
Modeling Human Muscular Dystrophies in Zebrafish: Mutant Lines, Transgenic Fluorescent Biosensors, and Phenotyping Assays

 , ,
, ,  and
and 
Abstract
:1. Introduction
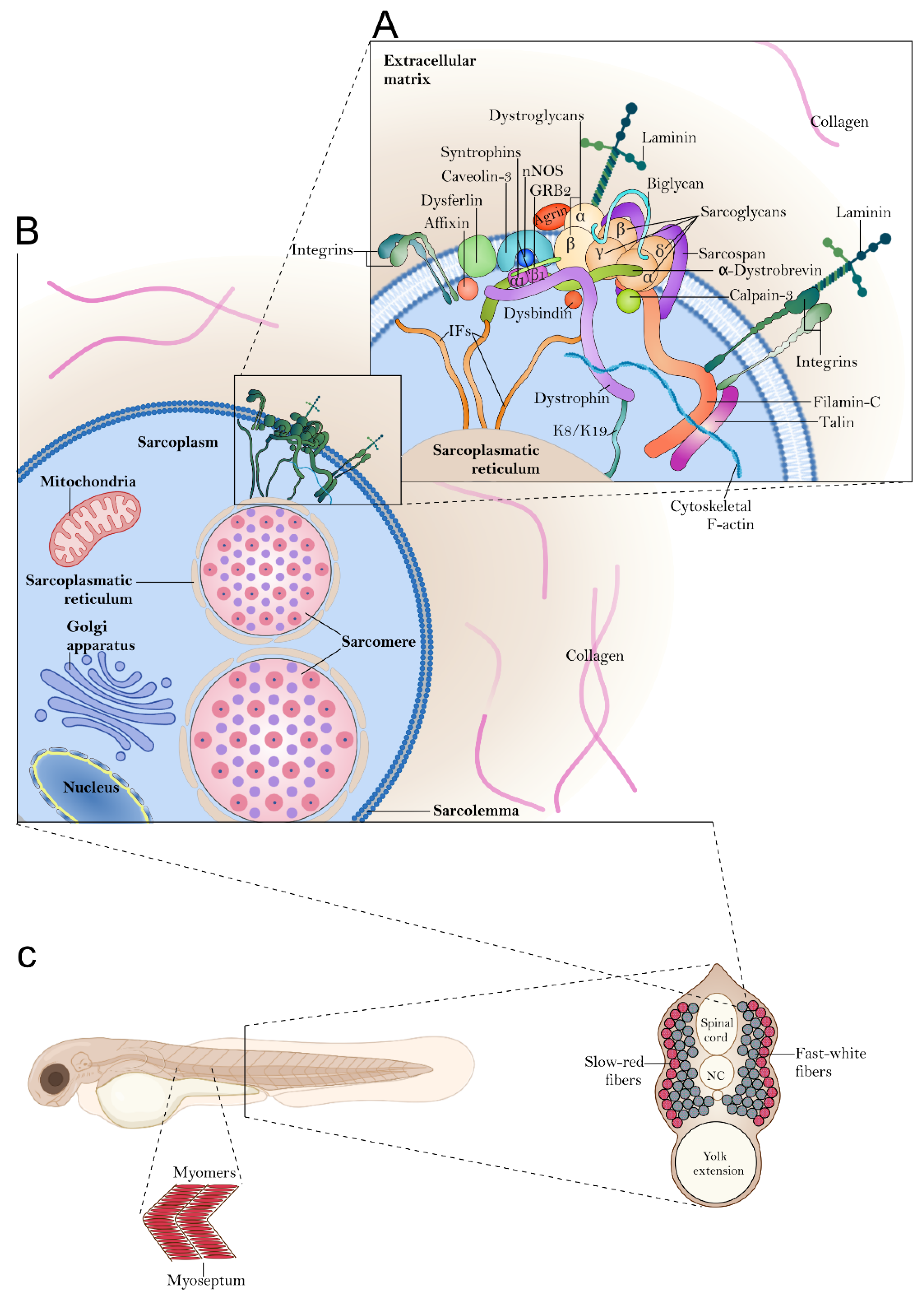
1.1. Skeletal Muscle Organization in Zebrafish
2. Models of Muscular Dystrophy in Zebrafish
2.1. Basal Membrane and Extracellular Matrix Protein
2.2. Cytosolic Proteins
2.3. Dystroglycan and α-DG Glycosylation-Related Protein
2.4. Nuclear Envelope and Cytoskeleton Proteins
2.5. Membrane Proteins
3. Functional and Genetic Tools for Investigating Skeletal Muscle Development and Functions
3.1. Motor Behavior
3.2. Muscle Force (Electrophysiology)
3.3. Muscle Structure
3.4. Calcium Homeostasis and Oxidative Stress (Use of Fluorescent Probes)
3.5. Mitochondrial and Metabolic Function
4. Transgenic Fluorescent Zebrafish Lines
4.1. Zebrafish Reporter Lines Targeting In Vivo Signaling Pathway Activities Involved in Muscle Development

{kind=link}
| Line | Transgenic ID | Responsive Element (RE) | Signaling Pathway | Features | Ref. |
|---|---|---|---|---|---|
| Tg(hsp70l:dnBmpr-GFP)w30 | ZDB-ALT-050503-2 | BRE | BMP signaling | Heat shock (hsp70) promoter GFP expression | [164] |
| Tg(BmpRE:mRFP)cj100 | ZDB-ALT-110705-4 | BRE | BMP signaling | Membrane-bound red fluorescent protein (RFP) | [167,168] |
| Tg(BmpRE-AAVmlp:eGFP)mw29 | ZDB-ALT-110308-1 | BRE | BMP signaling | GFP expression | [166] |
| Tg(BRE-AAVmlp:d2GFP)mw30 | ZDB-ALT-110310-1 | BRE | BMP signaling | Destabilized d2GFP | [166] |
| Tg(BRE-AAVmlp:dmKO2)mw40 | ZDB-ALT-110310-2 | BRE | BMP signaling | Destabilized monomeric Kusabira-Orange (dmKO2) | [166] |
| Tg(BMPRE:NLS-mCherry)ia17 | ZDB-ALT-130115-2 | BRE | BMP signaling | Nuclear localization signal mCherry | [165] |
| Tg(7xTCF-Xla.Siam:GFP)ia4 | ZDB-ALT-110113-1 | TCF | Wnt signaling | GFP expression | [163] |
| Tg(7xTCF-Xla.Siam:nlsmCherry)ia5 | ZDB-ALT-110113-2 | TCF | Wnt signaling | Nuclear localization signal mCherry | [163] |
| Tg(TOP:GFP)w25 | ZDB-ALT-020621-4 | LEF binding sites | Wnt signaling | GFP expression | [161,162] |
| Tg(6xCRE:eGFP) | CRE | CREB signaling | Enhanced GFP expression | [169] | |
| Tg(4xHRE-TATA:eGFP)ia21 | ZDB-ALT-131030-1 | HRE | Hypoxia signaling | Enhanced GFP expression | [156] |
| Tg(12xgli-HSV.Ul23:GFP)ia11 | ZDB-ALT-120404-2 | gli | Shh signaling | GFP expression | [159] |
| Tg(8xGliBS:mCherry-NLS-Odc1)st1002 | ZDB-ALT-141030-2 | gli | Shh signaling | Nuclear-localized fluorescent protein tagged with an ornithine decarboxylase-derived destabilizing peptide | [160] |
| Tg(9xGCRE-HSV.Ul23:eGFP)ia20 | ZDB-ALT-130123-1 | GRE | Glucocorticoid | Enhanced GFP expression | [171] |
| Tg(8XAORE:GFP)ia201 | ZDB-ALT-211202-1 | ARE | Nrf2/ARE | GFP expression | [173] |
4.2. Zebrafish Reporter Lines Targeting the Skeletal Muscle
| Line | Transgenic ID | Promoter | Cells Tagged | Features | Ref. |
|---|---|---|---|---|---|
| Tg(myf5:YFP)CLGY237 Tg(−80.0myf5: EGFP)zf37 | ZDB-ALT-150512-2 ZDB-ALT-070730-1 | Myogenic factor 5 | Quiescent and activated satellite cells | Yellow fluorescent protein Enhanced GFP expression | [176,192] |
| TgBAC(pax7a:GFP)i131 | ZDB-ALT-111118-32 | Paired box 7a | Muscle progenitors | GFP expression | [175] |
| TgBAC(pax3a:GFP)i150 | ZDB-ALT-111118-34 | Paired box 3a | Muscle progenitors | GFP expression | [175] |
| Tg(myog:GFP)pc27 | ZDB-ALT-171003-14 | Myogenin | Muscle progenitors | eGFP expression | [174] |
| TgBAC(myod:GFP)i124 | ZDB-ALT-111118-35 | Myogenic differentiation 1 | Muscle progenitors | GFP expression | [175] |
| Tg(-10en2a:EGFP)i233 | ZDB-ALT-110223-3 | Engrailed homeobox 2a | Muscle progenitors and medial fast fibers | Enhanced GFP expression | [178] |
| Tg(-2.2mylz2:GFP)i135 Tg(mylz2:GFP)gz8 Tg(mylpfa:mCherry)cz3327 | ZDB-ALT-081112-1 ZDB-ALT-080207-2 ZDB-ALT-130923-2 | Myosin light chain, phosphorylatable, fast skeletal muscle | Fast skeletal muscle | GFP expression or mCherry | [181,182,183] |
| TgPAC(prdm1a:EGFP)i106 | ZDB-ALT-080923-6 | PR domain containing 1a, with ZNF domain | Primary and secondary slow fibers | Enhanced GFP expression | [193] |
| Tg(tnnc1b:eGFP)i305 | ZDB-ALT-150723-4 | Troponin C type 1b | Slow-twitch muscle fibers | Enhanced GFP expression | [146] |
| Tg(Tru.Myhz1.1:EGFP)kj100 | ZDB-ALT-130823-1 | Myosin, heavy polypeptide 1.1, skeletal muscle | Slow muscle-specific heavy chain | Enhanced GFP expression | [194] |
| Tg(smyhc1:GFP)i104 Tg(smyhc1:LY-Tomato)oz29 | ZDB-ALT-080923-4 ZDB-ALT-191024-1 | Slow myosin heavy chain 1 | Slow-twitch muscle fibers | GFP or tomato expression | [146,193,195] |
| Tg(acta1: mCherryCAAX)pc22 TgBAC(actc1b: GFP)zf13: zf13Tg Tg(acta1:lifeact-GFP)pc21 | ZDB-ALT-150224-2 ZDB-ALT-060221-2 ZDB-ALT-150224-1 | Actin alpha cardiac muscle 1b | Myofibrils, sarcolemma, and t-tubules of the myofibers | Membrane-tethered mCherry or GFP | [184,185] |
| Tg(ubi: zebrabow-M)a131 Tg(msgn1: CreERT2)pc9 | ZDB-ALT-130816-2 ZDB-ALT-141117-7 | Mesogenin 1 | Muscle progenitors | dTomato, mCerulean and eYFP) | [196,197] |
| Tg(hsp70l:lamp1-RFP)pd1064 | ZDB-ALT-130409-7 | Lysosomal-associated membrane protein 1b | Skeletal muscle lysosomes | RFP expression | [186,187] |
| Tg(hsp70l:RFP-Rno.Map1lc3b)pd1065 | ZDB-ALT-130410-5 | Microtubule-associated protein 1 light chain 3 beta | Skeletal muscle lysosomes | RFP expression | [186,187] |
| Tg(h2ax:EGFP-rab7a)mw7 | ZDB-ALT-111017-6 | RAB7a, member RAS oncogene family | Skeletal muscle Late endosome | Enhanced GFP expression | [188] |
| Tg(Xla.Eef1a1:mlsEGFP) cms1 | ZDB-ALT-090309-2 | Cytochrome c oxidase subunit 8A | Mitochondria | Enhanced GFP expression | [190,191] |
5. Zebrafish Models of MDs Used for Drug Screening
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 7, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.C.; Sheehan, D.W.; Prochoroff, A.; Birnkrant, D.J. Muscular Dystrophies. Clin. Chest Med. 2018, 39, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef]
- Hussain, I.; Patni, N.; Ueda, M.; Sorkina, E.; Valerio, C.M.; Cochran, E.; Brown, R.J.; Peeden, J.; Tikhonovich, Y.; Tiulpakov, A.; et al. A Novel Generalized Lipodystrophy-Associated Progeroid Syndrome Due to Recurrent Heterozygous LMNA p.T10I Mutation. J. Clin. Endocrinol. Metab. 2018, 103, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Yukina, M.; Nuralieva, N.; Sorkina, E.; Troshina, E.; Tiulpakov, A.; Belaya, Z.; Melnichenko, G. Atypical progeroid syndrome (p.E262K LMNA mutation): A rare cause of short stature and osteoporosis. Endocrinol. Diabetes Metab. Case Rep. 2021, 2021, 20-0188. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Streisinger, G.; Walker, C.; Dower, N.; Knauber, D.; Singer, F. Production of clones of homozygous diploid zebra fish (Brachydanio rerio). Nature 1981, 291, 293–296. [Google Scholar] [CrossRef]
- Hall, T.E.; Bryson-Richardson, R.J.; Berger, S.; Jacoby, A.S.; Cole, N.J.; Hollway, G.E.; Berger, J.; Currie, P.D. The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin alpha2-deficient congenital muscular dystrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 7092–7097. [Google Scholar] [CrossRef]
- Vacaru, A.M.; Unlu, G.; Spitzner, M.; Mione, M.; Knapik, E.W.; Sadler, K.C. In vivo cell biology in zebrafish—Providing insights into vertebrate development and disease. J. Cell Sci. 2014, 127 Pt 3, 485–495. [Google Scholar] [CrossRef]
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef]
- Charvet, B.; Malbouyres, M.; Pagnon-Minot, A.; Ruggiero, F.; Le Guellec, D. Development of the zebrafish myoseptum with emphasis on the myotendinous junction. Cell Tissue Res. 2011, 346, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.A.; McNulty, I.M.; Durst, W.A.; Munchel, S.E.; Amacher, S.L. Interactions between muscle fibers and segment boundaries in zebrafish. Dev. Biol. 2005, 287, 346–360. [Google Scholar] [CrossRef] [PubMed]
- Stickney, H.L.; Barresi, M.J.; Devoto, S.H. Somite development in zebrafish. Dev. Dyn. 2000, 219, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Hollway, G.E.; Bryson-Richardson, R.J.; Berger, S.; Cole, N.J.; Hall, T.E.; Currie, P.D. Whole-somite rotation generates muscle progenitor cell compartments in the developing zebrafish embryo. Dev. Cell 2007, 12, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Altringham, J.D.; Ellerby, D.J. Fish swimming: Patterns in muscle function. J. Exp. Biol. 1999, 202 Pt 23, 3397–3403. [Google Scholar] [CrossRef] [PubMed]
- Squire, J.M. Architecture and function in the muscle sarcomere. Curr. Opin. Struct. Biol. 1997, 7, 247–257. [Google Scholar] [CrossRef]
- Dube, D.K.; Dube, S.; Abbott, L.; Wang, J.; Fan, Y.; Alshiekh-Nasany, R.; Shah, K.K.; Rudloff, A.P.; Poiesz, B.J.; Sanger, J.M.; et al. Identification, characterization, and expression of sarcomeric tropomyosin isoforms in zebrafish. Cytoskeleton 2017, 74, 125–142. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef]
- Lapidos, K.A.; Kakkar, R.; McNally, E.M. The dystrophin glycoprotein complex: Signaling strength and integrity for the sarcolemma. Circ. Res. 2004, 94, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Barresi, R.; Campbell, K.P. Dystroglycan: From biosynthesis to pathogenesis of human disease. J. Cell Sci. 2006, 119 Pt 2, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Muntoni, F.; Voit, T. The congenital muscular dystrophies in 2004: A century of exciting progress. Neuromuscul. Disord. 2004, 14, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; de Sa Moreira, E.; Piluso, G.; Vainzof, M.; Belsito, A.; Politano, L.; Puca, A.A.; Passos-Bueno, M.R.; Zatz, M. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta-sarcoglycan gene. Nat. Genet. 1996, 14, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Helbling-Leclerc, A.; Zhang, X.; Topaloglu, H.; Cruaud, C.; Tesson, F.; Weissenbach, J.; Tome, F.M.; Schwartz, K.; Fardeau, M.; Tryggvason, K.; et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet. 1995, 11, 216–218. [Google Scholar] [CrossRef] [PubMed]
- Moreira, E.S.; Wiltshire, T.J.; Faulkner, G.; Nilforoushan, A.; Vainzof, M.; Suzuki, O.T.; Valle, G.; Reeves, R.; Zatz, M.; Passos-Bueno, M.R.; et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat. Genet. 2000, 24, 163–166. [Google Scholar] [CrossRef]
- Anderson, L.V.; Davison, K.; Moss, J.A.; Young, C.; Cullen, M.J.; Walsh, J.; Johnson, M.A.; Bashir, R.; Britton, S.; Keers, S.; et al. Dysferlin is a plasma membrane protein and is expressed early in human development. Hum. Mol. Genet. 1999, 8, 855–861. [Google Scholar] [CrossRef]
- Bertini, E.; Pepe, G. Collagen type VI and related disorders: Bethlem myopathy and Ullrich scleroatonic muscular dystrophy. Eur. J. Paediatr. Neurol. 2002, 6, 193–198. [Google Scholar] [CrossRef]
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef]
- Genschel, J.; Schmidt, H.H. Mutations in the LMNA gene encoding lamin A/C. Hum. Mutat. 2000, 16, 451–459. [Google Scholar] [CrossRef]
- Mahdy, M.A.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef]
- Van Ry, P.M.; Fontelonga, T.M.; Barraza-Flores, P.; Sarathy, A.; Nunes, A.M.; Burkin, D.J. ECM-Related Myopathies and Muscular Dystrophies: Pros and Cons of Protein Therapies. Compr. Physiol. 2017, 7, 1519–1536. [Google Scholar] [PubMed]
- Zambon, A.A.; Muntoni, F. Congenital muscular dystrophies: What is new? Neuromuscul. Disord. 2021, 31, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Wang, J.C.; Gupta, V.A.; Dowling, J.J. A novel early onset phenotype in a zebrafish model of merosin deficient congenital muscular dystrophy. PLoS ONE 2017, 12, e0172648. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Arner, A. Immobilization of Dystrophin and Laminin alpha2-Chain Deficient Zebrafish Larvae In Vivo Prevents the Development of Muscular Dystrophy. PLoS ONE 2015, 10, e0139483. [Google Scholar]
- Hall, T.E.; Wood, A.J.; Ehrlich, O.; Li, M.; Sonntag, C.S.; Cole, N.J.; Huttner, I.G.; Sztal, T.E.; Currie, P.D. Cellular rescue in a zebrafish model of congenital muscular dystrophy type 1A. npj Regen. Med. 2019, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Horstick, E.J.; Davidson, A.E.; Dowling, J. Analysis of Zebrafish Larvae Skeletal Muscle Integrity with Evans Blue Dye. J. Vis. Exp. 2015, e53183. [Google Scholar] [CrossRef]
- Smith, S.J.; Fabian, L.; Sheikh, A.; Noche, R.; Cui, X.; Moore, S.A.; Dowling, J.J. Lysosomes and the pathogenesis of merosin-deficient congenital muscular dystrophy. Hum. Mol. Genet. 2022, 31, 733–747. [Google Scholar] [CrossRef]
- Gupta, V.A.; Kawahara, G.; Myers, J.A.; Chen, A.T.; Hall, T.E.; Manzini, M.C.; Currie, P.D.; Zhou, Y.; Zon, L.I.; Kunkel, L.M.; et al. A splice site mutation in laminin-alpha2 results in a severe muscular dystrophy and growth abnormalities in zebrafish. PLoS ONE 2012, 7, e43794. [Google Scholar] [CrossRef]
- Sztal, T.E.; Sonntag, C.; Hall, T.E.; Currie, P.D. Epistatic dissection of laminin-receptor interactions in dystrophic zebrafish muscle. Hum. Mol. Genet. 2012, 21, 4718–4731. [Google Scholar] [CrossRef]
- Fabian, L.; Dowling, J.J. Zebrafish Models of LAMA2-Related Congenital Muscular Dystrophy (MDC1A). Front. Mol. Neurosci. 2020, 13, 122. [Google Scholar] [CrossRef]
- Mienaltowski, M.J.; Birk, D.E. Structure, physiology, and biochemistry of collagens. Adv. Exp. Med. Biol. 2014, 802, 5–29. [Google Scholar]
- Lampe, A.K.; Bushby, K.M. Collagen VI related muscle disorders. J. Med. Genet. 2005, 42, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Guiraud, A.; Malbouyres, M.; Zwolanek, D.; Guillon, E.; Bretaud, S.; Monnot, C.; Schulze, J.; Bader, H.L.; Allard, B.; et al. Knockdown of col22a1 gene in zebrafish induces a muscular dystrophy by disruption of the myotendinous junction. Development 2013, 140, 4602–4613. [Google Scholar] [CrossRef] [PubMed]
- Tonelotto, V.; Consorti, C.; Facchinello, N.; Trapani, V.; Sabatelli, P.; Giraudo, C.; Spizzotin, M.; Cescon, M.; Bertolucci, C.; Bonaldo, P. Collagen VI ablation in zebrafish causes neuromuscular defects during developmental and adult stages. Matrix Biol. 2022, 112, 39–61. [Google Scholar] [CrossRef] [PubMed]
- Radev, Z.; Hermel, J.M.; Elipot, Y.; Bretaud, S.; Arnould, S.; Duchateau, P.; Ruggiero, F.; Joly, J.S.; Sohm, F. A TALEN-Exon Skipping Design for a Bethlem Myopathy Model in Zebrafish. PLoS ONE 2015, 10, e0133986. [Google Scholar] [CrossRef]
- Telfer, W.R.; Busta, A.S.; Bonnemann, C.G.; Feldman, E.L.; Dowling, J.J. Zebrafish models of collagen VI-related myopathies. Hum. Mol. Genet. 2010, 19, 2433–2444. [Google Scholar] [CrossRef]
- Malbouyres, M.; Guiraud, A.; Lefrancois, C.; Salamito, M.; Nauroy, P.; Bernard, L.; Sohm, F.; Allard, B.; Ruggiero, F. Lack of the myotendinous junction marker col22a1 results in posture and locomotion disabilities in zebrafish. Matrix Biol. 2022, 109, 1–18. [Google Scholar] [CrossRef]
- Parsons, M.J.; Campos, I.; Hirst, E.M.; Stemple, D.L. Removal of dystroglycan causes severe muscular dystrophy in zebrafish embryos. Development 2002, 129, 3505–3512. [Google Scholar] [CrossRef]
- Bassett, D.I.; Bryson-Richardson, R.J.; Daggett, D.F.; Gautier, P.; Keenan, D.G.; Currie, P.D. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 2003, 130, 5851–5860. [Google Scholar] [CrossRef]
- Granato, M.; van Eeden, F.J.; Schach, U.; Trowe, T.; Brand, M.; Furutani-Seiki, M.; Haffter, P.; Hammerschmidt, M.; Heisenberg, C.P.; Jiang, Y.J.; et al. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 1996, 123, 399–413. [Google Scholar] [CrossRef]
- Bolanos-Jimenez, F.; Bordais, A.; Behra, M.; Strahle, U.; Sahel, J.; Rendon, A. Dystrophin and Dp71, two products of the DMD gene, show a different pattern of expression during embryonic development in zebrafish. Mech. Dev. 2001, 102, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.R.; Goswami, J.; Jun, S.J.; Thorne, M.; Howell, M.; Pusack, T.; Kawahara, G.; Steffen, L.S.; Galdzicki, M.; Kunkel, L.M. Genetic isolation and characterization of a splicing mutant of zebrafish dystrophin. Hum. Mol. Genet. 2009, 18, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, H.; Mitsuhashi, S.; Lynn-Jones, T.; Kawahara, G.; Kunkel, L.M. Expression of DUX4 in zebrafish development recapitulates facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2013, 22, 568–577. [Google Scholar] [CrossRef]
- Pakula, A.; Lek, A.; Widrick, J.; Mitsuhashi, H.; Bugda Gwilt, K.M.; Gupta, V.A.; Rahimov, F.; Criscione, J.; Zhang, Y.; Gibbs, D.; et al. Transgenic zebrafish model of DUX4 misexpression reveals a developmental role in FSHD pathogenesis. Hum. Mol. Genet. 2019, 28, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.M.; Garwick, S.E.; Mei, W.; Belayew, A.; Coppee, F.; Ladner, K.J.; Guttridge, D.; Yang, J.; Harper, S.Q. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann. Neurol. 2011, 69, 540–552. [Google Scholar] [CrossRef]
- Hinman, M.N.; Richardson, J.I.; Sockol, R.A.; Aronson, E.D.; Stednitz, S.J.; Murray, K.N.; Berglund, J.A.; Guillemin, K. Zebrafish mbnl mutants model physical and molecular phenotypes of myotonic dystrophy. Dis. Model Mech. 2021, 14, dmm045773. [Google Scholar] [CrossRef]
- Machuca-Tzili, L.E.; Buxton, S.; Thorpe, A.; Timson, C.M.; Wigmore, P.; Luther, P.K.; Brook, J.D. Zebrafish deficient for Muscleblind-like 2 exhibit features of myotonic dystrophy. Dis. Model Mech. 2011, 4, 381–392. [Google Scholar] [CrossRef]
- Todd, P.K.; Ackall, F.Y.; Hur, J.; Sharma, K.; Paulson, H.L.; Dowling, J.J. Transcriptional changes and developmental abnormalities in a zebrafish model of myotonic dystrophy type 1. Dis. Model Mech. 2014, 7, 143–155. [Google Scholar] [CrossRef]
- Kustermann, M.; Manta, L.; Paone, C.; Kustermann, J.; Lausser, L.; Wiesner, C.; Eichinger, L.; Clemen, C.S.; Schroder, R.; Kestler, H.A.; et al. Loss of the novel Vcp (valosin containing protein) interactor Washc4 interferes with autophagy-mediated proteostasis in striated muscle and leads to myopathy in vivo. Autophagy 2018, 14, 1911–1927. [Google Scholar] [CrossRef]
- Voisard, P.; Diofano, F.; Glazier, A.A.; Rottbauer, W.; Just, S. CRISPR/Cas9-Mediated Constitutive Loss of VCP (Valosin-Containing Protein) Impairs Proteostasis and Leads to Defective Striated Muscle Structure and Function In Vivo. Int. J. Mol. Sci. 2022, 23, 6722. [Google Scholar] [CrossRef]
- Buhrdel, J.B.; Hirth, S.; Kessler, M.; Westphal, S.; Forster, M.; Manta, L.; Wiche, G.; Schoser, B.; Schessl, J.; Schroder, R.; et al. In vivo characterization of human myofibrillar myopathy genes in zebrafish. Biochem. Biophys. Res. Commun. 2015, 461, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.R.; Mosley, A.N.; Zhou, Y.; O’Brien, K.F.; Sheng, X.; Chiang, K.; Davidson, A.J.; Volinski, J.M.; Zon, L.I.; Kunkel, L.M. The dystrophin associated protein complex in zebrafish. Hum. Mol. Genet. 2003, 12, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.P.; Nixon, S.J.; Hall, T.E.; Cowling, B.S.; Ferguson, C.; Morgan, G.P.; Schieber, N.L.; Fernandez-Rojo, M.A.; Bastiani, M.; Floetenmeyer, M.; et al. The caveolin-cavin system plays a conserved and critical role in mechanoprotection of skeletal muscle. J. Cell Biol. 2015, 210, 833–849. [Google Scholar] [CrossRef] [PubMed]
- Vieira, N.M.; Naslavsky, M.S.; Licinio, L.; Kok, F.; Schlesinger, D.; Vainzof, M.; Sanchez, N.; Kitajima, J.P.; Gal, L.; Cavacana, N.; et al. A defect in the RNA-processing protein HNRPDL causes limb-girdle muscular dystrophy 1G (LGMD1G). Hum. Mol. Genet. 2014, 23, 4103–4110. [Google Scholar] [CrossRef]
- Zhang, R.; Yang, J.; Zhu, J.; Xu, X. Depletion of zebrafish Tcap leads to muscular dystrophy via disrupting sarcomere-membrane interaction, not sarcomere assembly. Hum. Mol. Genet. 2009, 18, 4130–4140. [Google Scholar] [CrossRef]
- Steffen, L.S.; Guyon, J.R.; Vogel, E.D.; Howell, M.H.; Zhou, Y.; Weber, G.J.; Zon, L.I.; Kunkel, L.M. The zebrafish runzel muscular dystrophy is linked to the titin gene. Dev. Biol. 2007, 309, 180–192. [Google Scholar] [CrossRef]
- Schindler, R.F.; Scotton, C.; Zhang, J.; Passarelli, C.; Ortiz-Bonnin, B.; Simrick, S.; Schwerte, T.; Poon, K.L.; Fang, M.; Rinne, S.; et al. POPDC1(S201F) causes muscular dystrophy and arrhythmia by affecting protein trafficking. J. Clin. Investig. 2016, 126, 239–253. [Google Scholar] [CrossRef]
- Osborn, D.P.S.; Pond, H.L.; Mazaheri, N.; Dejardin, J.; Munn, C.J.; Mushref, K.; Cauley, E.S.; Moroni, I.; Pasanisi, M.B.; Sellars, E.A.; et al. Mutations in INPP5K Cause a Form of Congenital Muscular Dystrophy Overlapping Marinesco-Sjogren Syndrome and Dystroglycanopathy. Am. J. Hum. Genet. 2017, 100, 537–545. [Google Scholar] [CrossRef]
- Wiessner, M.; Roos, A.; Munn, C.J.; Viswanathan, R.; Whyte, T.; Cox, D.; Schoser, B.; Sewry, C.; Roper, H.; Phadke, R.; et al. Mutations in INPP5K, Encoding a Phosphoinositide 5-Phosphatase, Cause Congenital Muscular Dystrophy with Cataracts and Mild Cognitive Impairment. Am. J. Hum. Genet. 2017, 100, 523–536. [Google Scholar] [CrossRef]
- Ruparelia, A.A.; Oorschot, V.; Vaz, R.; Ramm, G.; Bryson-Richardson, R.J. Zebrafish models of BAG3 myofibrillar myopathy suggest a toxic gain of function leading to BAG3 insufficiency. Acta Neuropathol. 2014, 128, 821–833. [Google Scholar] [CrossRef]
- Kessler, M.; Kieltsch, A.; Kayvanpour, E.; Katus, H.A.; Schoser, B.; Schessl, J.; Just, S.; Rottbauer, W. A zebrafish model for FHL1-opathy reveals loss-of-function effects of human FHL1 mutations. Neuromuscul. Disord. 2018, 28, 521–531. [Google Scholar] [CrossRef]
- van der Meer, D.L.; Marques, I.J.; Leito, J.T.; Besser, J.; Bakkers, J.; Schoonheere, E.; Bagowski, C.P. Zebrafish cypher is important for somite formation and heart development. Dev. Biol. 2006, 299, 356–372. [Google Scholar] [CrossRef]
- Jahncke, J.N.; Wright, K.M. The many roles of dystroglycan in nervous system development and function: Dystroglycan and neural circuit development: Dystroglycan and neural circuit development. Dev. Dyn. 2023, 252, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Vannoy, C.H.; Leroy, V.; Lu, Q.L. Dose-Dependent Effects of FKRP Gene-Replacement Therapy on Functional Rescue and Longevity in Dystrophic Mice. Mol. Ther. Methods Clin. Dev. 2018, 11, 106–120. [Google Scholar] [CrossRef]
- Gupta, V.; Kawahara, G.; Gundry, S.R.; Chen, A.T.; Lencer, W.I.; Zhou, Y.; Zon, L.I.; Kunkel, L.M.; Beggs, A.H. The zebrafish dag1 mutant: A novel genetic model for dystroglycanopathies. Hum. Mol. Genet. 2011, 20, 1712–1725. [Google Scholar] [CrossRef]
- Serafini, P.R.; Feyder, M.J.; Hightower, R.M.; Garcia-Perez, D.; Vieira, N.M.; Lek, A.; Gibbs, D.E.; Moukha-Chafiq, O.; Augelli-Szafran, C.E.; Kawahara, G.; et al. A limb-girdle muscular dystrophy 2I model of muscular dystrophy identifies corrective drug compounds for dystroglycanopathies. JCI Insight 2018, 3, e120493. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.J.; Lin, C.H.; Li, M.; Nishtala, K.; Alaei, S.; Rossello, F.; Sonntag, C.; Hersey, L.; Miles, L.B.; Krisp, C.; et al. FKRP-dependent glycosylation of fibronectin regulates muscle pathology in muscular dystrophy. Nat. Commun. 2021, 12, 2951. [Google Scholar] [CrossRef] [PubMed]
- Avsar-Ban, E.; Ishikawa, H.; Manya, H.; Watanabe, M.; Akiyama, S.; Miyake, H.; Endo, T.; Tamaru, Y. Protein O-mannosylation is necessary for normal embryonic development in zebrafish. Glycobiology 2010, 20, 1089–1102. [Google Scholar] [CrossRef]
- Manzini, M.C.; Tambunan, D.E.; Hill, R.S.; Yu, T.W.; Maynard, T.M.; Heinzen, E.L.; Shianna, K.V.; Stevens, C.R.; Partlow, J.N.; Barry, B.J.; et al. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am. J. Hum. Genet. 2012, 91, 541–547. [Google Scholar] [CrossRef]
- Lin, Y.Y.; White, R.J.; Torelli, S.; Cirak, S.; Muntoni, F.; Stemple, D.L. Zebrafish Fukutin family proteins link the unfolded protein response with dystroglycanopathies. Hum. Mol. Genet. 2011, 20, 1763–1775. [Google Scholar] [CrossRef]
- Wood, A.J.; Muller, J.S.; Jepson, C.D.; Laval, S.H.; Lochmuller, H.; Bushby, K.; Barresi, R.; Straub, V. Abnormal vascular development in zebrafish models for fukutin and FKRP deficiency. Hum. Mol. Genet. 2011, 20, 4879–4890. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, P.; Bassett, D.; Lochmuller, H.; Bushby, K.; Straub, V. Developmental defects in a zebrafish model for muscular dystrophies associated with the loss of fukutin-related protein (FKRP). Brain 2008, 131 Pt 6, 1551–1561. [Google Scholar] [CrossRef]
- Kawahara, G.; Guyon, J.R.; Nakamura, Y.; Kunkel, L.M. Zebrafish models for human FKRP muscular dystrophies. Hum. Mol. Genet. 2010, 19, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Roscioli, T.; Kamsteeg, E.J.; Buysse, K.; Maystadt, I.; van Reeuwijk, J.; van den Elzen, C.; van Beusekom, E.; Riemersma, M.; Pfundt, R.; Vissers, L.E.; et al. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of alpha-dystroglycan. Nat. Genet. 2012, 44, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Praissman, J.L.; Willer, T.; Sheikh, M.O.; Toi, A.; Chitayat, D.; Lin, Y.Y.; Lee, H.; Stalnaker, S.H.; Wang, S.; Prabhakar, P.K.; et al. The functional O-mannose glycan on alpha-dystroglycan contains a phospho-ribitol primed for matriglycan addition. eLife 2016, 5, e14473. [Google Scholar] [CrossRef]
- Stevens, E.; Carss, K.J.; Cirak, S.; Foley, A.R.; Torelli, S.; Willer, T.; Tambunan, D.E.; Yau, S.; Brodd, L.; Sewry, C.A.; et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 2013, 92, 354–365. [Google Scholar] [CrossRef]
- Buysse, K.; Riemersma, M.; Powell, G.; van Reeuwijk, J.; Chitayat, D.; Roscioli, T.; Kamsteeg, E.J.; van den Elzen, C.; van Beusekom, E.; Blaser, S.; et al. Missense mutations in beta-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome. Hum. Mol. Genet. 2013, 22, 1746–1754. [Google Scholar] [CrossRef]
- Ardiccioni, C.; Clarke, O.B.; Tomasek, D.; Issa, H.A.; von Alpen, D.C.; Pond, H.L.; Banerjee, S.; Rajashankar, K.R.; Liu, Q.; Guan, Z.; et al. Structure of the polyisoprenyl-phosphate glycosyltransferase GtrB and insights into the mechanism of catalysis. Nat. Commun. 2016, 7, 10175. [Google Scholar] [CrossRef]
- Marchese, M.; Pappalardo, A.; Baldacci, J.; Verri, T.; Doccini, S.; Cassandrini, D.; Bruno, C.; Fiorillo, C.; Garcia-Gil, M.; Bertini, E.; et al. Dolichol-phosphate mannose synthase depletion in zebrafish leads to dystrophic muscle with hypoglycosylated alpha-dystroglycan. Biochem. Biophys. Res. Commun. 2016, 477, 137–143. [Google Scholar] [CrossRef]
- Di Costanzo, S.; Balasubramanian, A.; Pond, H.L.; Rozkalne, A.; Pantaleoni, C.; Saredi, S.; Gupta, V.A.; Sunu, C.M.; Yu, T.W.; Kang, P.B.; et al. POMK mutations disrupt muscle development leading to a spectrum of neuromuscular presentations. Hum. Mol. Genet. 2014, 23, 5781–5792. [Google Scholar] [CrossRef]
- Carss, K.J.; Stevens, E.; Foley, A.R.; Cirak, S.; Riemersma, M.; Torelli, S.; Hoischen, A.; Willer, T.; van Scherpenzeel, M.; Moore, S.A.; et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of alpha-dystroglycan. Am. J. Hum. Genet. 2013, 93, 29–41. [Google Scholar] [CrossRef]
- Henderson, C.A.; Gomez, C.G.; Novak, S.M.; Mi-Mi, L.; Gregorio, C.C. Overview of the Muscle Cytoskeleton. Compr. Physiol. 2017, 7, 891–944. [Google Scholar]
- Capetanaki, Y.; Bloch, R.J.; Kouloumenta, A.; Mavroidis, M.; Psarras, S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp. Cell Res. 2007, 313, 2063–2076. [Google Scholar] [CrossRef]
- Ramspacher, C.; Steed, E.; Boselli, F.; Ferreira, R.; Faggianelli, N.; Roth, S.; Spiegelhalter, C.; Messaddeq, N.; Trinh, L.; Liebling, M.; et al. Developmental Alterations in Heart Biomechanics and Skeletal Muscle Function in Desmin Mutants Suggest an Early Pathological Root for Desminopathies. Cell Rep. 2015, 11, 1564–1576. [Google Scholar] [CrossRef]
- Paulin, D.; Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 2004, 301, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Andersson-Lendahl, M.; Sejersen, T.; Arner, A. Knockdown of desmin in zebrafish larvae affects interfilament spacing and mechanical properties of skeletal muscle. J. Gen. Physiol. 2013, 141, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Kayman Kurekci, G.; Kural Mangit, E.; Koyunlar, C.; Unsal, S.; Saglam, B.; Ergin, B.; Gizer, M.; Uyanik, I.; Boustanabadimaralan Duz, N.; Korkusuz, P.; et al. Knockout of zebrafish desmin genes does not cause skeletal muscle degeneration but alters calcium flux. Sci. Rep. 2021, 11, 7505. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, H.A.; Hua, K.; Quigley, H.; Ivare, J.; Tesson, F.; Akimenko, M.A. A CRISPR/Cas9 zebrafish lamin A/C mutant model of muscular laminopathy. Dev. Dyn. 2022, 251, 645–661. [Google Scholar] [CrossRef] [PubMed]
- Birely, J.; Schneider, V.A.; Santana, E.; Dosch, R.; Wagner, D.S.; Mullins, M.C.; Granato, M. Genetic screens for genes controlling motor nerve-muscle development and interactions. Dev. Biol. 2005, 280, 162–176. [Google Scholar] [CrossRef]
- Ruparelia, A.A.; Zhao, M.; Currie, P.D.; Bryson-Richardson, R.J. Characterization and investigation of zebrafish models of filamin-related myofibrillar myopathy. Hum. Mol. Genet. 2012, 21, 4073–4083. [Google Scholar] [CrossRef]
- Koshimizu, E.; Imamura, S.; Qi, J.; Toure, J.; Valdez, D.M., Jr.; Carr, C.E.; Hanai, J.; Kishi, S. Embryonic senescence and laminopathies in a progeroid zebrafish model. PLoS ONE 2011, 6, e17688. [Google Scholar] [CrossRef] [PubMed]
- Sarparanta, J.; Jonson, P.H.; Golzio, C.; Sandell, S.; Luque, H.; Screen, M.; McDonald, K.; Stajich, J.M.; Mahjneh, I.; Vihola, A.; et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 2012, 44, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Nam, T.S.; Li, W.; Heo, S.H.; Lee, K.H.; Cho, A.; Shin, J.H.; Kim, Y.O.; Chae, J.H.; Kim, D.S.; Kim, M.K.; et al. A novel mutation in DNAJB6, p.(Phe91Leu), in childhood-onset LGMD1D with a severe phenotype. Neuromuscul. Disord. 2015, 25, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Guo, X.F.; Yang, X.Y.; Chong, M.; Cheng, J.; Li, G.; Gui, Y.H.; Lu, D.R. Delta-sarcoglycan is necessary for early heart and muscle development in zebrafish. Biochem. Biophys. Res. Commun. 2006, 344, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.R.; Mosley, A.N.; Jun, S.J.; Montanaro, F.; Steffen, L.S.; Zhou, Y.; Nigro, V.; Zon, L.I.; Kunkel, L.M. Delta-sarcoglycan is required for early zebrafish muscle organization. Exp. Cell Res. 2005, 304, 105–115. [Google Scholar] [CrossRef]
- Parton, R.G. Caveolae: Structure, Function, and Relationship to Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 111–136. [Google Scholar] [CrossRef]
- McNally, E.M.; de Sa Moreira, E.; Duggan, D.J.; Bonnemann, C.G.; Lisanti, M.P.; Lidov, H.G.; Vainzof, M.; Passos-Bueno, M.R.; Hoffman, E.P.; Zatz, M.; et al. Caveolin-3 in muscular dystrophy. Hum. Mol. Genet. 1998, 7, 871–877. [Google Scholar] [CrossRef]
- Nixon, S.J.; Wegner, J.; Ferguson, C.; Mery, P.F.; Hancock, J.F.; Currie, P.D.; Key, B.; Westerfield, M.; Parton, R.G. Zebrafish as a model for caveolin-associated muscle disease; caveolin-3 is required for myofibril organization and muscle cell patterning. Hum. Mol. Genet. 2005, 14, 1727–1743. [Google Scholar] [CrossRef]
- Postel, R.; Vakeel, P.; Topczewski, J.; Knoll, R.; Bakkers, J. Zebrafish integrin-linked kinase is required in skeletal muscles for strengthening the integrin-ECM adhesion complex. Dev. Biol. 2008, 318, 92–101. [Google Scholar] [CrossRef]
- Kawahara, G.; Serafini, P.R.; Myers, J.A.; Alexander, M.S.; Kunkel, L.M. Characterization of zebrafish dysferlin by morpholino knockdown. Biochem. Biophys. Res. Commun. 2011, 413, 358–363. [Google Scholar] [CrossRef]
- Roostalu, U.; Strahle, U. In vivo imaging of molecular interactions at damaged sarcolemma. Dev. Cell 2012, 22, 515–529. [Google Scholar] [CrossRef]
- De Oliveira, A.A.S.; Brigante, T.A.V.; Oliveira, D.P. Tail coiling assay in zebrafish (Danio rerio) embryos: Stage of development, promising positive control candidates, and selection of an appropriate organic solvent for screening of developmental neurotoxicity (DNT). Water 2021, 2, 119. [Google Scholar] [CrossRef]
- Ogungbemi, A.O.; Massei, R.; Altenburger, R.; Scholz, S.; Kuster, E. Assessing Combined Effects for Mixtures of Similar and Dissimilar Acting Neuroactive Substances on Zebrafish Embryo Movement. Toxics 2021, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Sztal, T.E.; Ruparelia, A.A.; Williams, C.; Bryson-Richardson, R.J. Using Touch-evoked Response and Locomotion Assays to Assess Muscle Performance and Function in Zebrafish. J. Vis. Exp. 2016, 116, e54431. [Google Scholar]
- Brustein, E.; Saint-Amant, L.; Buss, R.R.; Chong, M.; McDearmid, J.R.; Drapeau, P. Steps during the development of the zebrafish locomotor network. J. Physiol. Paris 2003, 97, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Drapeau, P.; Saint-Amant, L.; Buss, R.R.; Chong, M.; McDearmid, J.R.; Brustein, E. Development of the locomotor network in zebrafish. Prog. Neurobiol. 2002, 68, 85–111. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, M.; Zulian, A.; Menazza, S.; Petronilli, V.; Argenton, F.; Merlini, L.; Sabatelli, P.; Bernardi, P. Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 2017, 125 Pt B, 122–131. [Google Scholar] [CrossRef]
- Stocco, A.; Smolina, N.; Sabatelli, P.; Sileikyte, J.; Artusi, E.; Mouly, V.; Cohen, M.; Forte, M.; Schiavone, M.; Bernardi, P. Treatment with a triazole inhibitor of the mitochondrial permeability transition pore fully corrects the pathology of sapje zebrafish lacking dystrophin. Pharmacol. Res. 2021, 165, 105421. [Google Scholar] [CrossRef]
- Zulian, A.; Rizzo, E.; Schiavone, M.; Palma, E.; Tagliavini, F.; Blaauw, B.; Merlini, L.; Maraldi, N.M.; Sabatelli, P.; Braghetta, P.; et al. NIM811, a cyclophilin inhibitor without immunosuppressive activity, is beneficial in collagen VI congenital muscular dystrophy models. Hum. Mol. Genet. 2014, 23, 5353–5363. [Google Scholar] [CrossRef]
- Mrinalini, R.; Tamilanban, T.; Naveen Kumar, V.; Manasa, K. Zebrafish—The Neurobehavioural Model in Trend. Neuroscience 2022, in press. [Google Scholar] [CrossRef]
- Ahmad, F.; Noldus, L.P.; Tegelenbosch, R.A.; Richardson, M.K. Zebrafish embryos and larvae in behavioural assays. Behaviour 2012, 149, 1241–1281. [Google Scholar]
- Orger, M.B.; de Polavieja, G.G. Zebrafish Behavior: Opportunities and Challenges. Annu. Rev. Neurosci. 2017, 40, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Rosa, J.G.S.; Lima, C.; Lopes-Ferreira, M. Zebrafish Larvae Behavior Models as a Tool for Drug Screenings and Pre-Clinical Trials: A Review. Int. J. Mol. Sci. 2022, 23, 6647. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Tsao, C.W.; Fang, Y.H. A Miniature Intermittent-Flow Respirometry System with a 3D-Printed, Palm-Sized Zebrafish Treadmill for Measuring Rest and Activity Metabolic Rates. Sensors 2020, 20, 5088. [Google Scholar] [CrossRef] [PubMed]
- Masse, A.J.; Thomas, J.K.; Janz, D.M. Reduced swim performance and aerobic capacity in adult zebrafish exposed to waterborne selenite. Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2013, 157, 266–271. [Google Scholar] [CrossRef]
- Palstra, A.P.; Kals, J.; Bohm, T.; Bastiaansen, J.W.M.; Komen, H. Swimming Performance and Oxygen Consumption as Non-lethal Indicators of Production Traits in Atlantic Salmon and Gilthead Seabream. Front. Physiol. 2020, 11, 759. [Google Scholar] [CrossRef]
- Wakamatsu, Y.; Ogino, K.; Hirata, H. Swimming capability of zebrafish is governed by water temperature, caudal fin length and genetic background. Sci. Rep. 2019, 9, 16307. [Google Scholar] [CrossRef]
- Cresci, A.; De Rosa, R.; Agnisola, C. Assessing the Influence of Personality on Sensitivity to Magnetic Fields in Zebrafish. Available online: https://app.jove.com/it/v/59229/assessing-the-influence-of-personality-on-sensitivity-to-magnetic-fields-in-zebrafish (accessed on 27 April 2023).
- Sloboda, D.D.; Claflin, D.R.; Dowling, J.J.; Brooks, S.V. Force measurement during contraction to assess muscle function in zebrafish larvae. J. Vis. Exp. 2013, e50539. [Google Scholar] [CrossRef]
- Widrick, J.J.; Alexander, M.S.; Sanchez, B.; Gibbs, D.E.; Kawahara, G.; Beggs, A.H.; Kunkel, L.M. Muscle dysfunction in a zebrafish model of Duchenne muscular dystrophy. Physiol. Genom. 2016, 48, 850–860. [Google Scholar] [CrossRef]
- Dowling, J.J.; Vreede, A.P.; Low, S.E.; Gibbs, E.M.; Kuwada, J.Y.; Bonnemann, C.G.; Feldman, E.L. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009, 5, e1000372. [Google Scholar] [CrossRef]
- Telfer, W.R.; Nelson, D.D.; Waugh, T.; Brooks, S.V.; Dowling, J.J. Neb: A zebrafish model of nemaline myopathy due to nebulin mutation. Dis. Model Mech. 2012, 5, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Saint-Amant, L.; Waterbury, J.; Cui, W.; Zhou, W.; Li, Q.; Goldman, D.; Granato, M.; Kuwada, J.Y. accordion, a zebrafish behavioral mutant, has a muscle relaxation defect due to a mutation in the ATPase Ca2+ pump SERCA1. Development 2004, 131, 5457–5468. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Beggs, A.H.; Gupta, V.A. Analysis of Skeletal Muscle Defects in Larval Zebrafish by Birefringence and Touch-Evoke Escape Response Assays. Available online: https://app.jove.com/it/v/50925/analysis-of-skeletal-muscle-defects-in-larval-zebrafish-by-birefringence-and-touch-evoke-escape-response-assays (accessed on 27 April 2023).
- Berger, J.; Sztal, T.; Currie, P.D. Quantification of birefringence readily measures the level of muscle damage in zebrafish. Biochem. Biophys. Res. Commun. 2012, 423, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Li, M.; Berger, S.; Meilak, M.; Rientjes, J.; Currie, P.D. Effect of Ataluren on dystrophin mutations. J. Cell. Mol. Med. 2020, 24, 6680–6689. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, G.; Karpf, J.A.; Myers, J.A.; Alexander, M.S.; Guyon, J.R.; Kunkel, L.M. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2011, 108, 5331–5336. [Google Scholar] [CrossRef] [PubMed]
- Moro, E.; Vettori, A.; Porazzi, P.; Schiavone, M.; Rampazzo, E.; Casari, A.; Ek, O.; Facchinello, N.; Astone, M.; Zancan, I.; et al. Generation and application of signaling pathway reporter lines in zebrafish. Mol. Genet. Genom. 2013, 288, 231–242. [Google Scholar] [CrossRef]
- Goody, M.; Henry, C. Phalloidin Staining and Immunohistochemistry of Zebrafish Embryos. Bio-Protocol 2013, 3, e786. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, A.; Huang, L.; Zhu, X.; Hu, Q.; Zhang, Y.; Chen, X.; Li, F.; Wang, Q.; Wang, H.; et al. Illuminating NAD(+) Metabolism in Live Cells and In Vivo Using a Genetically Encoded Fluorescent Sensor. Dev. Cell 2020, 53, 240–252.e7. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, A.; Shi, M.; Chen, X.; Liu, R.; Li, T.; Zhang, C.; Zhang, Z.; Zhu, L.; Ju, Z.; et al. Analysis of redox landscapes and dynamics in living cells and in vivo using genetically encoded fluorescent sensors. Nat. Protoc. 2018, 13, 2362–2386. [Google Scholar] [CrossRef]
- Scott, C.A.; Carney, T.J.; Amaya, E. Aerobic glycolysis is important for zebrafish larval wound closure and tail regeneration. Wound Repair Regen. 2022, 30, 665–680. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. Data on metabolic-dependent antioxidant response in the cardiovascular tissues of living zebrafish under stress conditions. Data Brief 2017, 12, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Lackmann, C.; Santos, M.M.; Rainieri, S.; Barranco, A.; Hollert, H.; Spirhanzlova, P.; Velki, M.; Seiler, T.B. Novel procedures for whole organism detection and quantification of fluorescence as a measurement for oxidative stress in zebrafish (Danio rerio) larvae. Chemosphere 2018, 197, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Mugoni, V.; Camporeale, A.; Santoro, M.M. Analysis of oxidative stress in zebrafish embryos. J. Vis. Exp. 2014, 51328. [Google Scholar] [CrossRef]
- Jackson, H.E.; Ono, Y.; Wang, X.; Elworthy, S.; Cunliffe, V.T.; Ingham, P.W. The role of Sox6 in zebrafish muscle fiber type specification. Skelet. Muscle 2015, 5, 2. [Google Scholar] [CrossRef]
- Kettunen, P. Calcium Imaging in the Zebrafish. Adv. Exp. Med. Biol. 2020, 1131, 901–942. [Google Scholar]
- Okamoto, S.I.; Hatta, K. Ca2+-imaging and photo-manipulation of the simple gut of zebrafish larvae in vivo. Sci. Rep. 2022, 12, 2018. [Google Scholar] [CrossRef]
- Tsuruwaka, Y.; Shimada, E.; Tsutsui, K.; Ogawa, T. Ca2+ dynamics in zebrafish morphogenesis. PeerJ 2017, 5, e2894. [Google Scholar] [CrossRef]
- Vanwalleghem, G.; Constantin, L.; Scott, E.K. Calcium Imaging and the Curse of Negativity. Front. Neural. Circuits 2020, 14, 607391. [Google Scholar] [CrossRef]
- Kole, K.; Voesenek, B.J.B.; Brinia, M.E.; Petersen, N.; Kole, M.H.P. Parvalbumin basket cell myelination accumulates axonal mitochondria to internodes. Nat. Commun. 2022, 13, 7598. [Google Scholar] [CrossRef]
- Bond, S.T.; McEwen, K.A.; Yoganantharajah, P.; Gibert, Y. Live Metabolic Profile Analysis of Zebrafish Embryos Using a Seahorse XF 24 Extracellular Flux Analyzer. Methods Mol. Biol. 2018, 1797, 393–401. [Google Scholar]
- Housley, M.P.; Njaine, B.; Ricciardi, F.; Stone, O.A.; Holper, S.; Kruger, M.; Kostin, S.; Stainier, D.Y. Cavin4b/Murcb Is Required for Skeletal Muscle Development and Function in Zebrafish. PLoS Genet. 2016, 12, e1006099. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, M.; Cafora, M.; Bragato, C.; Capitanio, D.; Marasca, F.; Bodega, B.; De Palma, C.; Mora, M.; Gelfi, C.; Marozzi, A.; et al. Targeting HDAC8 to ameliorate skeletal muscle differentiation in Duchenne muscular dystrophy. Pharmacol. Res. 2021, 170, 105750. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, N.; Vanbeselaere, J.; Chang, L.Y.; Yu, S.Y.; Ducrocq, L.; Harduin-Lepers, A.; Kurata, J.; Aoki-Kinoshita, K.F.; Sato, C.; Khoo, K.H.; et al. Systems glycomics of adult zebrafish identifies organ-specific sialylation and glycosylation patterns. Nat. Commun. 2018, 9, 4647. [Google Scholar] [CrossRef] [PubMed]
- Vettori, A.; Greenald, D.; Wilson, G.K.; Peron, M.; Facchinello, N.; Markham, E.; Sinnakaruppan, M.; Matthews, L.C.; McKeating, J.A.; Argenton, F.; et al. Glucocorticoids promote Von Hippel Lindau degradation and Hif-1alpha stabilization. Proc. Natl. Acad. Sci. USA 2017, 114, 9948–9953. [Google Scholar] [CrossRef]
- Barresi, M.J.; Stickney, H.L.; Devoto, S.H. The zebrafish slow-muscle-omitted gene product is required for Hedgehog signal transduction and the development of slow muscle identity. Development 2000, 127, 2189–2199. [Google Scholar] [CrossRef]
- Sasaki, H.; Hui, C.; Nakafuku, M.; Kondoh, H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 1997, 124, 1313–1322. [Google Scholar] [CrossRef]
- Corallo, D.; Schiavinato, A.; Trapani, V.; Moro, E.; Argenton, F.; Bonaldo, P. Emilin3 is required for notochord sheath integrity and interacts with Scube2 to regulate notochord-derived Hedgehog signals. Development 2013, 140, 4594–4601. [Google Scholar] [CrossRef]
- Mich, J.K.; Payumo, A.Y.; Rack, P.G.; Chen, J.K. In vivo imaging of Hedgehog pathway activation with a nuclear fluorescent reporter. PLoS ONE 2014, 9, e103661. [Google Scholar] [CrossRef]
- Barolo, S. Transgenic Wnt/TCF pathway reporters: All you need is Lef? Oncogene 2006, 25, 7505–7511. [Google Scholar] [CrossRef]
- Dorsky, R.I.; Sheldahl, L.C.; Moon, R.T. A transgenic Lef1/beta-catenin-dependent reporter is expressed in spatially restricted domains throughout zebrafish development. Dev. Biol. 2002, 241, 229–237. [Google Scholar] [CrossRef]
- Moro, E.; Ozhan-Kizil, G.; Mongera, A.; Beis, D.; Wierzbicki, C.; Young, R.M.; Bournele, D.; Domenichini, A.; Valdivia, L.E.; Lum, L.; et al. In vivo Wnt signaling tracing through a transgenic biosensor fish reveals novel activity domains. Dev. Biol. 2012, 366, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Pyati, U.J.; Webb, A.E.; Kimelman, D. Transgenic zebrafish reveal stage-specific roles for Bmp signaling in ventral and posterior mesoderm development. Development 2005, 132, 2333–2343. [Google Scholar] [CrossRef]
- Casari, A.; Schiavone, M.; Facchinello, N.; Vettori, A.; Meyer, D.; Tiso, N.; Moro, E.; Argenton, F. A Smad3 transgenic reporter reveals TGF-beta control of zebrafish spinal cord development. Dev. Biol. 2014, 396, 81–93. [Google Scholar] [CrossRef]
- Collery, R.F.; Link, B.A. Dynamic smad-mediated BMP signaling revealed through transgenic zebrafish. Dev. Dyn. 2011, 240, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Ramel, M.C.; Hill, C.S. The ventral to dorsal BMP activity gradient in the early zebrafish embryo is determined by graded expression of BMP ligands. Dev. Biol. 2013, 378, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Ramel, M.C.; Howell, M.; Hill, C.S. SNW1 is a critical regulator of spatial BMP activity, neural plate border formation, and neural crest specification in vertebrate embryos. PLoS Biol. 2011, 9, e1000593. [Google Scholar] [CrossRef] [PubMed]
- Giuliodori, A.; Beffagna, G.; Marchetto, G.; Fornetto, C.; Vanzi, F.; Toppo, S.; Facchinello, N.; Santimaria, M.; Vettori, A.; Rizzo, S.; et al. Loss of cardiac Wnt/beta-catenin signalling in desmoplakin-deficient AC8 zebrafish models is rescuable by genetic and pharmacological intervention. Cardiovasc. Res. 2018, 114, 1082–1097. [Google Scholar] [CrossRef]
- Merkulov, V.M.; Merkulova, T.I. Structural variants of glucocorticoid receptor binding sites and different versions of positive glucocorticoid responsive elements: Analysis of GR-TRRD database. J. Steroid Biochem. Mol. Biol. 2009, 115, 1–8. [Google Scholar] [CrossRef]
- Benato, F.; Colletti, E.; Skobo, T.; Moro, E.; Colombo, L.; Argenton, F.; Dalla Valle, L. A living biosensor model to dynamically trace glucocorticoid transcriptional activity during development and adult life in zebrafish. Mol. Cell. Endocrinol. 2014, 392, 60–72. [Google Scholar] [CrossRef]
- Wang, X.J.; Hayes, J.D.; Wolf, C.R. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006, 66, 10983–10994. [Google Scholar] [CrossRef]
- Badenetti, L.; Manzoli, R.; Rubin, M.; Cozza, G.; Moro, E. Monitoring Nrf2/ARE Pathway Activity with a New Zebrafish Reporter System. Int. J. Mol. Sci. 2023, 24, 6804. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.D.; Gurevich, D.B.; Sonntag, C.; Hersey, L.; Alaei, S.; Nim, H.T.; Siegel, A.; Hall, T.E.; Rossello, F.J.; Boyd, S.E.; et al. Muscle Stem Cells Undergo Extensive Clonal Drift during Tissue Growth via Meox1-Mediated Induction of G2 Cell-Cycle Arrest. Cell Stem Cell 2017, 21, 107–119.e6. [Google Scholar] [CrossRef] [PubMed]
- Seger, C.; Hargrave, M.; Wang, X.; Chai, R.J.; Elworthy, S.; Ingham, P.W. Analysis of Pax7 expressing myogenic cells in zebrafish muscle development, injury, and models of disease. Dev. Dyn. 2011, 240, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Ellingsen, S.; Laplante, M.A.; Konig, M.; Kikuta, H.; Furmanek, T.; Hoivik, E.A.; Becker, T.S. Large-scale enhancer detection in the zebrafish genome. Development 2005, 132, 3799–3811. [Google Scholar] [CrossRef]
- Ingham, P.W.; Kim, H.R. Hedgehog signalling and the specification of muscle cell identity in the zebrafish embryo. Exp. Cell Res. 2005, 306, 336–342. [Google Scholar] [CrossRef]
- Maurya, A.K.; Tan, H.; Souren, M.; Wang, X.; Wittbrodt, J.; Ingham, P.W. Integration of Hedgehog and BMP signalling by the engrailed2a gene in the zebrafish myotome. Development 2011, 138, 755–765. [Google Scholar] [CrossRef]
- Nguyen, P.D.; Currie, P.D. Guidelines and best practices in successfully using Zebrabow for lineage tracing multiple cells within tissues. Methods 2018, 150, 63–67. [Google Scholar] [CrossRef]
- Gurevich, D.B.; Nguyen, P.D.; Siegel, A.L.; Ehrlich, O.V.; Sonntag, C.; Phan, J.M.; Berger, S.; Ratnayake, D.; Hersey, L.; Berger, J.; et al. Asymmetric division of clonal muscle stem cells coordinates muscle regeneration in vivo. Science 2016, 353, aad9969. [Google Scholar] [CrossRef]
- Gong, Z.; Wan, H.; Tay, T.L.; Wang, H.; Chen, M.; Yan, T. Development of transgenic fish for ornamental and bioreactor by strong expression of fluorescent proteins in the skeletal muscle. Biochem. Biophys. Res. Commun. 2003, 308, 58–63. [Google Scholar] [CrossRef]
- Ju, B.; Chong, S.W.; He, J.; Wang, X.; Xu, Y.; Wan, H.; Tong, Y.; Yan, T.; Korzh, V.; Gong, Z. Recapitulation of fast skeletal muscle development in zebrafish by transgenic expression of GFP under the mylz2 promoter. Dev. Dyn. 2003, 227, 14–26. [Google Scholar] [CrossRef]
- Moore, C.A.; Parkin, C.A.; Bidet, Y.; Ingham, P.W. A role for the Myoblast city homologues Dock1 and Dock5 and the adaptor proteins Crk and Crk-like in zebrafish myoblast fusion. Development 2007, 134, 3145–3153. [Google Scholar] [CrossRef]
- Berger, J.; Tarakci, H.; Berger, S.; Li, M.; Hall, T.E.; Arner, A.; Currie, P.D. Loss of Tropomodulin4 in the zebrafish mutant trage causes cytoplasmic rod formation and muscle weakness reminiscent of nemaline myopathy. Dis. Model Mech. 2014, 7, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Higashijima, S.; Okamoto, H.; Ueno, N.; Hotta, Y.; Eguchi, G. High-frequency generation of transgenic zebrafish which reliably express GFP in whole muscles or the whole body by using promoters of zebrafish origin. Dev. Biol. 1997, 192, 289–299. [Google Scholar] [CrossRef]
- Meneghetti, G.; Skobo, T.; Chrisam, M.; Facchinello, N.; Fontana, C.M.; Bellesso, S.; Sabatelli, P.; Raggi, F.; Cecconi, F.; Bonaldo, P.; et al. The epg5 knockout zebrafish line: A model to study Vici syndrome. Autophagy 2019, 15, 1438–1454. [Google Scholar] [CrossRef]
- Ellis, K.; Bagwell, J.; Bagnat, M. Notochord vacuoles are lysosome-related organelles that function in axis and spine morphogenesis. J. Cell Biol. 2013, 200, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Clark, B.S.; Winter, M.; Cohen, A.R.; Link, B.A. Generation of Rab-based transgenic lines for in vivo studies of endosome biology in zebrafish. Dev. Dyn. 2011, 240, 2452–2465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, T.; Jian, C.; Lei, L.; Han, P.; Lv, Q.; Yang, R.; Zhou, X.; Xu, J.; Hu, Y.; et al. Remodeling of Mitochondrial Flashes in Muscular Development and Dystrophy in Zebrafish. PLoS ONE 2015, 10, e0132567. [Google Scholar] [CrossRef]
- Kim, M.J.; Kang, K.H.; Kim, C.H.; Choi, S.Y. Real-time imaging of mitochondria in transgenic zebrafish expressing mitochondrially targeted GFP. Biotechniques 2008, 45, 331–334. [Google Scholar]
- Facchinello, N.; Laquatra, C.; Locatello, L.; Beffagna, G.; Branas Casas, R.; Fornetto, C.; Dinarello, A.; Martorano, L.; Vettori, A.; Risato, G.; et al. Efficient clofilium tosylate-mediated rescue of POLG-related disease phenotypes in zebrafish. Cell Death Dis. 2021, 12, 100. [Google Scholar] [CrossRef]
- Chen, Y.H.; Wang, Y.H.; Chang, M.Y.; Lin, C.Y.; Weng, C.W.; Westerfield, M.; Tsai, H.J. Multiple upstream modules regulate zebrafish myf5 expression. BMC Dev. Biol. 2007, 7, 1. [Google Scholar] [CrossRef]
- Elworthy, S.; Hargrave, M.; Knight, R.; Mebus, K.; Ingham, P.W. Expression of multiple slow myosin heavy chain genes reveals a diversity of zebrafish slow twitch muscle fibres with differing requirements for Hedgehog and Prdm1 activity. Development 2008, 135, 2115–2126. [Google Scholar] [CrossRef] [PubMed]
- Asaduzzaman, M.; Kinoshita, S.; Bhuiyan, S.S.; Asakawa, S.; Watabe, S. Stimulatory and inhibitory mechanisms of slow muscle-specific myosin heavy chain gene expression in fish: Transient and transgenic analysis of torafugu MYH(M86-2) promoter in zebrafish embryos. Exp. Cell Res. 2013, 319, 820–837. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.C.; Teets, E.M.; Ratnayake, D.; Duy, P.Q.; Currie, P.D.; Amacher, S.L. Muscle precursor cell movements in zebrafish are dynamic and require Six family genes. Development 2019, 146, dev171421. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.D.; Hollway, G.E.; Sonntag, C.; Miles, L.B.; Hall, T.E.; Berger, S.; Fernandez, K.J.; Gurevich, D.B.; Cole, N.J.; Alaei, S.; et al. Haematopoietic stem cell induction by somite-derived endothelial cells controlled by meox1. Nature 2014, 512, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.A.; Freundlich, T.; Weissman, T.A.; Schoppik, D.; Wang, X.C.; Zimmerman, S.; Ciruna, B.; Sanes, J.R.; Lichtman, J.W.; Schier, A.F. Zebrabow: Multispectral cell labeling for cell tracing and lineage analysis in zebrafish. Development 2013, 140, 2835–2846. [Google Scholar] [CrossRef]
- Wasala, N.B.; Chen, S.J.; Duan, D. Duchenne muscular dystrophy animal models for high-throughput drug discovery and precision medicine. Expert Opin. Drug Discov. 2020, 15, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef]
- Calyjur, P.C.; Almeida Cde, F.; Ayub-Guerrieri, D.; Ribeiro, A.F., Jr.; Fernandes Sde, A.; Ishiba, R.; Santos, A.L.; Onofre-Oliveira, P.; Vainzof, M. The mdx Mutation in the 129/Sv Background Results in a Milder Phenotype: Transcriptome Comparative Analysis Searching for the Protective Factors. PLoS ONE 2016, 11, e0150748. [Google Scholar] [CrossRef]
- Coley, W.D.; Bogdanik, L.; Vila, M.C.; Yu, Q.; Van Der Meulen, J.H.; Rayavarapu, S.; Novak, J.S.; Nearing, M.; Quinn, J.L.; Saunders, A.; et al. Effect of genetic background on the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 2016, 25, 130–145. [Google Scholar] [CrossRef]
- Hammers, D.W.; Hart, C.C.; Matheny, M.K.; Wright, L.A.; Armellini, M.; Barton, E.R.; Sweeney, H.L. The D2.mdx mouse as a preclinical model of the skeletal muscle pathology associated with Duchenne muscular dystrophy. Sci. Rep. 2020, 10, 14070. [Google Scholar] [CrossRef]
- Zon, L.I.; Peterson, R.T. In vivo drug discovery in the zebrafish. Nat. Rev. Drug Discov. 2005, 4, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Licitra, R.; Marchese, M.; Brogi, L.; Fronte, B.; Pitto, L.; Santorelli, F.M. Nutraceutical Screening in a Zebrafish Model of Muscular Dystrophy: Gingerol as a Possible Food Aid. Nutrients 2021, 13, 998. [Google Scholar] [CrossRef] [PubMed]
- Waugh, T.A.; Horstick, E.; Hur, J.; Jackson, S.W.; Davidson, A.E.; Li, X.; Dowling, J.J. Fluoxetine prevents dystrophic changes in a zebrafish model of Duchenne muscular dystrophy. Hum. Mol. Genet. 2014, 23, 4651–4662. [Google Scholar] [CrossRef] [PubMed]
- Farr, G.H., 3rd; Morris, M.; Gomez, A.; Pham, T.; Kilroy, E.; Parker, E.U.; Said, S.; Henry, C.; Maves, L. A novel chemical-combination screen in zebrafish identifies epigenetic small molecule candidates for the treatment of Duchenne muscular dystrophy. Skelet. Muscle 2020, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, J.M.; Lambert, M.R.; Gibbs, D.E.; Conner, J.R.; Krikorian, G.L.; Pareek, P.; Rago, C.; Kunkel, L.M. Effect of serotonin modulation on dystrophin-deficient zebrafish. Biol. Open 2020, 9, bio53363. [Google Scholar] [CrossRef]
- Lambert, M.R.; Spinazzola, J.M.; Widrick, J.J.; Pakula, A.; Conner, J.R.; Chin, J.E.; Owens, J.M.; Kunkel, L.M. PDE10A Inhibition Reduces the Manifestation of Pathology in DMD Zebrafish and Represses the Genetic Modifier PITPNA. Mol. Ther. 2021, 29, 1086–1101. [Google Scholar] [CrossRef]
- Ketley, A.; Chen, C.Z.; Li, X.; Arya, S.; Robinson, T.E.; Granados-Riveron, J.; Udosen, I.; Morris, G.E.; Holt, I.; Furling, D.; et al. High-content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC-independent pathway in myotonic dystrophy cell lines. Hum. Mol. Genet. 2014, 23, 1551–1562. [Google Scholar] [CrossRef]
- Roy, S.; Sileikyte, J.; Schiavone, M.; Neuenswander, B.; Argenton, F.; Aube, J.; Hedrick, M.P.; Chung, T.D.; Forte, M.A.; Bernardi, P.; et al. Discovery, Synthesis, and Optimization of Diarylisoxazole-3-carboxamides as Potent Inhibitors of the Mitochondrial Permeability Transition Pore. ChemMedChem 2015, 10, 1655–1671. [Google Scholar] [CrossRef]
- Sileikyte, J.; Devereaux, J.; de Jong, J.; Schiavone, M.; Jones, K.; Nilsen, A.; Bernardi, P.; Forte, M.; Cohen, M.S. Second-Generation Inhibitors of the Mitochondrial Permeability Transition Pore with Improved Plasma Stability. ChemMedChem 2019, 14, 1771–1782. [Google Scholar] [CrossRef]
| Gene Symbol | Protein | Technique | Ref. |
|---|---|---|---|
| lama2 | Laminin-α2 | ENU screen ENU screen | [8] [38] |
| col6a1 | Collagen, type VI, alpha 1 | CRISPR/Cas9 TALEN mut. MO | [44] [45] [46] |
| col6a3 | Collagen, type VI, alpha 3 | MO | [46] |
| col22a1 | Collagen, type XXII, alpha 1 | MO CRISPR/Cas9 | [43] [47] |
| Gene Symbol | Protein | Technique | Ref. |
|---|---|---|---|
| dmd | Dystrophin | Genetic screen ENU screen MO | [49] [52] [62] |
| dux4 | Double homeobox protein 4 | mRNA injection Tol2 transposon system Tol2 transposon system | [53] [54] [55] |
| cavin1a/b | Caveolae-associated protein 1 | MO | [63] |
| hnrnpdl | Heterogeneous nuclear ribonucleoprotein D-like | MO | [64] |
| mbnl1/2/3 | Muscleblind-like splicing regulator | CRISPR/Cas9 mRNA injection | [56] [58] |
| vcp | Valosin containing protein | CRISPR/Cas9 MO | [59] [60] [61] |
| mbnl2 | Muscleblind-like splicing regulator 2 | MO | [57] |
| tcap | Telethonin | MO | [65] |
| ttn | Titin | MO | [66] |
| bves | Blood vessel epicardial substance | MO; TALEN mut. | [67] |
| inpp5ka/b | Inositol polyphosphate 5-phosphatase K | MO | [68,69] |
| craa/b | Cristallin | MO | [61] |
| bag3 | BCL2 associated athanogene 3 | MO | [61,70] |
| fhl1a/b | Four and a half LIM domains 1 | MO | [61,71] |
| ldb3 | LIM domain binding 3 | MO | [61,72] |
| Gene Symbol | Protein | Technique | Ref. |
|---|---|---|---|
| Primary dystroglycanopathy | |||
| dag1 | Dystroglycan | MO ENU screen | [48] [75] |
| Secondary dystroglycanopathies | |||
| pomt1 | O-mannosyl transferase | MO | [78] |
| pomt2 | O-mannosyl transferase | MO | [78] |
| pomgnt2 | O-mannose β-1,4-Nacetylglucosaminyltransferase | MO | [79] |
| fktn | Fukutin | MO MO | [80] [81] |
| fkrp | Fukutin-related protein | TALEN mut.; Tol2 transposon system Zinc finger nuclease KO; CRISPR/Cas9 MO MO MO MO | [76] [77] [80] [81] [82] [83] |
| ispd | CDP-ribitol pyrophosphorylase | MO | [84] |
| rxylt1 | β−1,4-xylosyl transferase | MO | [85] |
| b3galnt2 | β−1,3-N-acetylgalactosaminyltransferase | MO | [86] |
| b4gat1 | β−1,4-glucuronyltransferase | MO | [87] |
| dpm1 | Dolichol–phosphate mannose synthase | MO | [88,89] |
| dpm2 | Dolichol–phosphate mannose synthase | MO | [89] |
| dpm3 | Dolichol–phosphate mannose synthase | MO | [89] |
| pomk | O-mannose kinase | MO | [90] |
| gmppb | GDP-mannose pyrophosphorylase | MO | [91] |
| Gene Symbol | Protein | Technique | Ref. |
|---|---|---|---|
| desma/b | Desmin | MO ENU screen MO | [61] [94] [96] |
| lmna | Lamin-A/C | CRISPR/Cas9 mut. MO | [98] [101] |
| myot | Myotilin | MO | [61] |
| dnajb6a/b | dnaJ homolog subfamily B member 6 | MO MO mRNA injection | [61] [102] [103] |
| pleca/b | Plectin | MO | [61] |
| flnca/b | Filamin C | ENU screen, MO; Genetic screen | [99] [100] |
| sgcd | δ-sarcoglycan | MO | [104,105] |
| dmpk | Myotonic dystrophy protein kinase | mRNA injection | [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tesoriero, C.; Greco, F.; Cannone, E.; Ghirotto, F.; Facchinello, N.; Schiavone, M.; Vettori, A. Modeling Human Muscular Dystrophies in Zebrafish: Mutant Lines, Transgenic Fluorescent Biosensors, and Phenotyping Assays. Int. J. Mol. Sci. 2023, 24, 8314. https://doi.org/10.3390/ijms24098314
Tesoriero C, Greco F, Cannone E, Ghirotto F, Facchinello N, Schiavone M, Vettori A. Modeling Human Muscular Dystrophies in Zebrafish: Mutant Lines, Transgenic Fluorescent Biosensors, and Phenotyping Assays. International Journal of Molecular Sciences. 2023; 24(9):8314. https://doi.org/10.3390/ijms24098314
Chicago/Turabian StyleTesoriero, Chiara, Francesca Greco, Elena Cannone, Francesco Ghirotto, Nicola Facchinello, Marco Schiavone, and Andrea Vettori. 2023. "Modeling Human Muscular Dystrophies in Zebrafish: Mutant Lines, Transgenic Fluorescent Biosensors, and Phenotyping Assays" International Journal of Molecular Sciences 24, no. 9: 8314. https://doi.org/10.3390/ijms24098314
APA StyleTesoriero, C., Greco, F., Cannone, E., Ghirotto, F., Facchinello, N., Schiavone, M., & Vettori, A. (2023). Modeling Human Muscular Dystrophies in Zebrafish: Mutant Lines, Transgenic Fluorescent Biosensors, and Phenotyping Assays. International Journal of Molecular Sciences, 24(9), 8314. https://doi.org/10.3390/ijms24098314