
A Concerted Vision to Advance the Knowledge of Diabetes Mellitus Related to Immune Checkpoint Inhibitors

Abstract
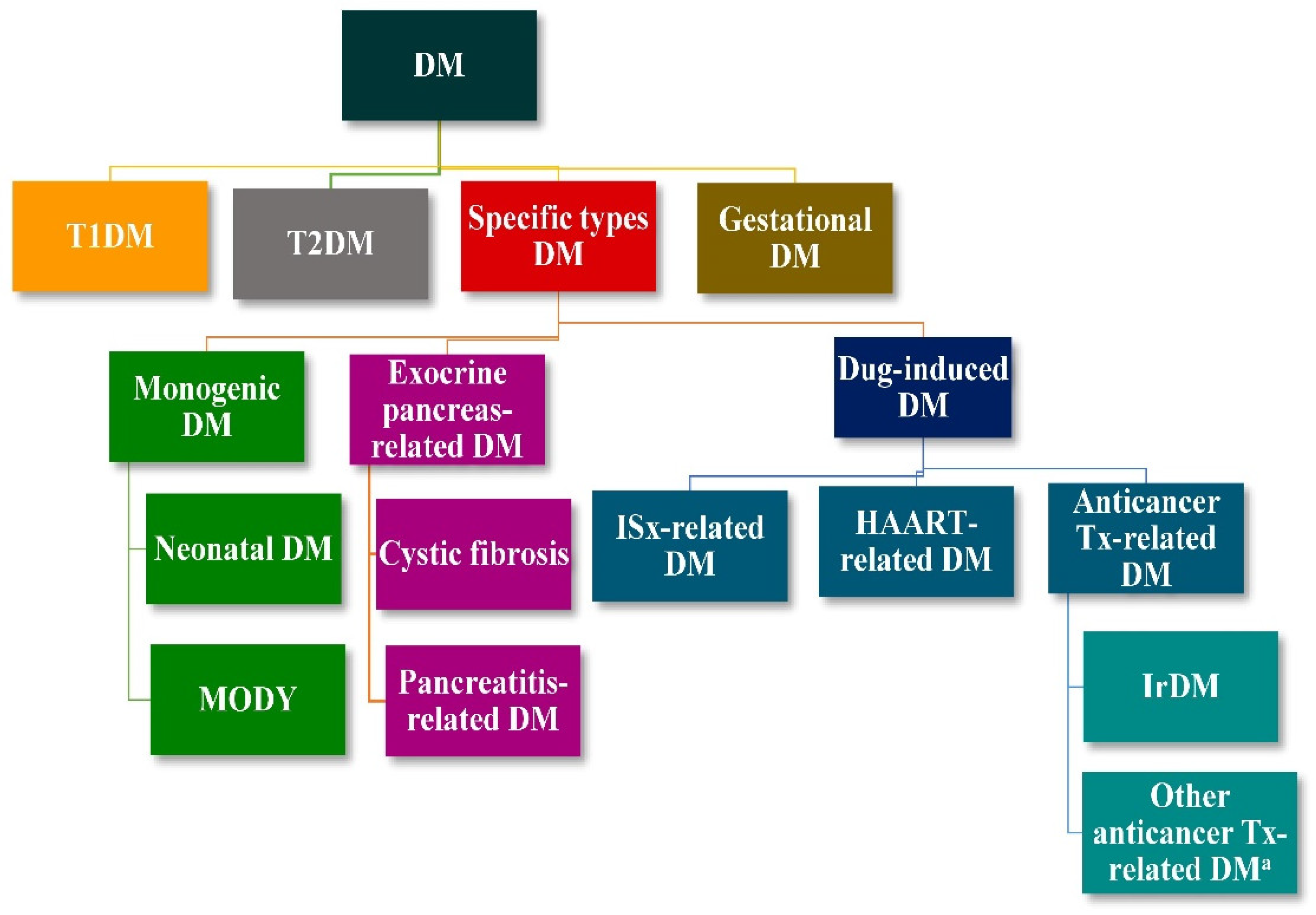
:1. Introduction
2. Methods of Data Collection
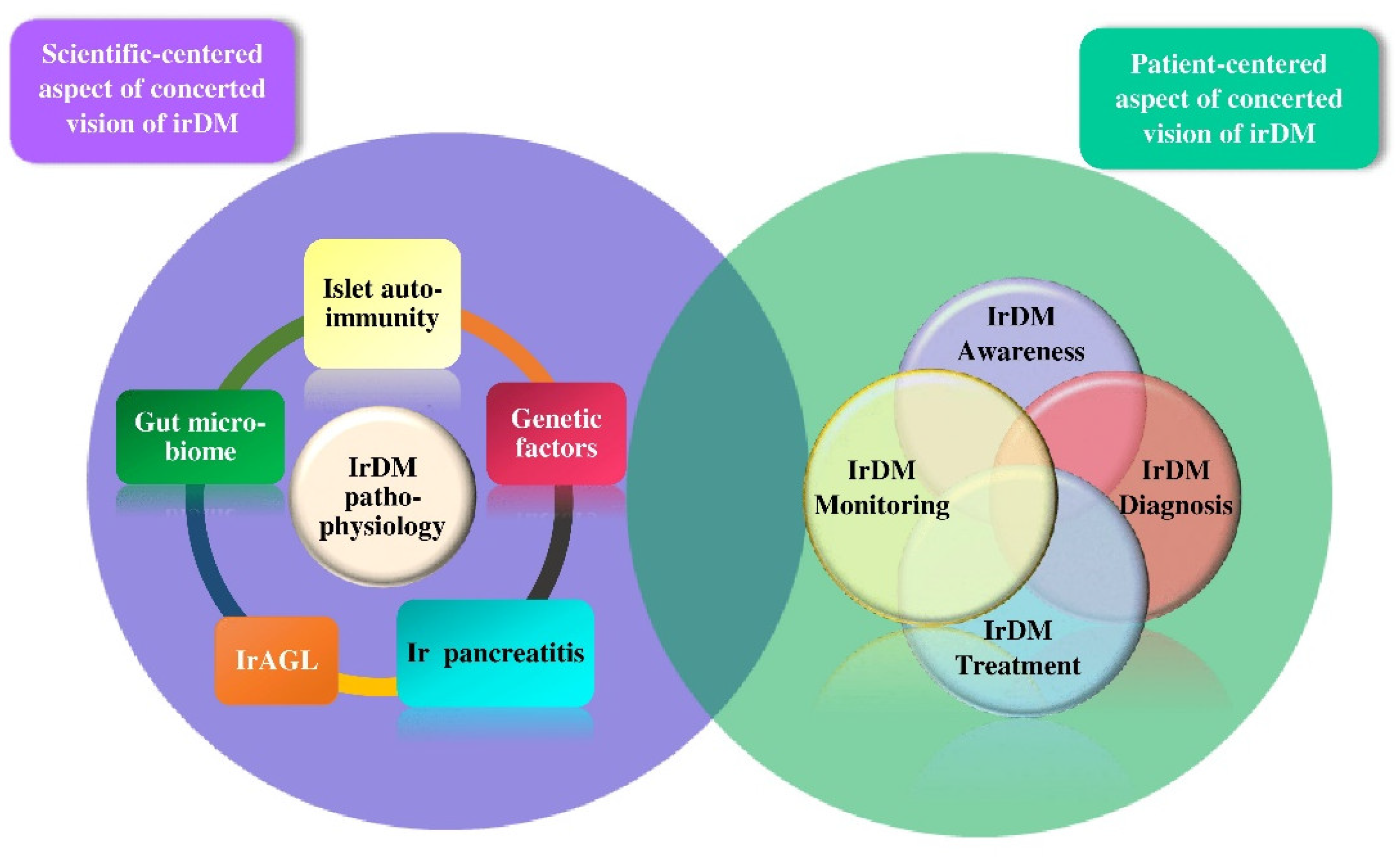
3. The Scientific-Centered Aspect of irDM
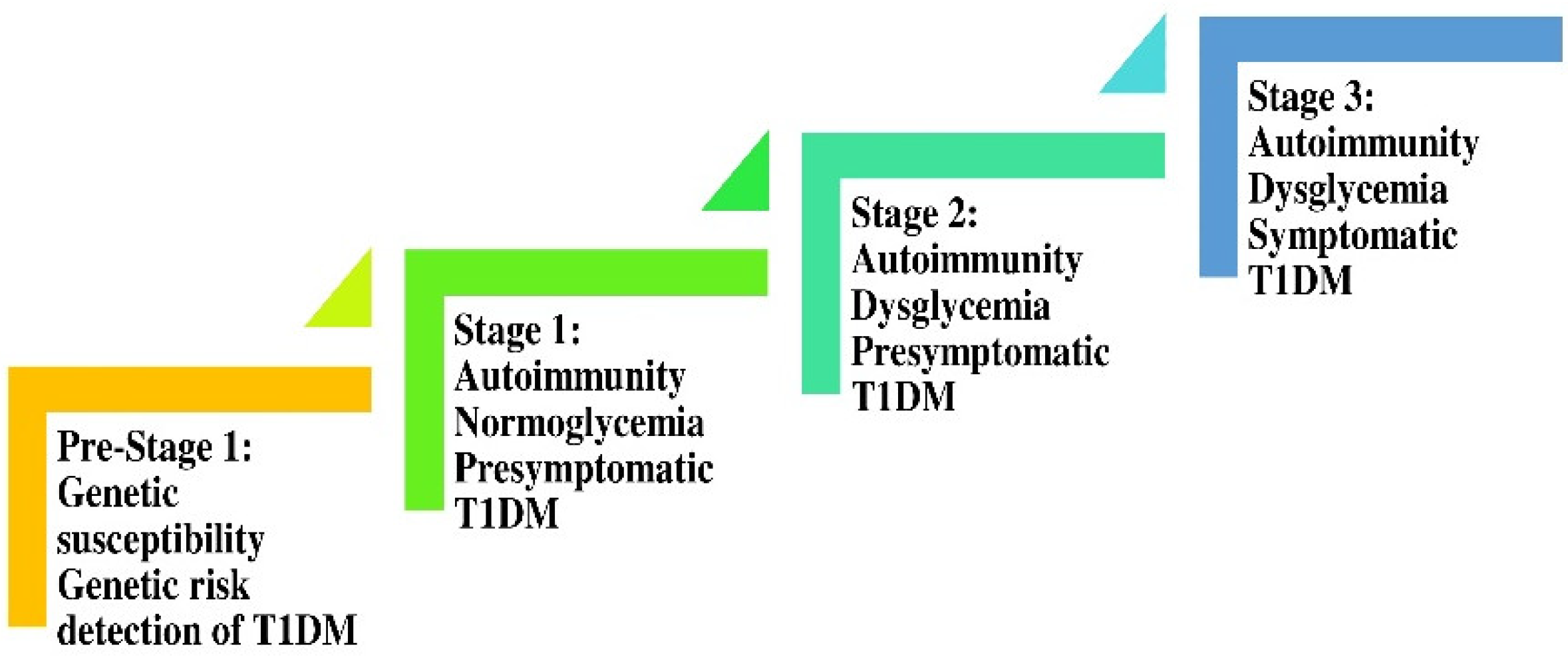
3.1. The Rationale for the Prevailing Hypothesis for the Pathophysiology of irT1DM
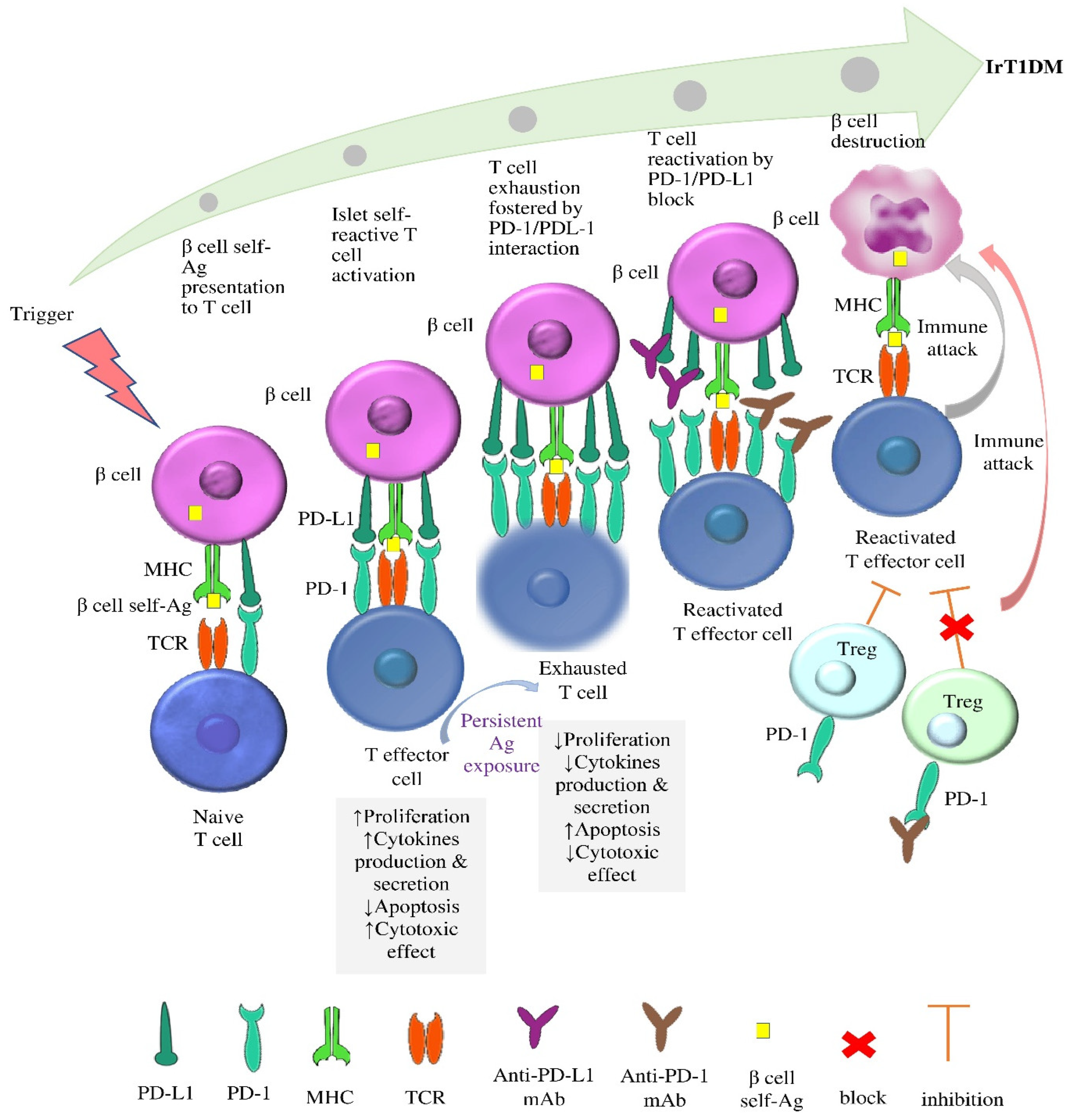
3.1.1. The Role of PD-1/PD-L1 Interaction in Autoimmunity
3.1.2. The Role of the Disinhibition of PD-1/PD-L1 Interaction in the Pathophysiology of irT1DM
3.1.3. Genetic Susceptibility to irDM
3.1.4. The Status of Islet Autoantibodies in the Setting of irT1DM
3.2. The Rationale for the Potential Involvement of the Gut Microbiome in the Pathophysiology of irT1DM
3.3. The Potential Involvement of Pancreatic Alpha (α) Cells and of the Exocrine Pancreas in the Pathophysiology of irDM
3.4. The Potential Involvement of irAGL in the Pathophysiology of irDM
4. The Patient-Centered Aspect of irDM
4.1. Pillar I: Awareness of irDM
4.2. Pillar II: Diagnosis of irDM
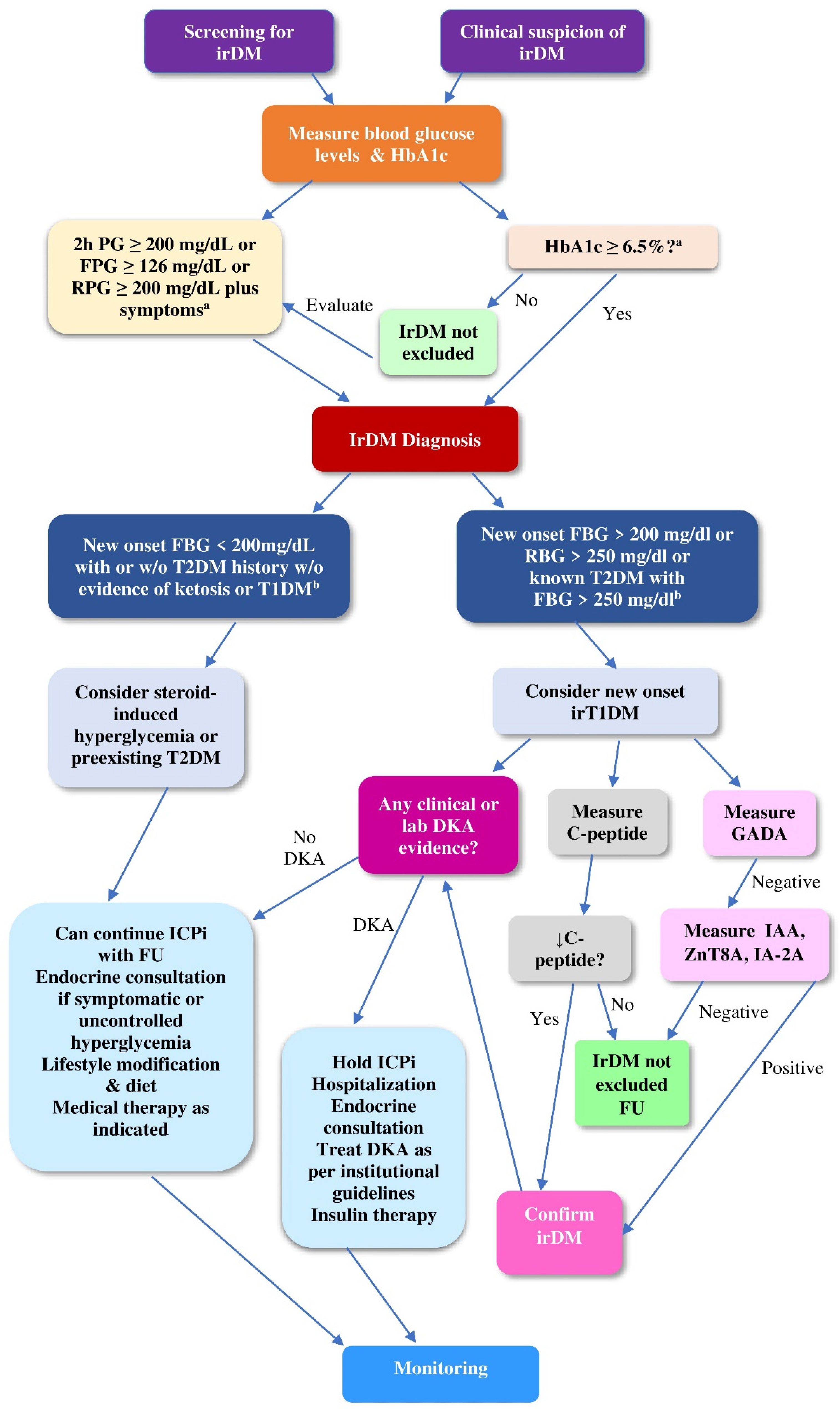
4.2.1. Screening of ICPi-Treated Patients for Hyperglycemia
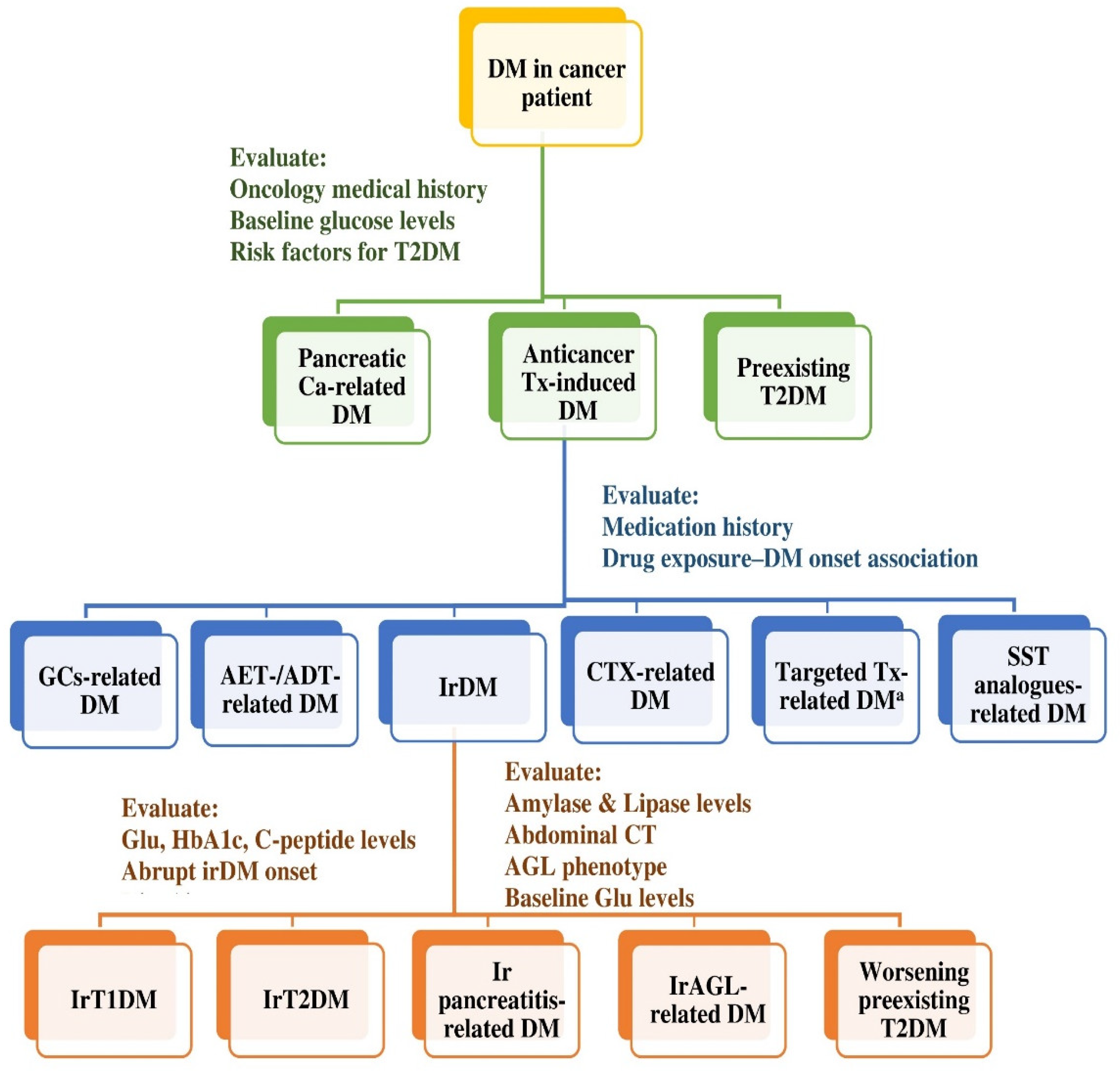
4.2.2. Diagnostic Work-Up of irDM
4.2.3. Differential Diagnosis of irDM
4.3. Pillar III: Treatment of irDM
4.4. Pillar IV: Monitoring of irDM
5. Current Challenges and Future Perspectives Regarding irDM
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Schnell, A.; Bod, L.; Madi, A.; Kuchroo, V.K. The yin and yang of co-inhibitory receptors: Toward anti-tumor immunity without autoimmunity. Cell Res. 2020, 30, 285–299. [Google Scholar] [CrossRef]
- Sharpe, A.; Pauken, K. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Ghosh, C.; Luong, G.; Sun, Y. A snapshot of the PD-1/PD-L1 pathway. J. Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef]
- Cancer Research Institute (CRI). FDA Approval Timeline of Active Immunotherapies. 2023. Available online: https://www.cancerresearch.org/fda-approval-timeline-of-active-immunotherapies (accessed on 15 January 2023).
- Thompson, J.A.; Schneider, B.J.; Brahmer, J.; Andrews, S.; Armand, P.; Bhatia, S.; Budde, L.E.; Costa, L.; Davies, M.; Dunnington, D.; et al. Management of Immunotherapy-Related Toxicities, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2019, 17, 255–289. [Google Scholar] [CrossRef]
- Raschi, E.; Mazzarella, A.; Antonazzo, I.C.; Bendinelli, N.; Forcesi, E.; Tuccori, M.; Moretti, U.; Poluzzi, E.; De Ponti, F. Toxicities with Immune Checkpoint Inhibitors: Emerging Priorities from Disproportionality Analysis of the FDA Adverse Event Reporting System. Target Oncol. 2019, 14, 205–221. [Google Scholar] [CrossRef]
- de Filette, J.; Andreescu, C.E.; Cools, F.; Bravenboer, B.; Velkeniers, B. A Systematic Review and Meta-Analysis of Endocrine-Related Adverse Events Associated with Immune Checkpoint Inhibitors. Horm. Metab. Res. 2019, 51, 145–156. [Google Scholar] [CrossRef]
- Deligiorgi, M.V.; Panayiotidis, M.I.; Trafalis, D.T. Endocrine adverse events related with immune checkpoint inhibitors: An update for clinicians. Immunotherapy 2020, 12, 481–510. [Google Scholar] [CrossRef]
- Quandt, Z.; Young, A.; Anderson, M. Immune checkpoint inhibitor diabetes mellitus: A novel form of autoimmune diabetes. Clin. Exp. Immunol. 2020, 200, 131–140. [Google Scholar] [CrossRef]
- Wu, L.; Tsang, V.H.M.; Sasson, S.C.; Menzies, A.M.; Carlino, M.S.; Brown, D.A.; Clifton-Bligh, R.; Gunton, J.E. Unravelling Checkpoint Inhibitor Associated Autoimmune Diabetes: From Bench to Bedside. Front. Endocrinol. 2021, 12, 764138. [Google Scholar] [CrossRef]
- Clotman, K.; Janssens, K.; Specenier, P.; Weets, I.; De Block, C.E.M. Programmed Cell Death-1 Inhibitor-Induced Type 1 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2018, 103, 3144–3154. [Google Scholar] [CrossRef]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes. Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43 (Suppl. S1), S14–S31. [Google Scholar] [CrossRef]
- Sayed Ahmed, A.; Abreo, M.; Thomas, A.; Chari, S.T. Type 3 autoimmune pancreatitis (immune checkpoint inhibitor-induced pancreatitis). Curr. Opin. Gastroenterol. 2022, 38, 516–520. [Google Scholar] [CrossRef]
- Abu-Sbeih, H.; Tang, T.; Lu, Y.; Thirumurthi, S.; Altan, M.; Jazaeri, A.A.; Dadu, R.; Coronel, E.; Wang, Y. Clinical characteristics and outcomes of immune checkpoint inhibitor-induced pancreatic injury. J. Immunother. Cancer 2019, 7, 31. [Google Scholar] [CrossRef]
- Gnanendran, S.S.; Miller, J.A.; Archer, C.A.; Jain, S.V.; Hwang, S.J.E.; Peters, G.; Miller, A. Acquired lipodystrophy associated with immune checkpoint inhibitors. Melanoma Res. 2020, 30, 599–602. [Google Scholar] [CrossRef]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Johnson, D.B.; Nebhan, C.A.; Moslehi, J.J.; Balko, J.M. Immune-checkpoint inhibitors: Long-term implications of toxicity. Nat. Rev. Clin. Oncol. 2022, 19, 254–267. [Google Scholar] [CrossRef]
- Wright, J.J.; Salem, J.E.; Johnson, D.B.; Lebrun-Vignes, B.; Stamatouli, A.; Thomas, J.W.; Herold, K.C.; Moslehi, J.; Powers, A.C. Increased reporting of immune checkpoint inhibitor-associated diabetes. Diabetes Care 2018, 41, e150–e151. [Google Scholar] [CrossRef]
- Lee, D.; Lee, H.J., Jr.; Farmer, J.R.; Reynolds, K.L. Mechanisms Driving Immune-Related Adverse Events in Cancer Patients Treated with Immune Checkpoint Inhibitors. Curr. Cardiol. Rep. 2021, 23, 98. [Google Scholar] [CrossRef]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef]
- Willsmore, Z.N.; Harris, R.J.; Crescioli, S.; Hussein, K.; Kakkassery, H.; Thapa, D.; Cheung, A.; Chauhan, J.; Bax, H.J.; Chenoweth, A.; et al. B Cells in Patients with Melanoma: Implications for Treatment with Checkpoint Inhibitor Antibodies. Front. Immunol. 2020, 11, 622442. [Google Scholar] [CrossRef] [PubMed]
- Pavan, A.; Calvetti, L.; Dal Maso, A.; Attili, I.; Del Bianco, P.; Pasello, G.; Guarneri, V.; Aprile, G.; Conte, P.; Bonanno, L. Peripheral Blood Markers Identify Risk of Immune-Related Toxicity in Advanced Non-Small Cell Lung Cancer Treated with Immune-Checkpoint Inhibitors. Oncologist 2019, 24, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Lee, J.H.; Gide, T.N.; Menzies, A.M.; Guminski, A.; Carlino, M.S.; Breen, E.J.; Yang, J.Y.H.; Ghazanfar, S.; Kefford, R.F.; et al. Circulating Cytokines Predict Immune-Related Toxicity in Melanoma Patients Receiving Anti-PD-1-based Immunotherapy. Clin. Cancer Res. 2019, 25, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.H.; Geng, L.Y.; Jiang, P.P.; Xu, H.; Nan, K.J.; Yao, Y.; Jiang, L.L.; Sun, H.; Qin, T.J.; Guo, H. The biomarkers related to immune related adverse events caused by immune checkpoint inhibitors. J. Exp. Clin. Cancer Res. 2020, 39, 284. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Strazza, M. Bridging the Gap: Connecting the Mechanisms of Immune-Related Adverse Events and Autoimmunity Through PD-1. Front. Cell Dev. Biol. 2022, 9, 790386. [Google Scholar] [CrossRef]
- Carr, A.L.J.; Evans-Molina, C.; Oram, R.A. Precision medicine in type 1 diabetes. Diabetologia 2022, 65, 1854–1866. [Google Scholar] [CrossRef]
- Mota Reyes, C.; Demir, E.; Çifcibaşı, K.; Istvanffy, R.; Friess, H.; Demir, I.E. Regulatory T Cells in Pancreatic Cancer: Of Mice and Men. Cancers 2022, 14, 4582. [Google Scholar] [CrossRef]
- Shevyrev, D.; Tereshchenko, V. Treg Heterogeneity, Function, and Homeostasis. Front. Immunol. 2020, 10, 3100. [Google Scholar] [CrossRef]
- Viisanen, T.; Gazali, A.M.; Ihantola, E.L.; Ekman, I.; Näntö-Salonen, K.; Veijola, R.; Toppari, J.; Knip, M.; Ilonen, J.; Kinnunen, T. FOXP3+ Regulatory T Cell Compartment Is Altered in Children with Newly Diagnosed Type 1 Diabetes but Not in Autoantibody-Positive at-Risk Children. Front. Immunol. 2019, 10, 19. [Google Scholar] [CrossRef]
- Anderson, A.M.; Landry, L.G.; Alkanani, A.A.; Pyle, L.; Powers, A.C.; Atkinson, M.A.; Mathews, C.E.; Roep, B.O.; Michels, A.W.; Nakayama, M. Human islet T cells are highly reactive to preproinsulin in type 1 diabetes. Proc. Natl. Acad. Sci. USA 2021, 118, e2107208118. [Google Scholar] [CrossRef]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, Å.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Ogando, J.; Saez, M.E.; Santos, J.; Nuevo-Tapioles, C.; Gut, M.; Esteve-Codina, A.; Heath, S.; Gonzalez-Perez, A.; Cuezva, J.M.; Lacalle, R.A.; et al. PD-1 signaling affects cristae morphology and leads to mitochondrial dysfunction in human CD8(+) T lymphocytes. J. Immunother. Cancer 2019, 7, 151. [Google Scholar] [CrossRef] [PubMed]
- Pichler, A.C.; Cannons, J.L.; Schwartzberg, P.L. The Road Less Taken: Less Appreciated Pathways for Manipulating CD8+ T Cell Exhaustion. Front. Immunol. 2022, 13, 926714. [Google Scholar] [CrossRef]
- He, X.; Xu, C. PD-1: A Driver or Passenger of T Cell Exhaustion? Mol. Cell 2020, 77, 930–931. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Feng, Y.; Xu, J.; Liang, J. T-cell exhaustion in immune-mediated inflammatory diseases: New implications for immunotherapy. Front. Immunol. 2022, 13, 977394. [Google Scholar] [CrossRef]
- Linsley, P.S.; Long, S.A. Enforcing the checkpoints: Harnessing T-cell exhaustion for therapy of T1D. Curr. Opin. Endocrinol. Diabetes Obes. 2019, 26, 213–218. [Google Scholar] [CrossRef]
- Colli, M.L.; Hill, J.L.E.; Marroquí, L.; Chaffey, J.; Dos Santos, R.S.; Leete, P.; Coomand de Brachѐne, A.; Paula, F.M.M.; Op de Beeck, A.; Castela, A.; et al. PDL1 is expressed in the islets of people with type 1 diabetes and is up-regulated by interferons-α and-γ via IRF1 induction. Ebiomedicine 2018, 36, 367–375. [Google Scholar] [CrossRef]
- Osum, K.C.; Burrack, A.L.; Martinov, T.; Sahli, N.L.; Mitchell, J.S.; Tucker, C.G.; Pauken, K.E.; Papas, K.; Appakalai, B.; Spanier, J.A.; et al. Interferon-gamma drives programmed death-ligand 1 expression on islet β cells to limit T cell function during autoimmune diabetes. Sci. Rep. 2018, 8, 8295. [Google Scholar] [CrossRef]
- Kwong, C.J.; Selck, C.; Tahija, K.; McAnaney, L.J.; Le, D.V.; Kay, T.W.; Thomas, H.E.; Krishnamurthy, B. Harnessing CD8+ T-cell exhaustion to treat type 1 diabetes. Immunol. Cell Biol. 2021, 99, 486–495. [Google Scholar] [CrossRef]
- Wong, F.S.; Wen, L. A predictive CD8+ T cell phenotype for T1DM progression. Nat. Rev. Endocrinol. 2020, 16, 198–199. [Google Scholar] [CrossRef]
- Wiedeman, A.E.; Muir, V.S.; Rosasco, M.G.; DeBerg, H.A.; Presnell, S.; Haas, B.; Dufort, M.J.; Speake, C.; Greenbaum, C.J.; Serti, E.; et al. Autoreactive Cd8+ T cell exhaustion distinguishes subjects with slow type 1 diabetes progression. J. Clin. Investig. 2020, 130, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Kong, Y.; Zhou, Y.; Guo, J.; Shi, Q.; Li, S.; Guo, H.; Huang, Y.; Ding, S.; Liu, C.; et al. Decreased expression of programmed death-1 on CD8+ effector memory T lymphocytes correlates with the pathogenesis of type 1 diabetes. Acta Diabetol. 2021, 58, 1239–1249. [Google Scholar] [CrossRef]
- Pellegrino, M.; Crinò, A.; Rosado, M.M.; Fierabracci, A. Identification and functional characterization of CD8+ T regulatory cells in type 1 diabetes patients. PLoS ONE 2019, 14, e0210839. [Google Scholar] [CrossRef] [PubMed]
- Zagouras, A.; Patil, P.D.; Yogi-Morren, D.; Pennell, N.A. Cases from the Immune-Related Adverse Event Tumor Board: Diagnosis and Management of Immune Checkpoint Blockade-Induced Diabetes. Oncologist 2020, 25, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, S.; Imagawa, A.; Hosokawa, Y.; Baden, M.Y.; Kimura, T.; Uno, S.; Fukui, K.; Goto, K.; Uemura, M.; Eguchi, H.; et al. T-Lymphocyte Infiltration to Islets in the Pancreas of a Patient Who Developed Type 1 Diabetes After Administration of Immune Checkpoint Inhibitors. Diabetes Care 2019, 42, e116–e118. [Google Scholar] [CrossRef]
- Mourad, D.; Azar, N.S.; Eid, A.A.; Azar, S.T. Immune Checkpoint Inhibitor-Induced Diabetes Mellitus: Potential Role of T Cells in the Underlying Mechanism. Int. J. Mol. Sci. 2021, 22, 2093. [Google Scholar] [CrossRef]
- Mazzucato, M.; Garelli, S.; Betterle, C.; Presotto, F. Checkpoint inhibitor develops histological autoimmune pancreatitis like type 1 diabetes. A case report. MOJ Clin. Med. Case Rep. 2020, 10, 78–79. [Google Scholar]
- Redondo, M.J.; Steck, A.K.; Pugliese, A. Genetics of type 1 diabetes. Pediatr. Diabetes 2018, 19, 346–353. [Google Scholar] [CrossRef]
- Zhao, L.P.; Papadopoulos, G.K.; Moustakas, A.K.; Bondinas, G.P.; Carlsson, A.; Larsson, H.E.; Ludvigsson, J.; Marcus, C.; Persson, M.; Samuelsson, U.; et al. Nine residues in HLA-DQ molecules determine with susceptibility and resistance to type 1 diabetes among young children in Sweden. Sci. Rep. 2021, 11, 8821. [Google Scholar] [CrossRef]
- Sticht, J.; Álvaro-Benito, M.; Konigorski, S. Type 1 Diabetes and the HLA Region: Genetic Association Besides Classical HLA Class II Genes. Front. Genet. 2021, 12, 683946. [Google Scholar] [CrossRef]
- Stamatouli, A.M.; Quandt, Z.; Perdigoto, A.L.; Clark, P.L.; Kluger, H.; Weiss, S.A.; Gettinger, S.; Sznol, M.; Young, A.; Rushakoff, R.; et al. Collateral damage: Insulin-dependent diabetes induced with checkpoint inhibitors. Diabetes 2018, 67, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Tsang, V.H.M.; McGrath, R.T.; Clifton-Bligh, R.J.; Scolyer, R.A.; Jakrot, V.; Guminski, A.D.; Long, G.V.; Menzies, A.M. Checkpoint Inhibitor-Associated Autoimmune Diabetes Is Distinct from Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 5499–5506. [Google Scholar] [CrossRef] [PubMed]
- Yun, K.; Daniels, G.; Gold, K.; McCowen, K.; Patel, S.P. Rapid Onset Type 1 Diabetes with Anti-PD-1 Directed Therapy. Oncotarget 2020, 11, 2740–2746. [Google Scholar] [CrossRef]
- Akturk, H.K.; Kahramangil, D.; Sarwal, A.; Hoffecker, L.; Murad, M.H.; Michels, A.W. Immune checkpoint inhibitor-induced Type 1 diabetes: A systematic review and meta-analysis. Diabet. Med. 2019, 36, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Lo Preiato, V.; Salvagni, S.; Ricci, C.; Ardizzoni, A.; Pagotto, U.; Pelusi, C. Diabetes Mellitus Induced by Immune Checkpoint Inhibitors: Type 1 Diabetes Variant or New Clinical Entity? Rev. Lit. Rev. Endocr. Metab. Disord. 2021, 22, 337–349. [Google Scholar] [CrossRef]
- Wen, S.; Jiang, W.; Zhou, L. Islet Autoantibodies in the Patients with Sjogren’s Syndrome and Thyroid Disease and Risk of Progression to Latent Autoimmune Diabetes in Adults: A Case Series. Diabetes Metab. Syndr. Obes. 2021, 14, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Mistry, S.; Gouripeddi, R.; Raman, V.; Facelli, J.C. Stratifying risk for onset of type 1 diabetes using islet autoantibody trajectory clustering. Diabetologia 2022, 66, 520–534. [Google Scholar] [CrossRef] [PubMed]
- Mallone, R.; Eizirik, D.L. Presumption of innocence for beta cells: Why are they vulnerable autoimmune targets in type 1 diabetes? Diabetologia 2020, 63, 1999–2006. [Google Scholar] [CrossRef]
- Hamadi, G.M. Immunological markers in type 1 diabetes mellitus in Thi-Qar province, southern Iraq. J. Med. Life 2022, 15, 1234–1239. [Google Scholar] [CrossRef]
- De Filette, J.M.K.; Pen, J.J.; Decoster, L.; Vissers, T.; Bravenboer, B.; van der Auwera, B.J.; Gorus, F.K.; Roep, B.O.; Aspeslagh, S.; Neyns, B.; et al. Immune Checkpoint Inhibitors and Type 1 Diabetes Mellitus: A Case Report and Systematic Review. Eur. J. Endocrinol. 2019, 181, 363–374. [Google Scholar] [CrossRef]
- Kotwal, A.; Haddox, C.; Block, M.; Kudva, Y.C. Immune Checkpoint Inhibitors: An Emerging Cause of Insulin-Dependent Diabetes. BMJ Open Diabetes Res. Care 2019, 7, e000591. [Google Scholar] [CrossRef]
- Tan, M.H.; Iyengar, R.; Mizokami-Stout, K.; Yentz, S.; MacEachern, M.P.; Shen, L.Y.; Redman, B.; Gianchandani, R. Spectrum of immune checkpoint inhibitors-induced endocrinopathies in cancer patients: A scoping review of case reports. Clin. Diabetes Endocrinol. 2019, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, M.; Wang, L.; Li, L.; Zhong, X. Sintilimab induced diabetic ketoacidosis in a patient with small cell lung cancer: A case report and literature review. Medicine 2021, 100, e25795. [Google Scholar] [CrossRef] [PubMed]
- Martinov, T.; Swanson, L.A.; Breed, E.R.; Tucker, C.G.; Dwyer, A.J.; Johnson, J.K.; Mitchell, J.S.; Sahli, N.L.; Wilson, J.C.; Singh, L.M.; et al. Programmed Death-1 Restrains the Germinal Center in Type 1 Diabetes. J. Immunol. 2019, 203, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Nakayamada, S.; Tanaka, Y. Differentiation, functions, and roles of T follicular regulatory cells in autoimmune diseases. Inflamm. Regen. 2021, 41, 14. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Shen, M.; Zhao, R.; Cai, Y.; Jiang, H.; Shen, Z.; Gao, R.; Xu, K.; Chen, H.; Yang, T. Follicular regulatory T cells are associated with β-cell autoimmunity and the development of type 1 diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 4199–4213. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Craft, J. T Follicular Regulatory Cells: Choreographers of Productive Germinal Center Responses. Front. Immunol. 2021, 12, 679909. [Google Scholar] [CrossRef]
- Matijašić, M.; Meštrović, T.; Paljetak, H.Č.; Perić, M.; Barešić, A.; Verbanac, D. Gut Microbiota beyond Bacteria-Mycobiome, Virome, Archaeome, and Eukaryotic Parasites in IBD. Int. J. Mol. Sci. 2020, 21, 2668. [Google Scholar] [CrossRef]
- Zhang, T.; Gao, G.; Sakandar, H.A.; Kwok, L.Y.; Sun, Z. Gut Dysbiosis in Pancreatic Diseases: A Causative Factor and a Novel Therapeutic Target. Front. Nutr. 2022, 9, 814269. [Google Scholar] [CrossRef]
- Shilo, S.; Godneva, A.; Rachmiel, M.; Korem, T.; Bussi, Y.; Kolobkov, D.; Karady, T.; Bar, N.; Wolf, B.C.; Glantz-Gashai, Y.; et al. The Gut Microbiome of Adults with Type 1 Diabetes and Its Association with the Host Glycemic Control. Diabetes Care 2022, 45, 555–563. [Google Scholar] [CrossRef]
- van Heck, J.I.P.; Gacesa, R.; Stienstra, R.; Fu, J.; Zhernakova, A.; Harmsen, H.J.M.; Weersma, R.K.; Joosten, L.A.B.; Tack, C.J. The Gut Microbiome Composition Is Altered in Long-standing Type 1 Diabetes and Associates with Glycemic Control and Disease-Related Complications. Diabetes Care 2022, 45, 2084–2094. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Zhang, J.; Wu, Q.; Fang, H.; Shi, C.; Li, Z.; Lin, C.; Tang, D.; Wang, D. Intestinal microbiota: A new force in cancer immunotherapy. Cell Commun. Signal 2020, 18, 90. [Google Scholar] [CrossRef] [PubMed]
- Oey, O.; Liu, Y.Y.; Sunjaya, A.F.; Simadibrata, D.M.; Khattak, M.A.; Gray, E. Gut microbiota diversity and composition in predicting immunotherapy response and immunotherapy-related colitis in melanoma patients: A systematic review. World J. Clin. Oncol. 2022, 13, 929–942. [Google Scholar] [CrossRef]
- Oh, B.; Boyle, F.; Pavlakis, N.; Clarke, S.; Eade, T.; Hruby, G.; Lamoury, G.; Carroll, S.; Morgia, M.; Kneebone, A.; et al. The Gut Microbiome and Cancer Immunotherapy: Can We Use the Gut Microbiome as a Predictive Biomarker for Clinical Response in Cancer Immunotherapy? Cancers 2021, 13, 4824. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.S.; Manzano, A.; Olivar, L.C.; Nava, M.; Salazar, J.; D’Marco, L.; Ortiz, R.; Chacín, M.; Guerrero-Wyss, M.; Cabrera de Bravo, M.; et al. The Role of the α Cell in the Pathogenesis of Diabetes: A World beyond the Mirror. Int. J. Mol. Sci. 2021, 22, 9504. [Google Scholar] [CrossRef]
- Ross, J.J.; Wasserfall, C.H.; Bacher, R.; Perry, D.J.; McGrail, K.; Posgai, A.L.; Dong, X.; Muir, A.; Li, X.; Campbell-Thompson, M.; et al. Exocrine Pancreatic Enzymes Are a Serological Biomarker for Type 1 Diabetes Staging and Pancreas Size. Diabetes 2021, 70, 944–954. [Google Scholar] [CrossRef]
- Ko, J.; Cho, J.; Petrov, M.S. Low serum amylase, lipase, and trypsin as biomarkers of metabolic disorders: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2020, 159, 107974. [Google Scholar] [CrossRef]
- Marchand, L.; Thivolet, A.; Dalle, S.; Chikh, K.; Reffet, S.; Vouillarmet, J.; Fabien, N.; Cugnet-Anceau, C.; Thivolet, C. Diabetes mellitus induced by PD-1 and PD-L1 inhibitors: Description of pancreatic endocrine and exocrine phenotype. Acta Diabetol. 2019, 56, 441–448. [Google Scholar] [CrossRef]
- Marchand, L.; Thivolet, A.; Saintigny, P.; Fabien, N.; Vouillarmet, J.; Thivolet, C. Anti-Programmed Death 1 (PD-1) Antibodies and the Pancreas: A Diabetic Storm Ahead? Diabetes Care 2018, 41, 638–639. [Google Scholar] [CrossRef]
- Nakano, R.; Shiomi, H.; Fujiwara, A.; Yoshihara, K.; Yoshioka, R.; Kawata, S.; Ota, S.; Yuri, Y.; Takashima, T.; Aizawa, N.; et al. Clinical Characteristics of ICI-Related Pancreatitis and Cholangitis Including Radiographic and Endoscopic Findings. Healthcare 2022, 10, 763. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Bajaj, D.; Sankaramangalam, K.; Yoo, J.W.; Joshi, N.S.; Gettinger, S.; Price, C.; Farrell, J.J. Incidence of pancreatitis with the use of immune checkpoint inhibitors (ICI) in advanced cancers: A systematic review and meta-analysis. Pancreatology 2019, 19, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Zhang, X.C.; Zhang, C.G.; Hou, Y.L.; Yao, Y.X.; Cao, B.W. Risk of Immune-Related Pancreatitis in Patients with Solid Tumors Treated with Immune Checkpoint Inhibitors: Systematic Assessment with Meta-Analysis. J. Immunol. Res. 2018, 2018, 1027323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, Y.; Shi, C.; Liu, X.; Lv, S.; Wang, X.; Li, W. Pancreatic injury following immune checkpoint inhibitors: A systematic review and meta-analysis. Front. Pharmacol. 2022, 13, 955701. [Google Scholar] [CrossRef]
- Corvillo, F.; Aparicio, V.; López-Lera, A.; Garrido, S.; Araújo-Vilar, D.; de Miguel, M.P.; López-Trascasa, M. Autoantibodies Against Perilipin 1 as a Cause of Acquired Generalized Lipodystrophy. Front. Immunol. 2018, 9, 2142. [Google Scholar] [CrossRef]
- Koethe, J.R.; Lagathu, C.; Lake, J.E.; Domingo, P.; Calmy, A.; Falutz, J.; Brown, T.T.; Capeau, J. HIV and antiretroviral therapy-related fat alterations. Nat. Rev. Dis. Primers 2020, 6, 48. [Google Scholar] [CrossRef]
- Falcao, C.K.; Cabral, M.C.S.; Mota, J.M.; Arbache, S.T.; Costa-Riquetto, A.D.; Muniz, D.Q.B.; Cury-Martins, J.; Almeida, M.Q.; Kaczemorska, P.C.; Nery, M.; et al. Acquired Lipodystrophy Associated with Nivolumab in a Patient With Advanced Renal Cell Carcinoma. J. Clin. Endocrinol. Metab. 2019, 104, 3245–3248. [Google Scholar] [CrossRef]
- Haddad, N.; Vidal-Trecan, T.; Baroudjian, B.; Zagdanski, A.M.; Arangalage, D.; Battistella, M.; Gautier, J.F.; Lebbe, C.; Delyon, J.; PATIO Group. Acquired generalized lipodystrophy under immune checkpoint inhibition. Br. J. Dermatol. 2020, 182, 477–480. [Google Scholar] [CrossRef]
- Bedrose, S.; Turin, C.G.; Lavis, V.R.; Kim, S.T.; Thosani, S.N. A Case of Acquired Generalized Lipodystrophy Associated with Pembrolizumab in a Patient with Metastatic Malignant Melanoma. AACE Clin. Case Rep. 2020, 6, e40–e45. [Google Scholar] [CrossRef]
- Jehl, A.; Cugnet-Anceau, C.; Vigouroux, C.; Legeay, A.L.; Dalle, S.; Harou, O.; Marchand, L.; Lascols, O.; Caussy, C.; Thivolet, C.; et al. Acquired Generalized Lipodystrophy: A New Cause of Anti-PD-1 Immune-Related Diabetes. Diabetes Care 2019, 42, 2008–2010. [Google Scholar] [CrossRef]
- Mandel-Brehm, C.; Vazquez, S.E.; Liverman, C.; Cheng, M.; Quandt, Z.; Kung, A.F.; Parent, A.; Miao, B.; Disse, E.; Cugnet-Anceau, C.; et al. Autoantibodies to Perilipin-1 Define a Subset of Acquired Generalized Lipodystrophy. Diabetes 2023, 72, 59–70. [Google Scholar] [CrossRef]
- Kalra, S. Post-immunotherapy new onset diabetes (PINOD)-under-recognized etiology, unexplored presentation. Ann. Transl. Med. 2018, 6, S84. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Sousa, R.; Barry, W.T.; Garrido-Castro, A.C.; Hodi, F.S.; Min, L.; Krop, I.E.; Tolaney, S.M. Incidence of Endocrine Dysfunction Following the Use of Different Immune Checkpoint Inhibitor Regimens a Systematic Review and Meta-Analysis. JAMA Oncol. 2018, 4, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, R.; Cheema, A.; Khan, T.; Amirpour, A.; Paul, A.; Chaughtai, S.; Patel, S.; Patel, T.; Bramson, J.; Gupta, V.; et al. Adverse Effects of Immune Checkpoint Inhibitors (Programmed Death-1 Inhibitors and Cytotoxic T-Lymphocyte-Associated Protein-4 Inhibitors): Results of a Retrospective Study. J. Clin. Med. Res. 2019, 11, 225–236. [Google Scholar] [CrossRef]
- Xu, H.; Tan, P.; Zheng, X.; Huang, Y.; Lin, T.; Wei, Q.; Ai, J.; Yang, L. Immune-related adverse events following administration of anti-cytotoxic T-lymphocyte-associated protein-4 drugs: A comprehensive systematic review and meta-analysis. Drug Des. Devel. Ther. 2019, 13, 2215–2234. [Google Scholar] [CrossRef]
- Lu, J.; Yang, J.; Liang, Y.; Meng, H.; Zhao, J.; Zhang, X. Incidence of Immune Checkpoint Inhibitor-Associated Diabetes: A Meta-Analysis of Randomized Controlled Studies. Front. Pharmacol. 2019, 10, 1453. [Google Scholar] [CrossRef]
- Ji, H.H.; Tang, X.W.; Dong, Z.; Song, L.; Jia, Y.T. Adverse Event Profiles of Anti-CTLA-4 and Anti-PD-1 Monoclonal Antibodies Alone or in Combination: Analysis of Spontaneous Reports Submitted to FAERS. Clin. Drug Investig. 2019, 9, 319–330. [Google Scholar] [CrossRef]
- Zhai, Y.; Ye, X.; Hu, F.; Xu, J.; Guo, X.; Zhuang, Y.; He, J. Endocrine toxicity of immune checkpoint inhibitors: A real-world study leveraging US Food and Drug Administration adverse events reporting system. J. Immunother. Cancer 2019, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Affinati, A.H.; Lee, Y.; Turcu, A.F.; Henry, N.L.; Schiopu, E.; Qin, A.; Othus, M.; Clauw, D.; Ramnath, N.; et al. Immune Checkpoint Inhibitors and Risk of Type 1 Diabetes. Diabetes Care 2022, 45, 1170–1176. [Google Scholar] [CrossRef]
- Perdigoto, A.L.; Quandt, Z.; Anderson, M.; Herold, K.C. Checkpoint inhibitor-induced insulin-dependent diabetes: An emerging syndrome. Lancet Diabetes Endocrinol. 2019, 7, 421–423. [Google Scholar] [CrossRef]
- Rodríguez de Vera-Gómez, P.; Piñar-Gutiérrez, A.; Guerrero-Vázquez, R.; Bellido, V.; Morales-Portillo, C.; Sancho-Márquez, M.P.; Espejo-García, P.; Gros-Herguido, N.; López-Gallardo, G.; Martínez-Brocca, M.A.; et al. Flash Glucose Monitoring and Diabetes Mellitus Induced by Immune Checkpoint Inhibitors: An Approach to Clinical Practice. J. Diabetes Res. 2022, 2022, 4508633. [Google Scholar] [CrossRef] [PubMed]
- Byun, D.J.; Braunstein, R.; Flynn, J.; Zheng, J.; Lefkowitz, R.A.; Kanbour, S.; Girotra, M. Immune Checkpoint Inhibitor–Associated Diabetes: A Single-Institution Experience. Diabetes Care 2020, 43, 3106–3109. [Google Scholar] [CrossRef]
- Liu, J.; Shi, Y.; Liu, X.; Zhang, D.; Zhang, H.; Chen, M.; Xu, Y.; Zhao, J.; Zhong, W.; Wang, M. Clinical characteristics and outcomes of immune checkpoint inhibitor-induced diabetes mellitus. Transl. Oncol. 2022, 24, 101473. [Google Scholar] [CrossRef] [PubMed]
- Kyriacou, A.; Melson, E.; Chen, W.; Kempegowda, P. Is immune checkpoint inhibitor-associated diabetes the same as fulminant type 1 diabetes mellitus? Clin. Med. 2020, 20, 417–423. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. National Comprehensive Cancer Network (2018). Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef]
- Smati, S.; Buffier, P.; Bouillet, B.; Archambeaud, F.; Vergès, B.; Cariou, B. Expert opinion on immunotherapy induced diabetes. Ann. Endocrinol. 2018, 79, 545–549. [Google Scholar] [CrossRef]
- Castinetti, F.; Albarel, F.; Archambeaud, F.; Bertherat, J.; Bouillet, B.; Buffier, P.; Briet, C.; Cariou, B.; Caron, P.; Chabre, O.; et al. French Endocrine Society Guidance on endocrine side effects of immunotherapy. Endocr. Relat. Cancer 2019, 26, G1–G18. [Google Scholar] [CrossRef]
- Holt, R.I.G.; DeVries, J.H.; Hess-Fischl, A.; Hirsch, I.B.; Kirkman, M.S.; Klupa, T.; Ludwig, B.; Nørgaard, K.; Pettus, J.; Renard, E.; et al. The Management of Type 1 Diabetes in Adults. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2021, 44, 2589–2625. [Google Scholar] [CrossRef]
- Schneider, B.J.; Naidoo, J.; Santomasso, B.D.; Lacchetti, C.; Adkins, S.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 4073–4126. [Google Scholar] [CrossRef] [PubMed]
- Dhatariya, K.K.; Joint British Diabetes Societies for Inpatient Care. The management of diabetic ketoacidosis in adults—An updated guideline from the Joint British Diabetes Society for Inpatient Care. Diabet. Med. 2022, 39, e14788. [Google Scholar] [CrossRef]
- Eledrisi, M.S.; Elzouki, A.N. Management of Diabetic Ketoacidosis in Adults: A Narrative Review. Saudi J. Med. Med. Sci. 2020, 8, 165–173. [Google Scholar] [PubMed]
- Imagawa, A.; Hanafusa, T. Fulminant type 1 diabetes—An important subtype in East Asia. Diabetes Metab. Res. Rev. 2011, 27, 959–964. [Google Scholar] [CrossRef]
- Roy, A.; Sahoo, J.; Kamalanathan, S.; Naik, D.; Mohan, P.; Kalayarasan, R. Diabetes and pancreatic cancer: Exploring the two-way traffic. World J. Gastroenterol. 2021, 27, 4939–4962. [Google Scholar] [CrossRef]
- Duan, X.; Wang, W.; Pan, Q.; Guo, L. Type 2 Diabetes Mellitus Intersects with Pancreatic Cancer Diagnosis and Development. Front. Oncol. 2021, 11, 730038. [Google Scholar] [CrossRef] [PubMed]
- Yim, C.; Mansell, K.; Hussein, N.; Arnason, T. Current cancer therapies and their influence on glucose control. World J. Diabetes. 2021, 12, 1010–1025. [Google Scholar] [CrossRef]
- Joharatnam-Hogan, N.; Carter, T.J.; Reynolds, N.; Ho, J.H.; Adam, S.; Board, R. Diabetes Mellitus in People with Cancer. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Pinheiro, L.C.; Kaur, H.; Nilo, D.; Safford, M.M.; DeRosa, A.P.; Kern, L.M. Determining the Impact of a Cancer Diagnosis on Diabetes Management: A Systematic Literature Review. Am. J. Clin. Oncol. 2019, 42, 870–883. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Standards of Medical Care in Diabetes—2020; Abridged for Primary Care Providers. Clin. Diabetes 2020, 38, 10–38. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Abu-Sbeih, H.; Ascierto, P.A.; Brufsky, J.; Cappelli, L.C.; Cortazar, F.B.; Gerber, D.E.; Hamad, L.; Hansen, E.; Johnson, D.B.; et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J. Immunother. Cancer 2021, 9, e002435. [Google Scholar] [CrossRef]
- Okubo, M.; Hataya, Y.; Fujimoto, K.; Iwakura, T.; Matsuoka, N. Recovery from insulin dependence in immune checkpoint inhibitor-associated diabetes mellitus: A case report. J. Diabetes Investig. 2023, 14, 147–150. [Google Scholar] [CrossRef]
- Chennamadhavuni, A.; Abushahin, L.; Jin, N.; Presley, C.J.; Manne, A. Risk Factors and Biomarkers for Immune-Related Adverse Events: A Practical Guide to Identifying High-Risk Patients and Rechallenging Immune Checkpoint Inhibitors. Front. Immunol. 2022, 13, 779691. [Google Scholar] [CrossRef]
- Allouchery, M.; Lombard, T.; Martin, M.; Rouby, F.; Sassier, M.; Bertin, C.; Atzenhoffer, M.; Miremont-Salame, G.; Perault-Pochat, M.C.; Puyade, M.; et al. Safety of immune checkpoint inhibitor rechallenge after discontinuation for grade ≥ 2 immune-related adverse events in patients with cancer. J. Immunother. Cancer 2020, 8, e001622. [Google Scholar] [CrossRef] [PubMed]
- Simonaggio, A.; Michot, J.M.; Voisin, A.L.; Le Pavec, J.; Collins, M.; Lallart, A.; Cengizalp, G.; Vozy, A.; Laparra, A.; Varga, A.; et al. Evaluation of Readministration of Immune Checkpoint Inhibitors After Immune-Related Adverse Events in Patients with Cancer. JAMA Oncol. 2019, 5, 1310–1317. [Google Scholar] [CrossRef] [PubMed]
- Haratani, K.; Hayashi, H.; Nakagawa, K. Association of immune-related adverse events with immune checkpoint inhibitor efficacy: Real or imaginary? BMC Med. 2020, 18, 111. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, H.; Murakami, E.; Oyanagi, J.; Shibaki, R.; Kaki, T.; Takase, E.; Tanaka, M.; Harutani, Y.; Yamagata, N.; Okuda, Y.; et al. Immune-Related Adverse Events by Immune Checkpoint Inhibitors Significantly Predict Durable Efficacy Even in Responders with Advanced Non-Small Cell Lung Cancer. Oncologist 2020, 25, e679–e683. [Google Scholar] [CrossRef]
- Zhou, X.; Yao, Z.; Yang, H.; Liang, N.; Zhang, X.; Zhang, F. Are immune-related adverse events associated with the efficacy of immune checkpoint inhibitors in patients with cancer? A systematic review and meta analysis. BMC Med. 2020, 18, 87. [Google Scholar] [CrossRef]
- Rogado, J.; Sánchez-Torres, J.M.; Romero-Laorden, N.; Ballesteros, A.I.; Pacheco-Barcia, V.; Ramos-Leví, A.; Arranz, R.; Lorenzo, A.; Gullón, P.; Donnay, O.; et al. Immune-related adverse events predict the therapeutic efficacy of anti-PD-1 antibodies in cancer patients. Eur. J. Cancer 2019, 109, 21–27. [Google Scholar] [CrossRef]
- Wang, D.; Chen, C.; Gu, Y.; Lu, W.; Zhan, P.; Liu, H.; Lv, T.; Song, Y.; Zhang, F. Immune-Related Adverse Events Predict the Efficacy of Immune Checkpoint Inhibitors in Lung Cancer Patients: A Meta-Analysis. Front. Oncol. 2021, 11, 631949. [Google Scholar] [CrossRef]
- Gunjur, A.; Klein, O.; Kee, D.; Cebon, J. Anti-programmed cell death protein 1 (anti-PD1) immunotherapy induced autoimmune polyendocrine syndrome type II (APS-2): A case report and review of the literature. J. Immunother. Cancer 2019, 7, 241. [Google Scholar] [CrossRef]
- Ebrahim, E.; Teklu, T.; Tajebe, F.; Wondmagegn, T.; Akelew, Y.; Fiseha, M. Association of Cytotoxic T-Lymphocyte Antigen-4 Gene Polymorphism with Type 1 Diabetes Mellitus: In silico Analysis of Biological Features of CTLA-4 Protein on Ethiopian Population. Diabetes Metab. Syndr. Obes. 2022, 15, 2733–2751. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, S.; Gu, Y.; Feng, Y.; Shi, Y.; Fu, Q.; Wang, Z.; Cai, Y.; Dai, H.; Zheng, S.; et al. CTLA-4 +49 G/A, a functional T1D risk SNP, affects CTLA-4 level in Treg subsets and IA-2A positivity, but not beta-cell function. Sci. Rep. 2018, 8, 10074. [Google Scholar] [CrossRef]
- Ren, D.; He, L.; Pang, X. Association of CLTA-4 Gene Polymorphisms with Diabetes Mellitus: A Study Based on the Han Population of Northern China. Diabetes Metab. Syndr Obes. 2022, 15, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.O.; Petrov, M.S.; Andersen, D.K.; Hart, P.A. Diabetes in chronic pancreatitis: Risk factors and natural history. Curr. Opin. Gastroenterol. 2021, 37, 526–531. [Google Scholar] [CrossRef]
- Xu, Y.; Fu, Y.; Zhu, B.; Wang, J.; Zhang, B. Predictive Biomarkers of Immune Checkpoint Inhibitors-Related Toxicities. Front. Immunol. 2020, 11, 2023. [Google Scholar] [CrossRef] [PubMed]
- Curran, C.S.; Sommers, C.L.; Young, H.A.; Bourcier, K.; Mancini, M.; Sharon, E. Report on the 2018 Cancer, Autoimmunity, and Immunology Conference. J. Immunol. 2019, 202, 2823–2828. [Google Scholar] [CrossRef]
- Nixon, A.B.; Schalper, K.A.; Jacobs, I.; Potluri, S.; Wang, I.M.; Fleener, C. Peripheral immune-based biomarkers in cancer immunotherapy: Can we realize their predictive potential? J. Immunother. Cancer 2019, 7, 325. [Google Scholar] [CrossRef]
- Januszewski, A.S.; Cho, Y.H.; Joglekar, M.V.; Farr, R.J.; Scott, E.S.; Wong, W.K.M.; Carroll, L.M.; Loh, Y.W.; Benitez-Aguirre, P.Z.; Keech, A.C.; et al. Insulin micro-secretion in Type 1 diabetes and related microRNA profiles. Sci. Rep. 2021, 11, 11727. [Google Scholar] [CrossRef]
- Tanvir Ahmed, K.; Cheng, S.; Li, Q.; Yong, J.; Zhang, W. Incomplete time-series gene expression in integrative study for islet autoimmunity prediction. Brief. Bioinform. 2023, 24, bbac537. [Google Scholar] [CrossRef]
- Gowen, M.F.; Giles, K.M.; Simpson, D.; Tchack, J.; Zhou, H.; Moran, U.; Dawood, Z.; Pavlick, A.C.; Hu, S.; Wilson, M.A.; et al. Baseline antibody profiles predict toxicity in melanoma patients treated with immune checkpoint inhibitors. J. Transl. Med. 2018, 16, 82. [Google Scholar] [CrossRef]
- Hassel, J.C.; Luke, J.J. Autoantibodies as Predictors for Clinical Outcome and Toxicity for Immunotherapy. Clin. Cancer Res. 2022, 28, 3914–3916. [Google Scholar] [CrossRef]
- Johannet, P.; Liu, W.; Fenyo, D.; Wind-Rotolo, M.; Krogsgaard, M.; Mehnert, J.M.; Weber, J.S.; Zhong, J.; Osman, I. Baseline Serum Autoantibody Signatures Predict Recurrence and Toxicity in Melanoma Patients Receiving Adjuvant Immune Checkpoint Blockade. Clin. Cancer Res. 2022, 28, 4121–4130. [Google Scholar] [CrossRef]
- Redondo, M.J.; Geyer, S.; Steck, A.K.; Sharp, S.; Wentworth, J.M.; Weedon, M.N.; Antinozzi, P.; Sosenko, J.; Atkinson, M.; Pugliese, A.; et al. Type 1 Diabetes TrialNet Study Group. A Type 1 Diabetes Genetic Risk Score Predicts Progression of Islet Autoimmunity and Development of Type 1 Diabetes in Individuals at Risk. Diabetes Care 2018, 41, 1887–1894. [Google Scholar] [CrossRef]
- Khan, Z.; Hammer, C.; Guardino, E.; Chandler, G.S.; Albert, M.L. Mechanisms of immune-related adverse events associated with immune checkpoint blockade: Using germline genetics to develop a personalized approach. Genome Med. 2019, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Huo, G.W.; Zhu, F.Y.; Yue, P.; Yuan, D.Q.; Chen, P. Safety and efficacy of immune checkpoint inhibitors in advanced cancer patients with autoimmune disease: A meta-analysis. Hum. Vaccin. Immunother. 2022, 18, 2145102. [Google Scholar] [CrossRef]
- Heinzerling, L.; Ascierto, P.A.; Dummer, R.; Gogas, H.; Grob, J.J.; Lebbe, C.; Long, G.V.; McArthur, G.; Moslehi, J.J.; Neilan, T.G.; et al. Adverse events 2.0-Let us get SERIOs: New reporting for adverse event outcomes needed in the era of immuno-oncology. Eur. J. Cancer 2019, 112, 29–31. [Google Scholar] [CrossRef]
- Nuzzo, P.V.; Pond, G.R.; Abou Alaiwi, S.; Nassar, A.H.; Flippot, R.; Curran, C.; Kilbridge, K.L.; Wei, X.X.; McGregor, B.A.; Choueiri, T.; et al. Conditional immune toxicity rate in patients with metastatic renal and urothelial cancer treated with immune checkpoint inhibitors. J. Immunother. Cancer 2020, 8, e000371. [Google Scholar] [CrossRef]
- Das, S.; Johnson, D.B. Immune-related adverse events, and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sykiotis, G.P.; Maillard, M.; Fraga, M.; Ribi, C.; Kuntzer, T.; Michielin, O.; Peters, S.; Coukos, G.; Spertini, F.; et al. New therapeutic perspectives to manage refractory immune checkpoint-related toxicities. Lancet Oncol. 2019, 20, e54–e64. [Google Scholar] [CrossRef]
- Arutyunyan, I.V.; Fatkhudinov, T.K.; Makarov, A.V.; Elchaninov, A.V.; Sukhikh, G.T. Regenerative medicine of pancreatic islets. World J. Gastroenterol. 2020, 26, 2948–2966. [Google Scholar] [CrossRef]
- Porter, J.M.; Guerassimoff, L.; Castiello, F.R.; Charette, A.; Tabrizian, M. INGAP-Peptide Variants as a Novel Therapy for Type 1 Diabetes: Effect on Human Islet Insulin Secretion and Gene Expression. Pharmaceutics 2022, 14, 1833. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Z.T.; Liu, L.; Cheng, M.Z.; Gao, Y.; Zhou, W.J. The Effects of 6 Common Antidiabetic Drugs on Anti-PD1 Immune Checkpoint Inhibitor in Tumor Treatment. J. Immunol. Res. 2022, 2022, 2651790. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Wang, Y.; Riese, M.J.; You, M. Uncoupling Therapeutic Efficacy from Immune-Related Adverse Events in Immune Checkpoint Blockade. iScience 2020, 23, 101580. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target Ther. 2022, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Thoreau, F.; Chudasama, V. Enabling the next steps in cancer immunotherapy: From antibody-based bispecifics to multispecifics, with an evolving role for bioconjugation chemistry. RSC Chem. Biol. 2021, 3, 140–169. [Google Scholar] [CrossRef]
- Jacobi, O.; Landman, Y.; Reinhorn, D.; Icht, O.; Sternschuss, M.; Rotem, O.; Finkel, I.; Allen, A.M.; Dudnik, E.; Goldstein, D.A.; et al. The Relationship of Diabetes Mellitus to Efficacy of Immune Checkpoint Inhibitors in Patients with Advanced Non-Small Cell Lung Cancer. Oncology 2021, 99, 555–561. [Google Scholar] [CrossRef]
- Shahid, R.K.; Ahmed, S.; Le, D.; Yadav, S. Diabetes and Cancer: Risk, Challenges, Management and Outcomes. Cancers 2021, 13, 5735. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Type [Ref] | Methods/Patients | Results |
|---|---|---|
| Systematic review [8] | Search of PubMed through 22 August 2017 regarding endocrine irAEs yielded 101early phase I/II, phase III experimental trials, prospective and retrospective observational studies, comprising 19,922 patients. |
|
| Pharmaco-vigilance study [19] | Pharmacovigilance analysis of the VigiBase from 2014 to April 2018 revealed 283 cases of irT1DM |
|
| Retrospective study [52] | Ρeview of cases of irDM occurring over a 6-year period (2012–2018) at two academic institutions yielded 27 patients with irDM. |
|
| Retrospective study [53] | Study of 538 patients with metastatic melanoma treated with anti-PD-1-based immunotherapy from March 2015 to March 2018 in a single quaternary melanoma center |
|
| Retrospective study [54] | Review of electronic medical record of 1327 adult patients who received anti-PD-(L)1 or anti-CTLA-4 mAbs from 2013 to 2018. |
|
| Case report of irDKA and systematic review [61] | Search of PubMed, Web of Science, and Cochrane, through November 2018 for cases of autoimmune DM related to ICPi yielded 90 irDM cases. |
|
| Retrospective study [62] | Review of 1444 patients treated with ICPi in a single center from January 2012 to December 2017 revealed 1163 patients treated with anti-PD-1 among whom 21 patients developed irDM |
|
| Systematic review and meta-analysis [94] | Search of PubMed through July 18, 2016 regarding endocrine irAEs yielded 38 randomized clinical trials comprising 7551 patients eligible for a meta-analysis. |
|
| Retrospective study [95] | Search of PubMed between January 2016 and April 2018 regarding irAEs yielded 101 publications, which reported 139 cases of irAEs. |
|
| Systematic review and meta-analysis [96] | Search of PubMed, Cochrane library, Web of Science, and ClinicalTrials.gov between January 1990 and March 2018 regarding studies reporting irAEs related to anti-CTLA-4 mAbs yielded a total of 11 clinical trials with 7088 patients of whom 10 clinical trials had data accessible on ClinicalTrials.gov. |
|
| Meta-analysis [97] | Search of PubMed, EMBASE, Cochrane Library databases, and ClinicalTrials.gov from the establishment of ICPi to March 2019 for RCTs regarding irDM yielded 40 trials reporting at least one irDM event, comprising 24,596 patients. |
|
| Pharmaco-vigilance study [98] | Detection of signals of irAEs using the US Food and Drug Administration (FDA) adverse events (AEs) Reporting System (FAERS) database from the respective FDA approval dates for each specific drug through the second quarter of 2017. |
|
| Pharmaco-vigilance study [100] | Review of Optum’s Clinformatics Data Mart database to assess T1DM incidence and characteristics in a large de-identified cohort of ICPi-treated patients between 2017 and 2020, encompassing 30,337 patients |
|
| Ref | Range of Time Interval between ICPi Initiation and irDM Diagnosis | Median Onset Time of irDM Diagnosis | Number of ICPi Cycles at the Time of irDM Diagnosis |
|---|---|---|---|
| [12] | 1 to 52 weeks | NA | Median: 3 cycles Range: 1 to 17 cycles |
| [19] | 5 to 790 days | 116 days | Median: 3 cycles Range: 1 to 24 cycles. |
| [53] | IQR: 17.5 to 34.5 weeks. | 25 weeks | NA |
| [54] | 20 to 972 days | NA | NA |
| [55] | 5 to 448 days | NA | NA |
| [63] | NA | 7.5 weeks | NA |
| [102] | 3.6 to 45 weeks | 8.14 weeks | Mean: 4.3 (SD: 2.6) Median: 3 cycles Range: 2 to 8 cycles. |
| [104] | 0 to 122 weeks | 12 weeks | NA |
| Parameter | Diagnosis | |
|---|---|---|
| Prediabetes | Diabetes Mellitus | |
| FPG a | IFG: 100–125 mg/dL (5.6–6.9 mmol/L) AND/OR | ≥126 mg/dL (7.0 mmol/L). OR |
| 2-h PG b | IGT: 140–199 mg/dL (7.8–11.0 mmol/L) AND/OR | ≥200 mg/dL (11.1 mmol/L) during OGTT. OR |
| HbA1c c | 5.7–6.4% (39–47 mmol/mol) or ≥10% increase in HbA1C | ≥6.5% (48 mmol/mol). OR |
| RPG | NA | ≥200 mg/dL (11.1 mmol/L) plus symptoms (polyuria, polyuria, polydipsia, weight loss, fatigue) |
| Expert Committees’ Guidelines for irDM Treatment | ||||
|---|---|---|---|---|
| (Year of Release) | ||||
| [Ref] | ||||
| Clinical practice guideline for the management of irAEs in patients treated with ICPi by ASCO in collaboration with NCCN (2018) [106] | Management of irAEs in Patients Treated with Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update (2021) [110] | Management of Immunotherapy-Related Toxicities, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. (2019) [6] | French Endocrine Society Guidance on endocrine side effects of immunotherapy. (2019) [108] | Expert opinion on immunotherapy induced diabetes (2018) [107] |
G1:
|
|
|
|
|
| Challenge | Future Perspective |
|---|---|
| Scientific-centered aspect of concerted vision for irDM | |
| Understanding the pathophysiology of irDM | |
|
|
| Patient-centered aspect of concerted vision for irDM | |
| Awareness and Diagnosis | |
|
|
|
|
|
|
| Treatment | |
|
|
| Monitoring | |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deligiorgi, M.V.; Trafalis, D.T. A Concerted Vision to Advance the Knowledge of Diabetes Mellitus Related to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2023, 24, 7630. https://doi.org/10.3390/ijms24087630
Deligiorgi MV, Trafalis DT. A Concerted Vision to Advance the Knowledge of Diabetes Mellitus Related to Immune Checkpoint Inhibitors. International Journal of Molecular Sciences. 2023; 24(8):7630. https://doi.org/10.3390/ijms24087630
Chicago/Turabian StyleDeligiorgi, Maria V., and Dimitrios T. Trafalis. 2023. "A Concerted Vision to Advance the Knowledge of Diabetes Mellitus Related to Immune Checkpoint Inhibitors" International Journal of Molecular Sciences 24, no. 8: 7630. https://doi.org/10.3390/ijms24087630