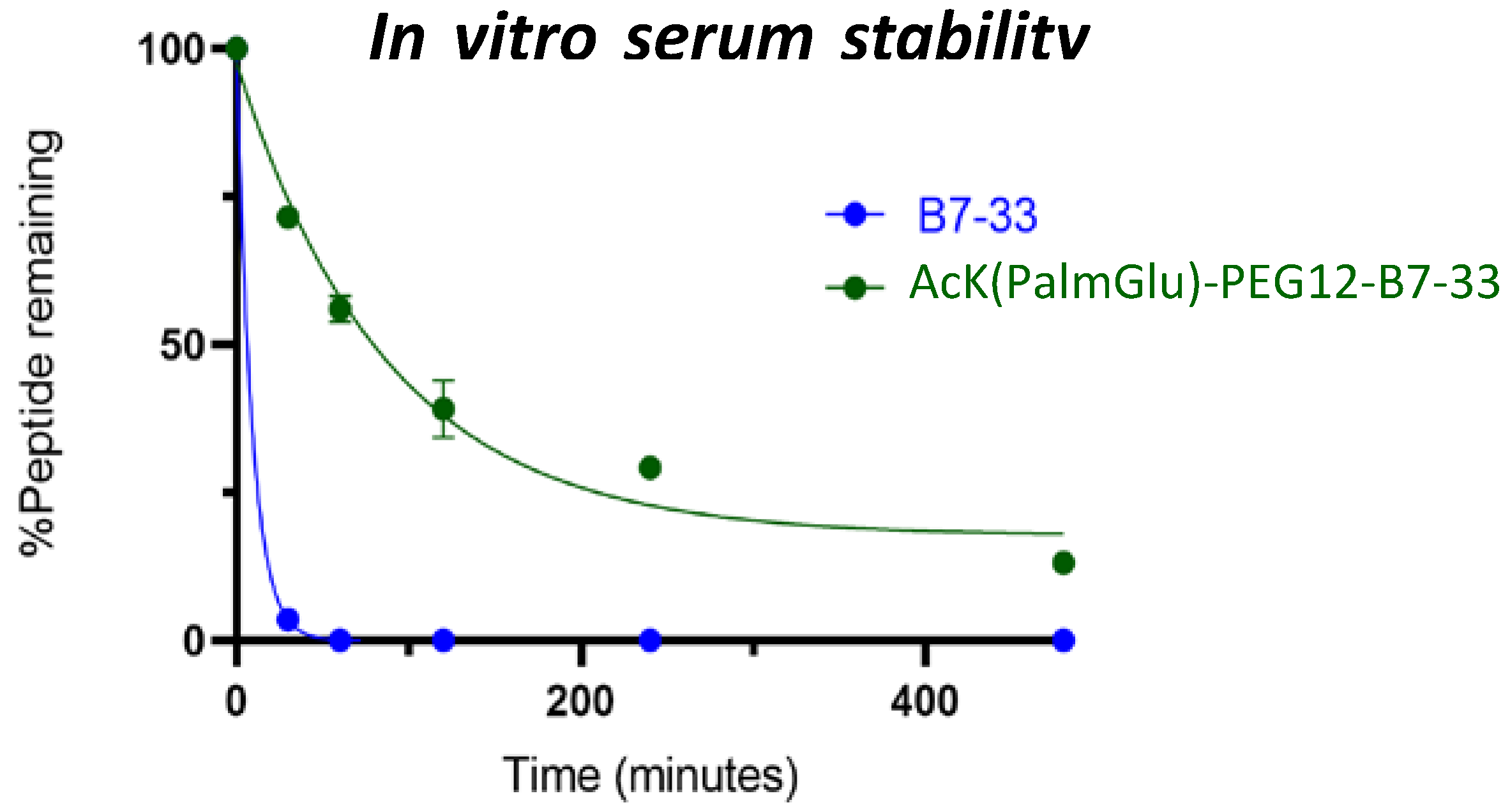
2.4. In Vitro Serum Stability of AcK(PalmGlu)-PEG12-B7-33
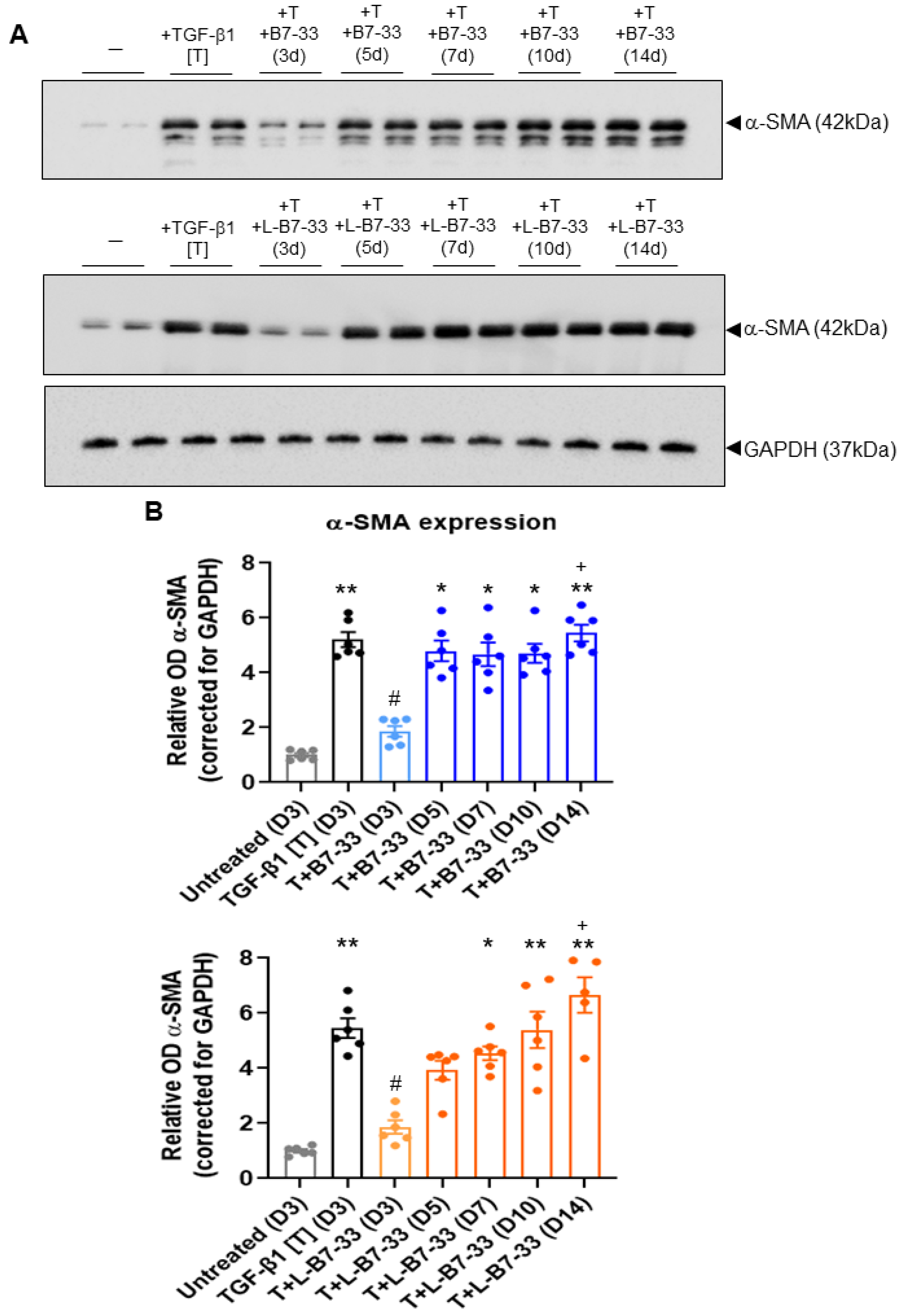
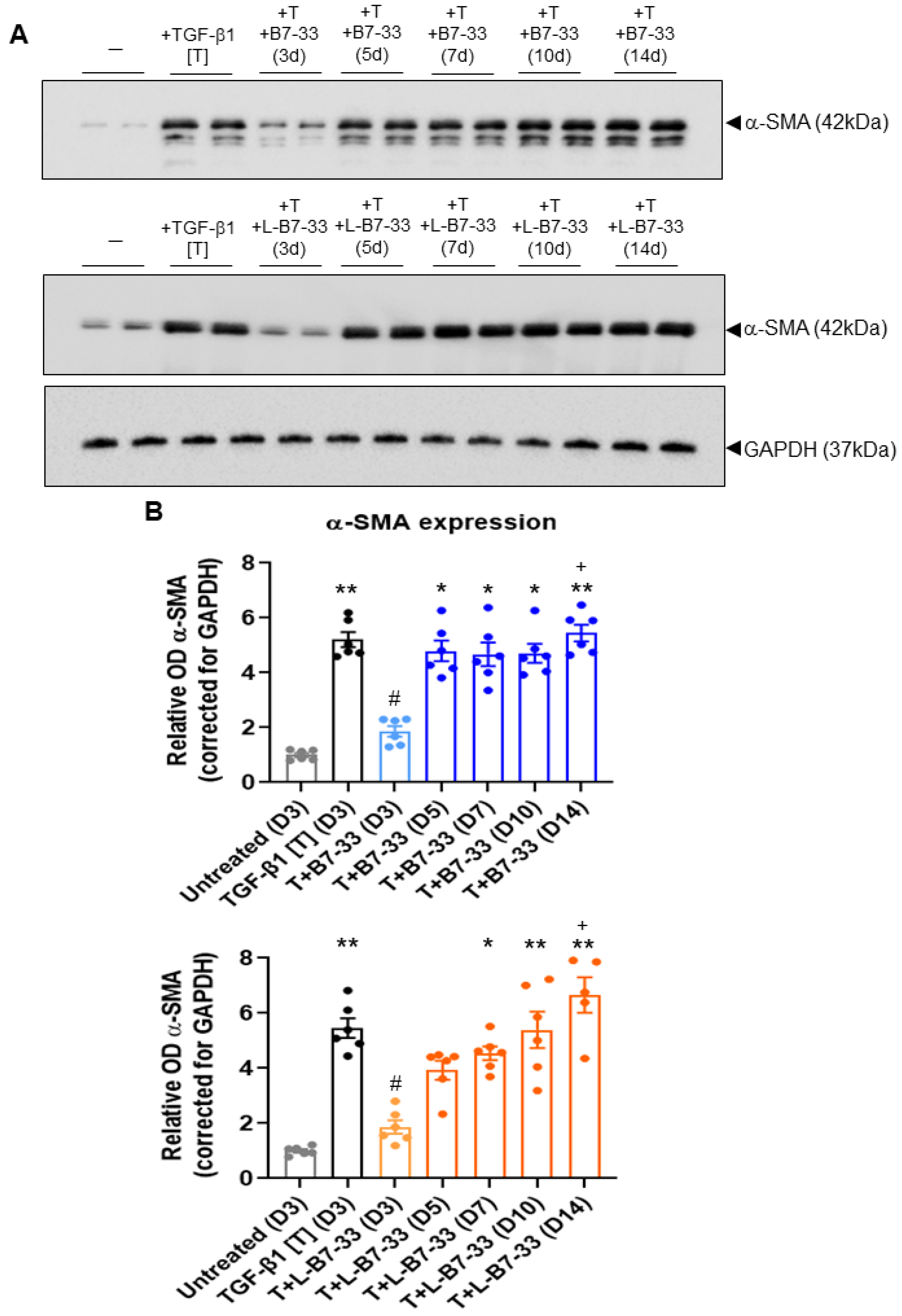
2.5. The Effects of Lipdated B7-33 (L-B7-33), AcK(PalmGlu)-PEG12-B7-33 on Myofibroblast Differentiation In Vitro
3. Materials and Methods
3.1. Materials
3.2. Solid-Phase Peptide Synthesis
3.3. Peptide Cleavage from Solid Support
3.4. Peptide Purification and Characterization
3.5. Amino Acid Analysis
3.6. HEK-7BP Binding Assays
3.7. In Vitro Serum Stability Assay
3.8. Determination of the Effects of Lipidated (L)-B7-33 on Myofibroblast Differentiation In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hisaw, F.L. Experimental relaxation of the pubic ligament of the guinea pig. Proc. Soc. Exp. Biol. Med. 1926, 23, 661–663. [Google Scholar] [CrossRef]
- Samuel, C.S.; Cendrawan, S.; Gao, X.M.; Ming, Z.; Zhao, C.; Kiriazis, H.; Xu, Q.; Tregear, G.W.; Bathgate, R.A.; Du, X.J. Relaxin remodels fibrotic healing following myocardial infarction. Lab. Investig. 2011, 91, 675–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dschietzig, T.B. Relaxin-2 for heart failure with preserved ejection fraction (HFpEF): Rationale for future clinical trials. Mol. Cell Endocrinol. 2019, 487, 54–58. [Google Scholar] [CrossRef]
- Samuel, C.S.; Bennett, R.G. Relaxin as an anti-fibrotic treatment: Perspectives, challenges and future directions. Biochem. Pharmacol. 2022, 197, 114884. [Google Scholar] [CrossRef]
- Hsu, S.Y.; Nakabayashi, K.; Nishi, S.; Kumagai, J.; Kudo, M.; Sherwood, O.D.; Hsueh, A.J. Activation of orphan receptors by the hormone relaxin. Science 2002, 295, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, J.; Hsu, S.Y.; Matsumi, H.; Roh, J.S.; Fu, P.; Wade, J.D.; Bathgate, R.A.; Hsueh, A.J. INSL3/Leydig insulin-like peptide activates the LGR8 receptor important in testis descent. J. Biol. Chem. 2002, 277, 31283–31286. [Google Scholar] [CrossRef] [Green Version]
- Dschietzig, T.; Bartsch, C.; Stangl, V.; Baumann, G.; Stangl, K. Identification of the pregnancy hormone relaxin as glucocorticoid receptor agonist. Faseb J. 2004, 18, 1536–1538. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Bruell, S.; Patil, N.; Hossain, M.A.; Scott, D.J.; Petrie, E.J.; Bathgate, R.A.D.; Gooley, P.R. The complex binding mode of the peptide hormone H2 relaxin to its receptor RXFP1. Nat. Commun. 2016, 7, 11344. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.A.; Kocan, M.; Yao, S.T.; Royce, S.G.; Nair, V.B.; Siwek, C.; Patil, N.A.; Harrison, I.P.; Rosengren, K.J.; Selemidis, S.; et al. A single-chain derivative of the relaxin hormone is a functionally selective agonist of the G protein-coupled receptor, RXFP1. Chem. Sci. 2016, 7, 3805–3819. [Google Scholar] [CrossRef] [Green Version]
- Devarakonda, T.; Mauro, A.G.; Guzman, G.; Hovsepian, S.; Cain, C.; Das, A.; Praveen, P.; Hossain, M.A.; Salloum, F.N. B7-33, a functionally selective relaxin receptor 1 agonist, attenuates myocardial infarction-related adverse cardiac remodeling in mice. J. Am. Heart Assoc. 2020, 9, e015748. [Google Scholar] [CrossRef]
- Bhuiyan, S.; Shen, M.; Chelvaretnam, S.; Tan, A.Y.; Ho, G.; Hossain, M.A.; Widdop, R.E.; Samuel, C.S. Assessment of renal fibrosis and anti-fibrotic agents using a novel diagnostic and stain-free second-harmonic generation platform. Faseb J. 2021, 35, e21595. [Google Scholar] [CrossRef]
- Alam, F.; Gaspari, T.A.; Kemp-Harper, B.K.; Low, E.; Aw, A.; Ferens, D.; Spizzo, I.; Jefferis, A.M.; Praveen, P.; Widdop, R.E.; et al. The single-chain relaxin mimetic, B7-33, maintains the cardioprotective effects of relaxin and more rapidly reduces left ventricular fibrosis compared to perindopril in an experimental model of cardiomyopathy. Biomed. Pharmacother. 2023, 160, 114370. [Google Scholar] [CrossRef]
- Bathgate, R.A.D.; Kocan, M.; Scott, D.J.; Hossain, M.A.; Good, S.V.; Yegorov, S.; Bogerd, J.; Gooley, P.R. The relaxin receptor as a therapeutic target—Perspectives from evolution and drug targeting. Pharmacol. Ther. 2018, 187, 114–132. [Google Scholar] [CrossRef]
- Chow, B.S.; Kocan, M.; Bosnyak, S.; Sarwar, M.; Wigg, B.; Jones, E.S.; Widdop, R.E.; Summers, R.J.; Bathgate, R.A.; Hewitson, T.D.; et al. Relaxin requires the angiotensin II type 2 receptor to abrogate renal interstitial fibrosis. Kidney Int. 2014, 86, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werle, M.; Bernkop-Schnürch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef] [Green Version]
- Knauf, M.J.; Bell, D.P.; Hirtzer, P.; Luo, Z.P.; Young, J.D.; Katre, N.V. Relationship of effective molecular size to systemic clearance in rats of recombinant interleukin-2 chemically modified with water-soluble polymers. J. Biol. Chem. 1988, 263, 15064–15070. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.A.; Perlman, A.J.; Spanski, N.; Peterson, C.M.; Sanders, S.W.; Jaffe, R.; Martin, M.; Yalcinkaya, T.; Cefalo, R.C.; Chescheir, N.C.; et al. The pharmacokinetics of recombinant human relaxin in nonpregnant women after intravenous, intravaginal, and intracervical administration. Pharm. Res. 1993, 10, 834–838. [Google Scholar] [CrossRef]
- Chen, S.A.; Reed, B.; Nguyen, T.; Gaylord, N.; Fuller, G.B.; Mordenti, J. The pharmacokinetics and absorption of recombinant human relaxin in nonpregnant rabbits and rhesus monkeys after intravenous and intravaginal administration. Pharm. Res. 1993, 10, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, J.R.; Cotter, G.; Davison, B.A.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; Ponikowski, P.; Unemori, E.; Voors, A.A.; Adams, K.F., Jr.; et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): A randomised, placebo-controlled trial. Lancet 2013, 381, 29–39. [Google Scholar] [CrossRef]
- Mallart, S.; Ingenito, R.; Bianchi, E.; Bresciani, A.; Esposito, S.; Gallo, M.; Magotti, P.; Monteagudo, E.; Orsatti, L.; Roversi, D.; et al. Identification of potent and long-acting single-chain peptide mimetics of human relaxin-2 for cardiovascular diseases. J. Med. Chem. 2021, 64, 2139–2150. [Google Scholar] [CrossRef]
- Kurtzhals, P.; Havelund, S.; Jonassen, I.; Kiehr, B.; Larsen, U.D.; Ribel, U.; Markussen, J. Albumin binding of insulins acylated with fatty acids: Characterization of the ligand-protein interaction and correlation between binding affinity and timing of the insulin effect in vivo. Biochem. J. 1995, 312 Pt 3, 725–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simard, J.R.; Zunszain, P.A.; Hamilton, J.A.; Curry, S. Location of high and low affinity fatty acid binding sites on human serum albumin revealed by NMR drug-competition analysis. J. Mol. Biol. 2006, 361, 336–351. [Google Scholar] [CrossRef]
- Hijazi, Y. Prediction of half-life extension of peptides via serum albumin binding: Current challenges. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Havelund, S.; Plum, A.; Ribel, U.; Jonassen, I.; Vølund, A.; Markussen, J.; Kurtzhals, P. The mechanism of protraction of insulin detemir, a long-acting, acylated analog of human insulin. Pharm. Res. 2004, 21, 1498–1504. [Google Scholar] [CrossRef]
- Jonassen, I.; Havelund, S.; Hoeg-Jensen, T.; Steensgaard, D.B.; Wahlund, P.O.; Ribel, U. Design of the novel protraction mechanism of insulin degludec, an ultra-long-acting basal insulin. Pharm. Res. 2012, 29, 2104–2114. [Google Scholar] [CrossRef] [Green Version]
- Guryanov, I.; Bondesan, A.; Visentini, D.; Orlandin, A.; Biondi, B.; Toniolo, C.; Formaggio, F.; Ricci, A.; Zanon, J.; Cabri, W. Innovative chemical synthesis and conformational hints on the lipopeptide liraglutide. J. Pept. Sci. 2016, 22, 471–479. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [Green Version]
- Zorzi, A.; Linciano, S.; Angelini, A. Non-covalent albumin-binding ligands for extending the circulating half-life of small biotherapeutics. Medchemcomm 2019, 10, 1068–1081. [Google Scholar] [CrossRef]
- Scott, D.J.; Rosengren, K.J.; Bathgate, R.A. The different ligand-binding modes of relaxin family peptide receptors RXFP1 and RXFP2. Mol. Endocrinol. 2012, 26, 1896–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabanpoor, F.; Bathgate, R.A.; Belgi, A.; Chan, L.J.; Nair, V.B.; Wade, J.D.; Hossain, M.A. Site-specific conjugation of a lanthanide chelator and its effects on the chemical synthesis and receptor binding affinity of human relaxin-2 hormone. Biochem. Biophys. Res. Commun. 2012, 420, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.S.; Chew, E.G.; Zhao, C.; Bathgate, R.A.; Hewitson, T.D.; Samuel, C.S. Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: The additional involvement of iNOS. PLoS ONE 2012, 7, e42714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.J.; Bathgate, R.A.; Samuel, C.S.; Dart, A.M.; Summers, R.J. Cardiovascular effects of relaxin: From basic science to clinical therapy. Nat. Rev. Cardiol. 2010, 7, 48–58. [Google Scholar] [CrossRef]
- Pinar, A.A.; Yuferov, A.; Gaspari, T.A.; Samuel, C.S. Relaxin can mediate its anti-fibrotic effects by targeting the myofibroblast NLRP3 inflammasome at the level of caspase-1. Front. Pharmacol. 2020, 11, 1201. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analogue | HEK-7BP Binding B.S.A. Free Eu-H2 pKi (n) |
|---|---|
| B7-33 | 7.28 ± 0.11 (7) |
| Decanoic acid-PEG6-B7-33 | 6.63 ± 0.14 (3) |
| Myristic acid-PEG6-B7-33 | 6.0 ± 0.11 (3) ** |
| Palmitic acid-PEG6-B7-33 | <5 (3) |
| K(Palm)-PEG6-B7-33 | 5.71 ± 0.14 (3) *** |
| B7-33_9K(Palm) | <5 (3) |
| Tag-B7-33 | <5 (3) |
| Tag-PEG4-B7-33 | <5 (3) |
| Tag-PEG6-B7-33 | <5 (3) |
| K(Palm)-(PEG6)2-B7-33 | 6.80 ± 0.16 (3) |
| PA-(PEG6)2-B7-33 | 6.40 ± 0.10 (3) ** |
| K(Palm)-PEG12-B7-33 | 6.56 ± 0.52 (3) ** |
| K(PalmGlu)-PEG12-B7-33 | 7.37 ± 0.10 (3) |
| Ac K(PalmGlu)-PEG12-B7-33 | 7.52 ± 0.13 (3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Praveen, P.; Wang, C.; Handley, T.N.G.; Wu, H.; Samuel, C.S.; Bathgate, R.A.D.; Hossain, M.A. A Lipidated Single-B-Chain Derivative of Relaxin Exhibits Improved In Vitro Serum Stability without Altering Activity. Int. J. Mol. Sci. 2023, 24, 6616. https://doi.org/10.3390/ijms24076616
Praveen P, Wang C, Handley TNG, Wu H, Samuel CS, Bathgate RAD, Hossain MA. A Lipidated Single-B-Chain Derivative of Relaxin Exhibits Improved In Vitro Serum Stability without Altering Activity. International Journal of Molecular Sciences. 2023; 24(7):6616. https://doi.org/10.3390/ijms24076616
Chicago/Turabian StylePraveen, Praveen, Chao Wang, Thomas N. G. Handley, Hongkang Wu, Chrishan S. Samuel, Ross A. D. Bathgate, and Mohammed Akhter Hossain. 2023. "A Lipidated Single-B-Chain Derivative of Relaxin Exhibits Improved In Vitro Serum Stability without Altering Activity" International Journal of Molecular Sciences 24, no. 7: 6616. https://doi.org/10.3390/ijms24076616
APA StylePraveen, P., Wang, C., Handley, T. N. G., Wu, H., Samuel, C. S., Bathgate, R. A. D., & Hossain, M. A. (2023). A Lipidated Single-B-Chain Derivative of Relaxin Exhibits Improved In Vitro Serum Stability without Altering Activity. International Journal of Molecular Sciences, 24(7), 6616. https://doi.org/10.3390/ijms24076616