
Epigenetic Alterations from Barrett’s Esophagus to Esophageal Adenocarcinoma

Abstract
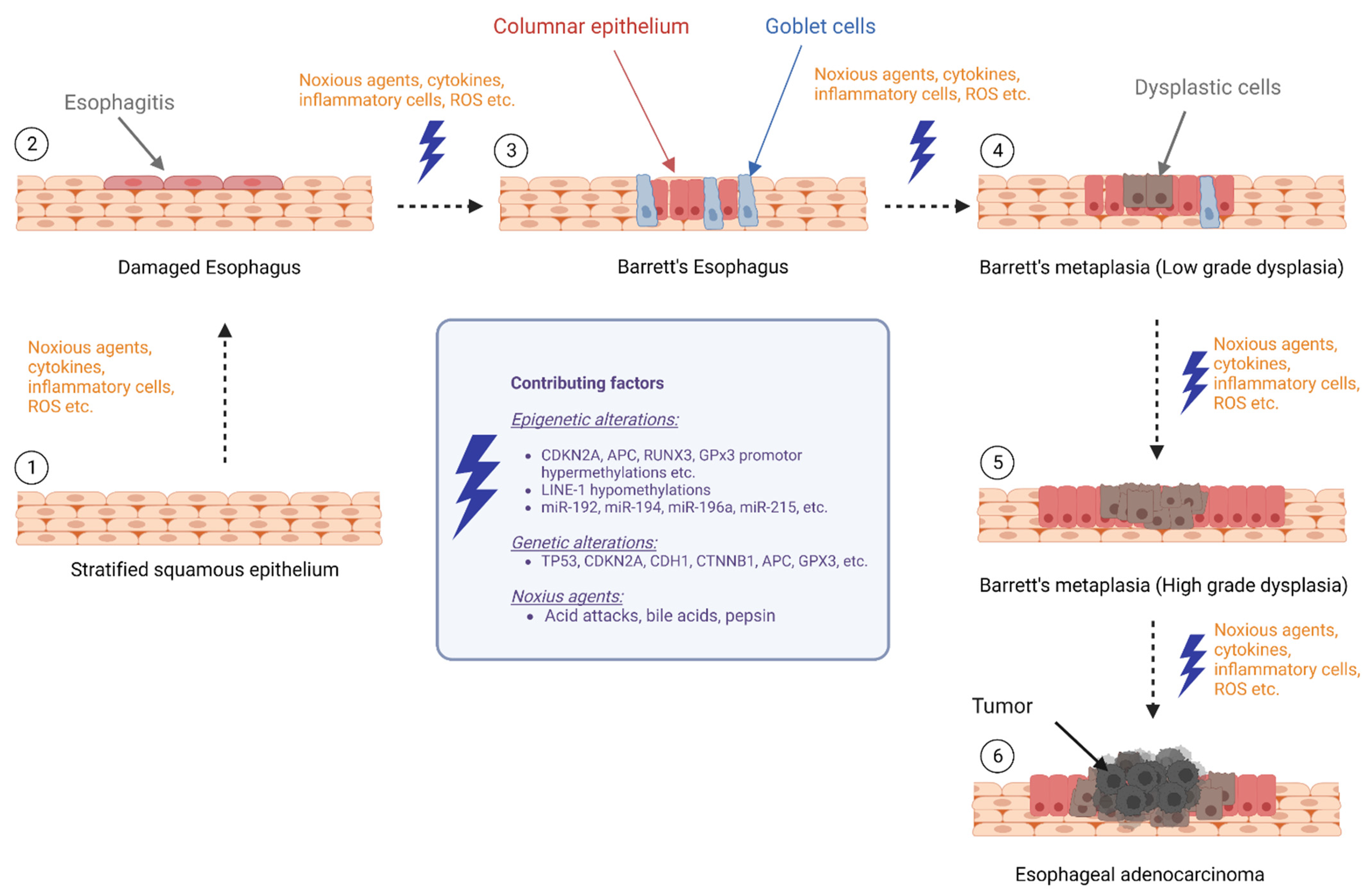
:1. Introduction
1.1. DNA Methylation as Biomarkers BE and EAC
1.2. DNA Hypermethylation Is a Frequent Event in BE and EAC
1.3. DNA Hypomethylation in BE and EAC
1.4. Histone Modifications in BE and EAC Pathogenesis
1.5. The Role of miRNAs as Biomarkers in BE and EAC
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Katzka, D.A.; Pandolfino, J.E.; Kahrilas, P.J. Phenotypes of Gastroesophageal Reflux Disease: Where Rome, Lyon, and Montreal Meet. Clin. Gastroenterol. Hepatol. 2020, 18, 767–776. [Google Scholar] [CrossRef]
- Barrett, N.R. Chronic Peptic Ulcerz of the Œophagus and ‘Œsophagitis’. Br. J. Surg. 2005, 38, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Eusebi, L.H.; Cirota, G.G.; Zagari, R.M.; Ford, A.C. Global Prevalence of Barrett’s Oesophagus and Oesophageal Cancer in Individuals with Gastro-Oesophageal Reflux: A Systematic Review and Meta-Analysis. Gut 2021, 70, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Bor, S.; Saritas Yuksel, E. How Is the Gastroesophageal Reflux Disease Prevalence, Incidence, and Frequency of Complications (Stricture/Esophagitis/Barrett’s Esophagus/Carcinoma) in Turkey Compared to Other Geographical Regions Globally? Turk. J. Gastroenterol. 2017, 28, 4–9. [Google Scholar] [CrossRef]
- Shimamura, Y.; Iwaya, Y.; Goda, K.; Teshima, C.W. Endoscopic Treatment of Barrett’s Esophagus: What Can We Learn from the Western Perspective? Dig. Endosc. 2018, 30, 182–191. [Google Scholar] [CrossRef]
- Snider, E.J.; Kaz, A.M.; Inadomi, J.M.; Grady, W.M. Chemoprevention of Esophageal Adenocarcinoma. Gastroenterol. Rep. 2020, 8, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Solaymani-Dodaran, M. Risk of Oesophageal Cancer in Barrett’s Oesophagus and Gastro-Oesophageal Reflux. Gut 2004, 53, 1070–1074. [Google Scholar] [CrossRef][Green Version]
- Haiyu, Z.; Xiaofeng, P.; Xiangqiong, M.; Junlan, Q.; Xiaobin, Z.; Shuncong, W.; Huanhuan, S.; Haiqing, M. Incidence and Survival Changes in Patients with Esophageal Adenocarcinoma during 1984–2013. Biomed. Res. Int. 2019, 2019, 7431850. [Google Scholar] [CrossRef][Green Version]
- Then, E.O.; Lopez, M.; Saleem, S.; Gayam, V.; Sunkara, T.; Culliford, A.; Gaduputi, V. Esophageal Cancer: An Updated Surveillance Epidemiology and End Results Database Analysis. World J. Oncol. 2020, 11, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Maslyonkina, K.S.; Konyukova, A.K.; Alexeeva, D.Y.; Sinelnikov, M.Y.; Mikhaleva, L.M. Barrett’s Esophagus: The Pathomorphological and Molecular Genetic Keystones of Neoplastic Progression. Cancer Med. 2022, 11, 447–478. [Google Scholar] [CrossRef]
- Yu, M.; Moinova, H.R.; Willbanks, A.; Cannon, V.K.; Wang, T.; Carter, K.; Kaz, A.; Reddi, D.; Inadomi, J.; Luebeck, G.; et al. Novel DNA Methylation Biomarker Panel for Detection of Esophageal Adenocarcinoma and High-Grade Dysplasia. Clin. Cancer Res. 2022, 28, 3761–3769. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Yao, Z.; Fang, W.; He, Q.; Xu, W.W.; Li, B. Epigenetics in Esophageal Cancer: From Mechanisms to Therapeutics. Small Methods 2020, 4, 2000391. [Google Scholar] [CrossRef]
- Dilworth, M.P.; Nieto, T.; Stockton, J.D.; Whalley, C.M.; Tee, L.; James, J.D.; Noble, F.; Underwood, T.J.; Hallissey, M.T.; Hejmadi, R.; et al. Whole Genome Methylation Analysis of Nondysplastic Barrett Esophagus That Progresses to Invasive Cancer. Ann. Surg. 2019, 269, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Kaz, A.M.; Grady, W.M.; Stachler, M.D.; Bass, A.J. Genetic and Epigenetic Alterations in Barrett’s Esophagus and Esophageal Adenocarcinoma. Gastroenterol. Clin. N. Am. 2015, 44, 473–489. [Google Scholar] [CrossRef][Green Version]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Kim, J.K.; Samaranayake, M.; Pradhan, S. Epigenetic Mechanisms in Mammals. Cell. Mol. Life Sci. 2009, 66, 596. [Google Scholar] [CrossRef][Green Version]
- Leighton, G.; Williams, D.C. The Methyl-CpG–Binding Domain 2 and 3 Proteins and Formation of the Nucleosome Remodeling and Deacetylase Complex. J. Mol. Biol. 2020, 432, 1624–1639. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA Methylation in Cancer: Too Much, but Also Too Little. Oncogene 2002, 21, 5400–5413. [Google Scholar] [CrossRef][Green Version]
- Xu, E.; Gu, J.; Hawk, E.T.; Wang, K.K.; Lai, M.; Huang, M.; Ajani, J.; Wu, X. Genome-Wide Methylation Analysis Shows Similar Patterns in Barrett’s Esophagus and Esophageal Adenocarcinoma. Carcinogenesis 2013, 34, 2750–2756. [Google Scholar] [CrossRef][Green Version]
- Kalatskaya, I. Overview of Major Molecular Alterations during Progression from Barrett’s Esophagus to Esophageal Adenocarcinoma. Ann. N. Y. Acad. Sci. 2016, 1381, 74–91. [Google Scholar] [CrossRef]
- Bian, Y.; Osterheld, M.; Fontolliet, C.; Bosman, F.T.; Benhattar, J. P16 Inactivation by Methylation of the CDKN2A Promoter Occurs Early during Neoplastic Progression in Barrett’s Esophagus. Gastroenterology 2002, 122, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Klump, B.; Hsieh, C.-J.; Holzmann, K.; Gregor, M.; Porschen, R. Hypermethylation of the CDKN2/P16 Promoter during Neoplastic Progression in Barrett’s Esophagus. Gastroenterology 1998, 115, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Maley, C.C.; Galipeau, P.C.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Reid, B.J. Selectively Advantageous Mutations and Hitchhikers in Neoplasms. Cancer Res. 2004, 64, 3414–3427. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schulmann, K.; Sterian, A.; Berki, A.; Yin, J.; Sato, F.; Xu, Y.; Olaru, A.; Wang, S.; Mori, Y.; Deacu, E.; et al. Inactivation of P16, RUNX3, and HPP1 Occurs Early in Barrett’s-Associated Neoplastic Progression and Predicts Progression Risk. Oncogene 2005, 24, 4138–4148. [Google Scholar] [CrossRef][Green Version]
- Vieth, M.; Schneider-Stock, R.; Röhrich, K.; May, A.; Ell, C.; Markwarth, A.; Roessner, A.; Stolte, M.; Tannapfel, A. INK4a-ARF Alterations in Barrett?S Epithelium, Intraepithelial Neoplasia and Barrett?S Adenocarcinoma. Virchows Arch. 2004, 445, 135–141. [Google Scholar] [CrossRef]
- Wong, D.J.; Barrett, M.T.; Stöger, R.; Emond, M.J.; Reid, B.J. P16INK4a Promoter Is Hypermethylated at a High Frequency in Esophageal Adenocarcinomas. Cancer Res. 1997, 57, 2619–2622. [Google Scholar]
- Sarbia, M.; Geddert, H.; Klump, B.; Kiel, S.; Iskender, E.; Gabbert, H.E. Hypermethylation of Tumor Suppressor Genes (P16INK4A,P14ARF AndAPC) in Adenocarcinomas of the Upper Gastrointestinal Tract. Int. J. Cancer 2004, 111, 224–228. [Google Scholar] [CrossRef]
- Jin, Z.; Hamilton, J.P.; Yang, J.; Mori, Y.; Olaru, A.; Sato, F.; Ito, T.; Kan, T.; Cheng, Y.; Paun, B.; et al. Hypermethylation of the AKAP12 Promoter Is a Biomarker of Barrett’s-Associated Esophageal Neoplastic Progression. Cancer Epidemiol. Biomark. Prev. 2008, 17, 111–117. [Google Scholar] [CrossRef][Green Version]
- Clément, G.; Braunschweig, R.; Pasquier, N.; Bosman, F.T.; Benhattar, J. Methylation OfAPC, TIMP3, AndTERT: A New Predictive Marker to Distinguish Barrett’s Oesophagus Patients at Risk for Malignant Transformation. J. Pathol. 2006, 208, 100–107. [Google Scholar] [CrossRef]
- Wang, J.S.; Guo, M.; Montgomery, E.A.; Thompson, R.E.; Cosby, H.; Hicks, L.; Wang, S.; Herman, J.G.; Canto, M.I. DNA Promoter Hypermethylation of P16 and APC Predicts Neoplastic Progression in Barrett’s Esophagus. Am. J. Gastroenterol. 2009, 104, 2153–2160. [Google Scholar] [CrossRef][Green Version]
- Eads, C.A.; Lord, R.V.; Kurumboor, S.K.; Wickramasinghe, K.; Skinner, M.L.; Long, T.I.; Peters, J.H.; DeMeester, T.R.; Danenberg, K.D.; Danenberg, P.V.; et al. Fields of Aberrant CpG Island Hypermethylation in Barrett’s Esophagus and Associated Adenocarcinoma. Cancer Res. 2000, 60, 5021–5026. [Google Scholar] [PubMed]
- Brock, M.V.; Gou, M.; Akiyama, Y.; Muller, A.; Wu, T.-T.; Montgomery, E.; Deasel, M.; Germonpré, P.; Rubinson, L.; Heitmiller, R.F.; et al. Prognostic Importance of Promoter Hypermethylation of Multiple Genes in Esophageal Adenocarcinoma. Clin. Cancer Res. 2003, 9, 2912–2919. [Google Scholar] [PubMed]
- Smith, E.; De Young, N.J.; Pavey, S.J.; Hayward, N.K.; Nancarrow, D.J.; Whiteman, D.C.; Smithers, B.M.; Ruszkiewicz, A.R.; Clouston, A.D.; Gotley, D.C.; et al. Similarity of Aberrant DNA Methylation in Barrett’s Esophagus and Esophageal Adenocarcinoma. Mol. Cancer 2008, 7, 75. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eads, C.A.; Lord, R.V.; Wickramasinghe, K.; Long, T.I.; Kurumboor, S.K.; Bernstein, L.; Peters, J.H.; DeMeester, S.R.; DeMeester, T.R.; Skinner, K.A.; et al. Epigenetic Patterns in the Progression of Esophageal Adenocarcinoma. Cancer Res. 2001, 61, 3410–3418. [Google Scholar]
- Kawakami, K.; Brabender, J.; Lord, R.V.; Groshen, S.; Greenwald, B.D.; Krasna, M.J. Hypermethylated APC DNA in Plasma and Prognosis of Patients With Esophageal Adenocarcinoma. J. Natl. Cancer Inst. 2000, 92, 1805–1811. [Google Scholar] [CrossRef][Green Version]
- Moinova, H.R.; LaFramboise, T.; Lutterbaugh, J.D.; Chandar, A.K.; Dumot, J.; Faulx, A.; Brock, W.; De la Cruz Cabrera, O.; Guda, K.; Barnholtz-Sloan, J.S.; et al. Identifying DNA Methylation Biomarkers for Non-Endoscopic Detection of Barrett’s Esophagus. Sci. Transl. Med. 2018, 10, eaao5848. [Google Scholar] [CrossRef][Green Version]
- Corn, P.G.; Heath, E.I.; Heitmiller, R.; Fogt, F.; Forastiere, A.A.; Herman, J.G.; Wu, T.T. Frequent Hypermethylation of the 5′ CpG Island of E-Cadherin in Esophageal Adenocarcinoma. Clin. Cancer Res. 2001, 7, 2765–2769. [Google Scholar]
- Schildhaus, H.-U.; Kröckel, I.; Lippert, H.; Malfertheiner, P.; Roessner, A.; Schneider-Stock, R. Promoter Hypermethylation of P16INK4a, E-Cadherin, O6-MGMT, DAPK and FHIT in Adenocarcinomas of the Esophagus, Esophagogastric Junction and Proximal Stomach. Int. J. Oncol. 2005, 26, 1493–1500. [Google Scholar] [CrossRef]
- Jin, Z.; Cheng, Y.; Olaru, A.; Kan, T.; Yang, J.; Paun, B.; Ito, T.; Hamilton, J.P.; David, S.; Agarwal, R.; et al. Promoter Hypermethylation of CDH13 Is a Common, Early Event in Human Esophageal Adenocarcinogenesis and Correlates with Clinical Risk Factors. Int. J. Cancer 2008, 123, 2331–2336. [Google Scholar] [CrossRef][Green Version]
- Chueca, E.; Valero, A.; Hördnler, C.; Puertas, A.; Carrera, P.; García-González, M.A.; Strunk, M.; Lanas, A.; Piazuelo, E. Quantitative Analysis of P16 Methylation in Barrett’s Carcinogenesis. Ann. Diagn. Pathol. 2020, 47, 151554. [Google Scholar] [CrossRef]
- Hardie, L.J.; Darnton, S.J.; Wallis, Y.L.; Chauhan, A.; Hainaut, P.; Wild, C.P.; Casson, A.G. P16 Expression in Barrett’s Esophagus and Esophageal Adenocarcinoma: Association with Genetic and Epigenetic Alterations. Cancer Lett. 2005, 217, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.; Hauge, T.; Jeanmougin, M.; Pharo, H.D.; Kresse, S.H.; Honne, H.; Winge, S.B.; Five, M.-B.; Kumar, T.; Mala, T.; et al. Targeted Genetic and Epigenetic Profiling of Esophageal Adenocarcinomas and Non-Dysplastic Barrett’s Esophagus. Clin. Epigenet. 2022, 14, 77. [Google Scholar] [CrossRef] [PubMed]
- Kuester, D.; Dar, A.A.; Moskaluk, C.C.; Krueger, S.; Meyer, F.; Hartig, R.; Stolte, M.; Malfertheiner, P.; Lippert, H.; Roessner, A.; et al. Early Involvement of Death-Associated Protein Kinase Promoter Hypermethylation in the Carcinogenesis of Barrett’s Esophageal Adenocarcinoma and Its Association with Clinical Progression. Neoplasia 2007, 9, 236–245. [Google Scholar] [CrossRef][Green Version]
- Zou, H.; Osborn, N.K.; Harrington, J.J.; Klatt, K.K.; Molina, J.R.; Burgart, L.J.; Ahlquist, D.A. Frequent Methylation of Eyes Absent 4 Gene in Barrett’s Esophagus and Esophageal Adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2005, 14, 830–834. [Google Scholar] [CrossRef][Green Version]
- Lee, O.-J.; Schneider-Stock, R.; McChesney, P.A.; Kuester, D.; Roessner, A.; Vieth, M.; Moskaluk, C.A.; El-Rifai, W. Hypermethylation, Loss of Expression of Glutathione Peroxidase-3 in Barrett’s Tumorigenesis. Neoplasia 2005, 7, 854–861. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peng, D.F.; Razvi, M.; Chen, H.; Washington, K.; Roessner, A.; Schneider-Stock, R.; El-Rifai, W. DNA Hypermethylation Regulates the Expression of Members of the Mu-Class Glutathione S-Transferases and Glutathione Peroxidases in Barrett’s Adenocarcinoma. Gut 2009, 58, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Kuester, D.; El-Rifai, W.; Peng, D.; Ruemmele, P.; Kroeckel, I.; Peters, B.; Moskaluk, C.A.; Stolte, M.; Mönkemüller, K.; Meyer, F.; et al. Silencing of MGMT Expression by Promoter Hypermethylation in the Metaplasia–Dysplasia–Carcinoma Sequence of Barrett’s Esophagus. Cancer Lett. 2009, 275, 117–126. [Google Scholar] [CrossRef][Green Version]
- Baumann, S.; Keller, G.; Pühringer, F.; Napieralski, R.; Feith, M.; Langer, R.; Höfler, H.; Stein, H.J.; Sarbia, M. The Prognostic Impact OfO6-Methylguanine-DNA Methyltransferase (MGMT) Promotor Hypermethylation in Esophageal Adenocarcinoma. Int. J. Cancer 2006, 119, 264–268. [Google Scholar] [CrossRef]
- Jin, Z.; Mori, Y.; Yang, J.; Sato, F.; Ito, T.; Cheng, Y.; Paun, B.; Hamilton, J.P.; Kan, T.; Olaru, A.; et al. Hypermethylation of the Nel-like 1 Gene Is a Common and Early Event and Is Associated with Poor Prognosis in Early-Stage Esophageal Adenocarcinoma. Oncogene 2007, 26, 6332–6340. [Google Scholar] [CrossRef][Green Version]
- Zou, H.; Molina, J.R.; Harrington, J.J.; Osborn, N.K.; Klatt, K.K.; Romero, Y.; Burgart, L.J.; Ahlquist, D.A. Aberrant Methylation of Secreted Frizzled-Related Protein Genes in Esophageal Adenocarcinoma and Barrett’s Esophagus. Int. J. Cancer 2005, 116, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Tischoff, I.; Hengge, U.R.; Vieth, M.; Ell, C.; Stolte, M.; Weber, A.; Schmidt, W.E.; Tannapfel, A. Methylation of SOCS-3 and SOCS-1 in the Carcinogenesis of Barrett’s Adenocarcinoma. Gut 2007, 56, 1047–1053. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jin, Z.; Olaru, A.; Yang, J.; Sato, F.; Cheng, Y.; Kan, T.; Mori, Y.; Mantzur, C.; Paun, B.; Hamilton, J.P.; et al. Hypermethylation of Tachykinin-1 Is a Potential Biomarker in Human Esophageal Cancer. Clin. Cancer Res. 2007, 13, 6293–6300. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Darnton, S.J.; Hardie, L.J.; Muc, R.S.; Wild, C.P.; Casson, A.G. Tissue Inhibitor of Metalloproteinase-3 (TIMP-3) Gene Is Methylated in the Development of Esophageal Adenocarcinoma: Loss of Expression Correlates with Poor Prognosis. Int. J. Cancer 2005, 115, 351–358. [Google Scholar] [CrossRef]
- Moinova, H.; Leidner, R.S.; Ravi, L.; Lutterbaugh, J.; Barnholtz-Sloan, J.S.; Chen, Y.; Chak, A.; Markowitz, S.D.; Willis, J.E. Aberrant Vimentin Methylation Is Characteristic of Upper Gastrointestinal Pathologies. Cancer Epidemiol. Biomark. Prev. 2012, 21, 594–600. [Google Scholar] [CrossRef][Green Version]
- Galipeau, P.C.; Li, X.; Blount, P.L.; Maley, C.C.; Sanchez, C.A.; Odze, R.D.; Ayub, K.; Rabinovitch, P.S.; Vaughan, T.L.; Reid, B.J. NSAIDs Modulate CDKN2A, TP53, and DNA Content Risk for Progression to Esophageal Adenocarcinoma. PLoS Med. 2007, 4, e67. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jin, Z.; Cheng, Y.; Gu, W.; Zheng, Y.; Sato, F.; Mori, Y.; Olaru, A.V.; Paun, B.C.; Yang, J.; Kan, T.; et al. A Multicenter, Double-Blinded Validation Study of Methylation Biomarkers for Progression Prediction in Barrett’s Esophagus. Cancer Res. 2009, 69, 4112–4115. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenführ, M.; Maertens, G.; Banck, M.; Zhou, M.-M.; Walsh, M.J.; et al. Histone Demethylase JMJD3 Contributes to Epigenetic Control of INK4a/ARF by Oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef][Green Version]
- Huang, Y.; Peters, C.J.; Fitzgerald, R.C.; Gjerset, R.A. Progressive Silencing of P14ARF in Oesophageal Adenocarcinoma. J. Cell. Mol. Med. 2009, 13, 398–409. [Google Scholar] [CrossRef]
- Antony, S.; Jiang, G.; Wu, Y.; Meitzler, J.L.; Makhlouf, H.R.; Haines, D.C.; Butcher, D.; Hoon, D.S.; Ji, J.; Zhang, Y.; et al. NADPH Oxidase 5 (NOX5)-Induced Reactive Oxygen Signaling Modulates Normoxic HIF-1α and P27 Kip1 Expression in Malignant Melanoma and Other Human Tumors. Mol. Carcinog. 2017, 56, 2643–2662. [Google Scholar] [CrossRef][Green Version]
- Fu, X.; Beer, D.G.; Behar, J.; Wands, J.; Lambeth, D.; Cao, W. CAMP-Response Element-Binding Protein Mediates Acid-Induced NADPH Oxidase NOX5-S Expression in Barrett Esophageal Adenocarcinoma Cells. J. Biol. Chem. 2006, 281, 20368–20382. [Google Scholar] [CrossRef][Green Version]
- Hong, J.; Resnick, M.; Behar, J.; Wang, L.J.; Wands, J.; DeLellis, R.A.; Souza, R.F.; Spechler, S.J.; Cao, W. Acid-Induced P16 Hypermethylation Contributes to Development of Esophageal Adenocarcinoma via Activation of NADPH Oxidase NOX5-S. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G697–G706. [Google Scholar] [CrossRef][Green Version]
- Hong, J.; Li, D.; Cao, W. Rho Kinase ROCK2 Mediates Acid-Induced NADPH Oxidase NOX5-S Expression in Human Esophageal Adenocarcinoma Cells. PLoS ONE 2016, 11, e0149735. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chuang, L.S.H.; Matsuo, J.; Douchi, D.; Bte Mawan, N.A.; Ito, Y. RUNX3 in Stem Cell and Cancer Biology. Cells 2023, 12, 408. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qin, X.; Wu, J.; Qi, B.; Tao, Y.; Wang, W.; Liu, F.; Li, H.; Zhao, B. Association of Promoter Methylation of RUNX3 Gene with the Development of Esophageal Cancer: A Meta Analysis. PLoS ONE 2014, 9, e107598. [Google Scholar] [CrossRef]
- Fukui, S.; Watari, J.; Tomita, T.; Yamasaki, T.; Okugawa, T.; Kondo, T.; Kono, T.; Tozawa, K.; Ikehara, H.; Ohda, Y.; et al. Localization of Specialized Intestinal Metaplasia and the Molecular Alterations in Barrett Esophagus in a Japanese Population: An Analysis of Biopsy Samples Based on the “Seattle” Biopsy Protocol. Hum. Pathol. 2016, 51, 32–40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, B.; Song, H.; Jiang, H.; Fu, Y.; Ding, X.; Zhou, C. Early Diagnostic Potential of APC hypermethylation in Esophageal Cancer. Cancer Manag. Res. 2018, 10, 181–198. [Google Scholar] [CrossRef][Green Version]
- Restucci, B.; Martano, M.; DE Vico, G.; Lo Muzio, L.; Maiolino, P. Expression of E-Cadherin, Beta-Catenin and APC Protein in Canine Colorectal Tumours. Anticancer Res. 2009, 29, 2919–2925. [Google Scholar]
- Petrova, Y.I.; Schecterson, L.; Gumbiner, B.M. Roles for E-Cadherin Cell Surface Regulation in Cancer. Mol. Biol. Cell 2016, 27, 3233–3244. [Google Scholar] [CrossRef]
- Pinheiro, H.; Bordeira-Carrico, R.; Seixas, S.; Carvalho, J.; Senz, J.; Oliveira, P.; Inacio, P.; Gusmao, L.; Rocha, J.; Huntsman, D.; et al. Allele-Specific CDH1 Downregulation and Hereditary Diffuse Gastric Cancer. Hum. Mol. Genet. 2010, 19, 943–952. [Google Scholar] [CrossRef][Green Version]
- Shenoy, S. CDH1 (E-Cadherin) Mutation and Gastric Cancer: Genetics, Molecular Mechanisms and Guidelines for Management. Cancer Manag. Res. 2019, 11, 10477–10486. [Google Scholar] [CrossRef][Green Version]
- Pecchillo Cimmino, T.; Ammendola, R.; Cattaneo, F.; Esposito, G. NOX Dependent ROS Generation and Cell Metabolism. Int. J. Mol. Sci. 2023, 24, 2086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Hormi-Carver, K.; Zhang, X.; Spechler, S.J.; Souza, R.F. In Benign Barrett’s Epithelial Cells, Acid Exposure Generates Reactive Oxygen Species That Cause DNA Double-Strand Breaks. Cancer Res. 2009, 69, 9083–9089. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jones, P.A.; Baylin, S.B. The Fundamental Role of Epigenetic Events in Cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Jammula, S.; Katz-Summercorn, A.C.; Li, X.; Linossi, C.; Smyth, E.; Killcoyne, S.; Biasci, D.; Subash, V.V.; Abbas, S.; Blasko, A.; et al. Identification of Subtypes of Barrett’s Esophagus and Esophageal Adenocarcinoma Based on DNA Methylation Profiles and Integration of Transcriptome and Genome Data. Gastroenterology 2020, 158, 1682–1697.e1. [Google Scholar] [CrossRef]
- Alvarez, H.; Opalinska, J.; Zhou, L.; Sohal, D.; Fazzari, M.J.; Yu, Y.; Montagna, C.; Montgomery, E.A.; Canto, M.; Dunbar, K.B.; et al. Widespread Hypomethylation Occurs Early and Synergizes with Gene Amplification during Esophageal Carcinogenesis. PLoS Genet. 2011, 7, e1001356. [Google Scholar] [CrossRef]
- Boldrin, E.; Curtarello, M.; Dallan, M.; Alfieri, R.; Realdon, S.; Fassan, M.; Saggioro, D. Detection of LINE-1 Hypomethylation in CfDNA of Esophageal Adenocarcinoma Patients. Int. J. Mol. Sci. 2020, 21, 1547. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, W.; Bhagat, T.D.; Yang, X.; Song, J.H.; Cheng, Y.; Agarwal, R.; Abraham, J.M.; Ibrahim, S.; Bartenstein, M.; Hussain, Z.; et al. Hypomethylation of Noncoding DNA Regions and Overexpression of the Long Noncoding RNA, AFAP1-AS1, in Barrett’s Esophagus and Esophageal Adenocarcinoma. Gastroenterology 2013, 144, 956–966.e4. [Google Scholar] [CrossRef][Green Version]
- Gilbert, N. Biophysical Regulation of Local Chromatin Structure. Curr. Opin. Genet. Dev. 2019, 55, 66–75. [Google Scholar] [CrossRef]
- Tessarz, P.; Kouzarides, T. Histone Core Modifications Regulating Nucleosome Structure and Dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef]
- Grady, W.M.; Yu, M.; Markowitz, S.D.; Hutchinson, F. Emerging Use for Biomarkers of Cancer. Gastroenterology 2022, 160, 690–709. [Google Scholar] [CrossRef]
- Damaskos, C.; Garmpis, N.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schizas, D.; Mastoraki, A.; Naar, L.; Spartalis, E.; Tsilimigras, D.I.; Karachaliou, G.S.; Bagias, G.; Moris, D. Concept of Histone Deacetylases in Cancer: Reflections on Esophageal Carcinogenesis and Treatment. World J. Gastroenterol. 2018, 24, 4635–4642. [Google Scholar] [CrossRef]
- Langer, R.; Mutze, K.; Becker, K.; Feith, M.; Ott, K.; Höfler, H.; Keller, G. Expression of Class I Histone Deacetylases (HDAC1 and HDAC2) in Oesophageal Adenocarcinomas: An Immunohistochemical Study. J. Clin. Pathol. 2010, 63, 994–998. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Calin, G.A.; Croce, C.M. MicroRNA Signatures in Human Cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Song, B.; Wang, Y.; Kudo, K.; Gavin, E.J.; Xi, Y.; Ju, J. MiR-192 Regulates Dihydrofolate Reductase and Cellular Proliferation through the P53-MicroRNA Circuit. Clin. Cancer Res. 2008, 14, 8080–8086. [Google Scholar] [CrossRef][Green Version]
- Sun, J.; Fan, Z.; Lu, S.; Yang, J.; Hao, T.; Huo, Q. MiR-192 Suppresses the Tumorigenicity of Prostate Cancer Cells by Targeting and Inhibiting Nin One Binding Protein. Int. J. Mol. Med. 2016, 37, 485–492. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. MicroRNAs as Oncogenes and Tumor Suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fassan, M.; Volinia, S.; Palatini, J.; Pizzi, M.; Baffa, R.; De Bernard, M.; Battaglia, G.; Parente, P.; Croce, C.M.; Zaninotto, G.; et al. MicroRNA Expression Profiling in Human Barrett’s Carcinogenesis. Int. J. Cancer 2011, 129, 1661–1670. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Kleeman, S.; Coburn, S.B.; Fumagalli, C.; Perner, J.; Jammula, S.; Pfeiffer, R.M.; Orzolek, L.; Hao, H.; Taylor, P.R.; et al. Selection and Application of Tissue MicroRNAs for Nonendoscopic Diagnosis of Barrett’s Esophagus. Gastroenterology 2018, 155, 771–783.e3. [Google Scholar] [CrossRef]
- Revilla-Nuin, B.; Parrilla, P.; Lozano, J.J.; De Haro, L.F.M.; Ortiz, A.; Martínez, C.; Munitiz, V.; De Angulo, D.R.; Bermejo, J.; Molina, J.; et al. Predictive Value of MicroRNAs in the Progression of Barrett Esophagus to Adenocarcinoma in a Long-Term Follow-up Study. Ann. Surg. 2013, 257, 886–893. [Google Scholar] [CrossRef]
- Chen, H.; Zhou, X. Serum Exosomal MiRNAs Expression as Novel Biomarkers for Detection of Esophageal Adenocarcinoma: 441. Off. J. Am. Coll. Gastroenterol. ACG 2016, 111, S198–S199. [Google Scholar] [CrossRef]
- Wijnhoven, B.P.L.; Hussey, D.J.; Watson, D.J.; Tsykin, A.; Smith, C.M.; Michael, M.Z. MicroRNA Profiling of Barrett’s Oesophagus and Oesophageal Adenocarcinoma. Br. J. Surg. 2010, 97, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ji, F.; Liu, G.; Wang, W.; Li, Z.; Yue, Y.; Wang, Z. Upregulation of Circulating Mir130a Is Correlated with Development of Barrett’s Esophagus and Esophageal Adenocarcinoma. OncoTargets Ther. 2019, 12, 1–7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fassan, M.; Realdon, S.; Cascione, L.; Hahne, J.C.; Munari, G.; Guzzardo, V.; Arcidiacono, D.; Lampis, A.; Brignola, S.; Dal Santo, L.; et al. Circulating MicroRNA Expression Profiling Revealed MiR-92a-3p as a Novel Biomarker of Barrett’s Carcinogenesis. Pathol. Res. Pract. 2020, 216, 152907. [Google Scholar] [CrossRef]
- Yao, C.; Li, Y.; Luo, L.; Xiong, Q.; Zhong, X.; Xie, F.; Feng, P. Identification of MiRNAs and Genes for Predicting Barrett’s Esophagus Progressing to Esophageal Adenocarcinoma Using MiRNAmRNA Integrated Analysis. PLoS ONE 2021, 16, e0260353. [Google Scholar] [CrossRef]
- Wu, X.; Ajani, J.A.; Gu, J.; Chang, D.W.; Tan, W.; Hildebrandt, M.A.T.; Huang, M.; Wang, K.K.; Hawk, E. MicroRNA Expression Signatures during Malignant Progression from Barrett’s Esophagus to Esophageal Adenocarcinoma. Cancer Prev. Res. 2013, 6, 196–205. [Google Scholar] [CrossRef][Green Version]
- Slaby, O.; Srovnal, J.; Radova, L.; Gregar, J.; Juracek, J.; Luzna, P.; Svoboda, M.; Hajduch, M.; Ehrmann, J. Dynamic Changes in MicroRNA Expression Profiles Reflect Progression of Barrett’s Esophagus to Esophageal Adenocarcinoma. Carcinogenesis 2015, 36, 521–527. [Google Scholar] [CrossRef][Green Version]
- Leidner, R.S.; Ravi, L.; Leahy, P.; Chen, Y.; Bednarchik, B.; Streppel, M.; Canto, M.; Wang, J.S.; Maitra, A.; Willis, J.; et al. The MicroRNAs, MiR-31 and MiR-375, as Candidate Markers in Barrett’s Esophageal Carcinogenesis. Genes Chromosomes Cancer 2012, 51, 473–479. [Google Scholar] [CrossRef][Green Version]
- Hezova, R.; Kovarikova, A.; Srovnal, J.; Zemanova, M.; Harustiak, T.; Ehrmann, J.; Hajduch, M.; Svoboda, M.; Sachlova, M.; Slaby, O. Diagnostic and Prognostic Potential of MiR-21, MiR-29c, MiR-148 and MiR-203 in Adenocarcinoma and Squamous Cell Carcinoma of Esophagus. Diagn. Pathol. 2015, 10, 42. [Google Scholar] [CrossRef][Green Version]
- Drahos, J.; Schwameis, K.; Orzolek, L.D.; Hao, H.; Birner, P.; Taylor, P.R.; Pfeiffer, R.M.; Schoppmann, S.F.; Cook, M.B. MicroRNA Profiles of Barrett’s Esophagus and Esophageal Adenocarcinoma: Differences in Glandular Non-Native Epithelium. Cancer Epidemiol. Biomark. Prev. 2016, 25, 429–437. [Google Scholar] [CrossRef][Green Version]
- Zhang, M.; Zhuang, Q.; Cui, L. MiR-194 Inhibits Cell Proliferation and Invasion via Repression of RAP2B in Bladder Cancer. Biomed. Pharmacother. 2016, 80, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Yen, Y.-T.; Yang, J.-C.; Chang, J.-B.; Tsai, S.-C. Down-Regulation of MiR-194-5p for Predicting Metastasis in Breast Cancer Cells. Int. J. Mol. Sci. 2021, 23, 325. [Google Scholar] [CrossRef]
- Bus, P.; Kestens, C.; Ten Kate, F.J.W.; Peters, W.; Drenth, J.P.H.; Roodhart, J.M.L.; Siersema, P.D.; van Baal, J.W.P.M. Profiling of Circulating MicroRNAs in Patients with Barrett’s Esophagus and Esophageal Adenocarcinoma. J. Gastroenterol. 2016, 51, 560–570. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hino, K.; Tsuchiya, K.; Fukao, T.; Kiga, K.; Okamoto, R.; Kanai, T.; Watanabe, M. Inducible Expression of MicroRNA-194 Is Regulated by HNF-1α during Intestinal Epithelial Cell Differentiation. Rna 2008, 14, 1433–1442. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cabibi, D.; Caruso, S.; Bazan, V.; Castiglia, M.; Bronte, G.; Ingrao, S.; Fanale, D.; Cangemi, A.; Calò, V.; Listì, A.; et al. Analysis of Tissue and Circulating MicroRNA Expression during Metaplastic Transformation of the Esophagus. Oncotarget 2016, 7, 47821–47830. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hou, Y.; Zhen, J.; Xu, X.; Zhen, K.; Zhu, B.; Pan, R.; Zhao, C. MiR-215 Functions as a Tumor Suppressor and Directly Targets ZEB2 in Human Non-Small Cell Lung Cancer. Oncol. Lett. 2021, 22, 600. [Google Scholar] [CrossRef]
- Li, N.; Zhang, Q.-Y.; Zou, J.-L.; Li, Z.-W.; Tian, T.-T.; Dong, B.; Liu, X.-J.; Ge, S.; Zhu, Y.; Gao, J.; et al. MiR-215 Promotes Malignant Progression of Gastric Cancer by Targeting RUNX1. Oncotarget 2016, 7, 4817–4828. [Google Scholar] [CrossRef][Green Version]
- Bansal, A.; Lee, I.H.; Hong, X.; Mathur, S.C.; Tawfik, O.; Rastogi, A.; Buttar, N.; Visvanathan, M.; Sharma, P.; Christenson, L.K. Discovery and Validation of Barrett’s Esophagus MicroRNA Transcriptome by Next Generation Sequencing. PLoS ONE 2013, 8, e54240. [Google Scholar] [CrossRef][Green Version]
- Fassan, M.; Volinia, S.; Palatini, J.; Pizzi, M.; Fernandez-Cymering, C.; Balistreri, M.; Realdon, S.; Battaglia, G.; Souza, R.; Odze, R.D.; et al. MicroRNA Expression Profiling in the Histological Subtypes of Barrett’s Metaplasia. Clin. Transl. Gastroenterol. 2013, 4, e34-7. [Google Scholar] [CrossRef]
- Inokuchi, K.; Ochiya, T.; Matsuzaki, J. Extracellular Mirnas for the Management of Barrett’s Esophagus and Esophageal Adenocarcinoma: A Systematic Review. J. Clin. Med. 2021, 10, 117. [Google Scholar] [CrossRef]
- Sonkoly, E.; Lovén, J.; Xu, N.; Meisgen, F.; Wei, T.; Brodin, P.; Jaks, V.; Kasper, M.; Shimokawa, T.; Harada, M.; et al. MicroRNA-203 Functions as a Tumor Suppressor in Basal Cell Carcinoma. Oncogenesis 2012, 1, e3. [Google Scholar] [CrossRef][Green Version]
- Zhang, A.; Lakshmanan, J.; Motameni, A.; Harbrecht, B.G. MicroRNA-203 Suppresses Proliferation in Liver Cancer Associated with PIK3CA, P38 MAPK, c-Jun, and GSK3 Signaling. Mol. Cell Biochem. 2018, 441, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Li, X.; Zhang, Y.; Guo, Y.; Zhou, J.; Gao, K.; Dai, J.; Hu, G.; Lv, L.; Du, J. The MicroRNA Feedback Regulation of P63 in Cancer Progression. Oncotarget 2015, 6, 8434–8453. [Google Scholar] [CrossRef][Green Version]
- Yuan, Y.; Zeng, Z.-Y.; Liu, X.-H.; Gong, D.-J.; Tao, J.; Cheng, H.-Z.; Huang, S.-D. MicroRNA-203 Inhibits Cell Proliferation by Repressing ΔNp63 Expression in Human Esophageal Squamous Cell Carcinoma. BMC Cancer 2011, 11, 57. [Google Scholar] [CrossRef][Green Version]
- Qi, Q.; Ling, Y.; Zhu, M.; Zhang, Y.; Zhou, L.; Wan, M.; Bao, Y.; Liu, Y. Hypermethylation and Low Expression of MiR-203 in Patients with Esophageal Cancer in Chinese Population. Int. J. Clin. Exp. Pathol. 2016, 9, 6245–6251. [Google Scholar]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The MiR-200 Family and MiR-205 Regulate Epithelial to Mesenchymal Transition by Targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Hezova, R.; Kovarikova, A.; Srovnal, J.; Zemanova, M.; Harustiak, T.; Ehrmann, J.; Hajduch, M.; Sachlova, M.; Svoboda, M.; Slaby, O. MiR-205 Functions as a Tumor Suppressor in Adenocarcinoma and an Oncogene in Squamous Cell Carcinoma of Esophagus. Tumor Biol. 2016, 37, 8007–8018. [Google Scholar] [CrossRef]
- Pan, F.; Mao, H.; Bu, F.; Tong, X.; Li, J.; Zhang, S.; Liu, X.; Wang, L.; Wu, L.; Chen, R.; et al. Sp1-Mediated Transcriptional Activation of MiR-205 Promotes Radioresistance in Esophageal Squamous Cell Carcinoma. Oncotarget 2017, 8, 5735–5752. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saller, J.; Jiang, K.; Xiong, Y.; Yoder, S.J.; Neill, K.; Pimiento, J.M.; Pena, L.; Corbett, F.S.; Magliocco, A.; Coppola, D. A MicroRNA Signature Identifies Patients at Risk of Barrett Esophagus Progression to Dysplasia and Cancer. Dig. Dis. Sci. 2022, 67, 516–523. [Google Scholar] [CrossRef]
- Zarrilli, G.; Galuppini, F.; Angerilli, V.; Munari, G.; Sabbadin, M.; Lazzarin, V.; Nicolè, L.; Biancotti, R.; Fassan, M. Mirnas Involved in Esophageal Carcinogenesis and MiRNA- Related Therapeutic Perspectives in Esophageal Carcinoma. Int. J. Mol. Sci. 2021, 22, 3640. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Hypermethylated Genes | Main Function | Classification | Hypermethylation Ranges | References |
|---|---|---|---|---|
| AKAP12 | a cell-growth-related protein | acts as a tumor suppressor | BE (39%), BD (53%), EAC (52%) NSE (0%) | [28] |
| APC | negative regulator that controls beta-catenin | tumor suppressor | BE (36–95%, mean: 67%) [7] BD (50–89%, mean: 61%) [3] EAC (42–95%, mean: 74%) [9] (additionally, 25% in plasma of EAC) [1] NSE (0–14%, mean: 3%) [5] | [24,27,29,30,31,32,33,34,35] In plasma [35] |
| CCNA1 | controls proliferative and survival activities in tumorigenesis | oncogenic | BE (81%), BD (68%), EAC (90%) NSE (1%) | [36] |
| CDH1 (e-cadherin) | an essential transmembrane protein within adherens junctions | tumor suppressor | EAC (10–84%, mean: 53%) [3] NSE (0–12%, mean: 6%) [2] | [32,37,38] |
| CDH13 | a member of the calcium-dependent cell adhesion molecule family | tumor suppressor | BE (70%), BD (78%), EAC (76%) NSE (0%) | [39] |
| CDKN2A | regulates the cell cycle | tumor suppressor | p16INK4A promotor: BE (3–77%, mean: 29%) [12] BD (11–75%, mean: 40%) [9] EAC (16–85%, mean: 51%) [14] NSE (0–43%, mean: 9%) [9] p14ARF promotor: BE (8%) [1] EAC (0–20%, mean: 7%) [3] NSE (0%) [1] | p16INK4A [21,22,24,25,27,30,31,32,33,34,40,41,42] p14ARF [24,25,27] |
| DAPK | positive mediator of gamma-interferon-induced programmed cell death | tumor suppressor | BE (50%) [1], BD (53%) [1], EAC (20–70%, mean: 50%) [3] NSE (5–20%, mean: 13) [2] | [32,38,43] |
| ESR1 | plays a role in growth, metabolism, sexual development, and gestation | tumor suppressor | BE (69%) [1] BD (67–100% mean: 84%) [2] EAC (51–100%, mean: 76%) [2] NSE (5–12%, mean: 9%) [2] | [31,32] |
| EYA4 | possesses phosphatase, hydrolase, and transcriptional activation functions | tumor suppressor | BE (77%), EAC (83%) NSE (3%) | [44] |
| FHIT | role in the regulation of apoptosis | tumor suppressor | EAC (70%) | [38] |
| GPx3 | H2O2 detoxification | acts as a tumor suppressor | BE (62–90%, mean: 76%) [2] BD (82–88%, mean: 85%) [2] EAC (62–88%, mean: 75) [2] NSE (8–17%, mean: 13%) [2] | [45,46] |
| GPx7 | catalyzes the reduction of H2O2 | acts as a tumor suppressor | BE (18%), BD (80%), EAC (67%) NSE (0%) | [46] |
| GSTM2 | detoxification of electrophilic compounds | acts as a tumor suppressor | BE (50%), BD (56%), EAC (69%) NSE (5%) | [46] |
| GSTM3 | detoxification of chemical substrates or electronic compounds | acts as a tumor suppressor | BE (13%), BD (38%), EAC (15%) NSE (0%) | [46] |
| HPP1/TMEFF2 | may play multiple roles in cell growth, maturation, and adhesion | tumor suppressor | BE (44–75%, mean: 60%) [2] BD (79–100%, mean: 90%) [2] EAC (71–83%, mean: 77%) [2] NSE (3–4% mean: 4%) [2] | [24,33] |
| ID4 | inhibits of DNA binding protein family | acts as a tumor suppressor | BE (78%), BD (86%), EAC (78%) NSE (21%) | [33] |
| MGMT | DNA repair enzyme that plays an important role in chemoresistance to alkylating agents | tumor suppressor | BE (25–89%, mean: 52%) [6] BD (89–100%, mean: 95%) [2] EAC (21–79%, mean: 51%) [8] NSE (17–55%, mean: 27%) [5] | [24,32,33,34,38,42,47,48] |
| NELL1 | encodes a cytoplasmic protein that contains epidermal growth factor-like repeats | promising tumor suppressor | BE (42%), BD (53%), EAC (48%) NSE (0%) | [49] |
| RBP1 | the carrier protein involved in the transport of retinol | tumor suppressor | BE (58%), BD (57%), EAC (70%) NSE (18%) | [33] |
| RUNX3 | activate or suppress transcription, role in the TGF-β signaling pathway | tumor suppressor | BE (25–48%, mean 37%) [2] BD (57%) 2] EAC (48–73%, mean: 61%) [2] NSE (2%) [2] | [24,33] |
| SFRP1 | affects cell growth, limits the cell cycle, and induces apoptosis | tumor suppressor | BE (81–100%, mean: 91%) [3] BD (100%) [1] EAC (91–95%, mean: 93%) [3] NSE (10–17%, mean: 14%) [2] | [29,33,50] |
| SOCS1 | potent inhibitor of the interferon gamma (IFNγ) pathway | acts as a tumor suppressor | BE (0%), BD (13%), EAC (42%) NSE (0%) | [51] |
| SOCS3 | regulates cytokine or hormone signaling | acts as a tumor suppressor | BE (13%), BD (46%), EAC (74%) NSE (0%) | [51] |
| TAC1 | encodes peptides that target nerve receptors, immune cells, stem cells, hematopoietic cells, and smooth muscle cells | acts as a tumor suppressor and oncogenic | BE (56%), BD (58%), EAC (61%) NSE (8%) | [52] |
| TERT | ensuring chromosomal stability by maintaining telomere length | acts as a tumor suppressor | BE (17%), BD (92%), EAC (64%) NSE (0%) | [29] |
| TIMP3 | Complexes with metalloproteinases and irreversibly inactivates them | tumor suppressor | BE (23–88%, mean: 63%) [6] BD (78%) [1] EAC (20–90%, mean: 63%) [7] NSE (0–19%, mean: 6%) [4] | [24,29,32,33,34,42,53] |
| VIM | supporting and anchoring the position of the organelles in the cytosol | Interacts with tumor suppressors | BE (91–100%, mean: 96%) [2] BD (63–100%, mean: 82) [2] EAC (77–81%, mean: 79%) [2] NSE (0–1%, mean: 1%) [2] | [36,54] |
| Deregulated microRNAs | Classification | Sample Types | References |
|---|---|---|---|
| miR-192 | tumor suppressor | BE, EAC (tissue and serum, cytosponge) | [88,91] |
| miR-194 | oncomir or tumor suppressor properties | BE, EAC (tissue and serum, cytosponge) | [89,90] |
| miR-215 | tumor suppressor | BE, EAC (tissue and serum, cytosponge) | [90,92] |
| miR-203 | oncomir | BE, EAC (tissue and serum) | [89,90] |
| miR-205 | tumor suppressor | BE, EAC (tissue and serum) | [89,90] |
| miR-130a | oncomir | BE, EAC (serum) | [93] |
| miR-92a-3p | oncomir | BE, EAC (tissue and serum) | [94] |
| miR-496, miR-214, miR-15b | tumor suppressor | BE, EAC (enrichment analysis *) | [95] |
| Let-7c | tumor suppressor | BE, EAC (tissue) | [88,96] |
| miR-196a | oncomir | BE, EAC (tissue and cytosponge) | [2,89,90,97] |
| miR-31 | oncomir | BE, EAC (tissue) | [98,99,100] |
| miR-375 | oncomir | BE, EAC (tissue) | [98] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ergun, P.; Kipcak, S.; Bor, S. Epigenetic Alterations from Barrett’s Esophagus to Esophageal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 7817. https://doi.org/10.3390/ijms24097817
Ergun P, Kipcak S, Bor S. Epigenetic Alterations from Barrett’s Esophagus to Esophageal Adenocarcinoma. International Journal of Molecular Sciences. 2023; 24(9):7817. https://doi.org/10.3390/ijms24097817
Chicago/Turabian StyleErgun, Pelin, Sezgi Kipcak, and Serhat Bor. 2023. "Epigenetic Alterations from Barrett’s Esophagus to Esophageal Adenocarcinoma" International Journal of Molecular Sciences 24, no. 9: 7817. https://doi.org/10.3390/ijms24097817