
Secretome Analyses Identify FKBP4 as a GBA1-Associated Protein in CSF and iPS Cells from Parkinson’s Disease Patients with GBA1 Mutations

 , , ,
, , , 
Abstract
1. Introduction
2. Results
2.1. Proteins Significantly Altered in GBA1-PD CSF
2.2. CSF CRKL Levels and UPDRS-III Scores Oppositely Correlate in GBA1-PD and iPD
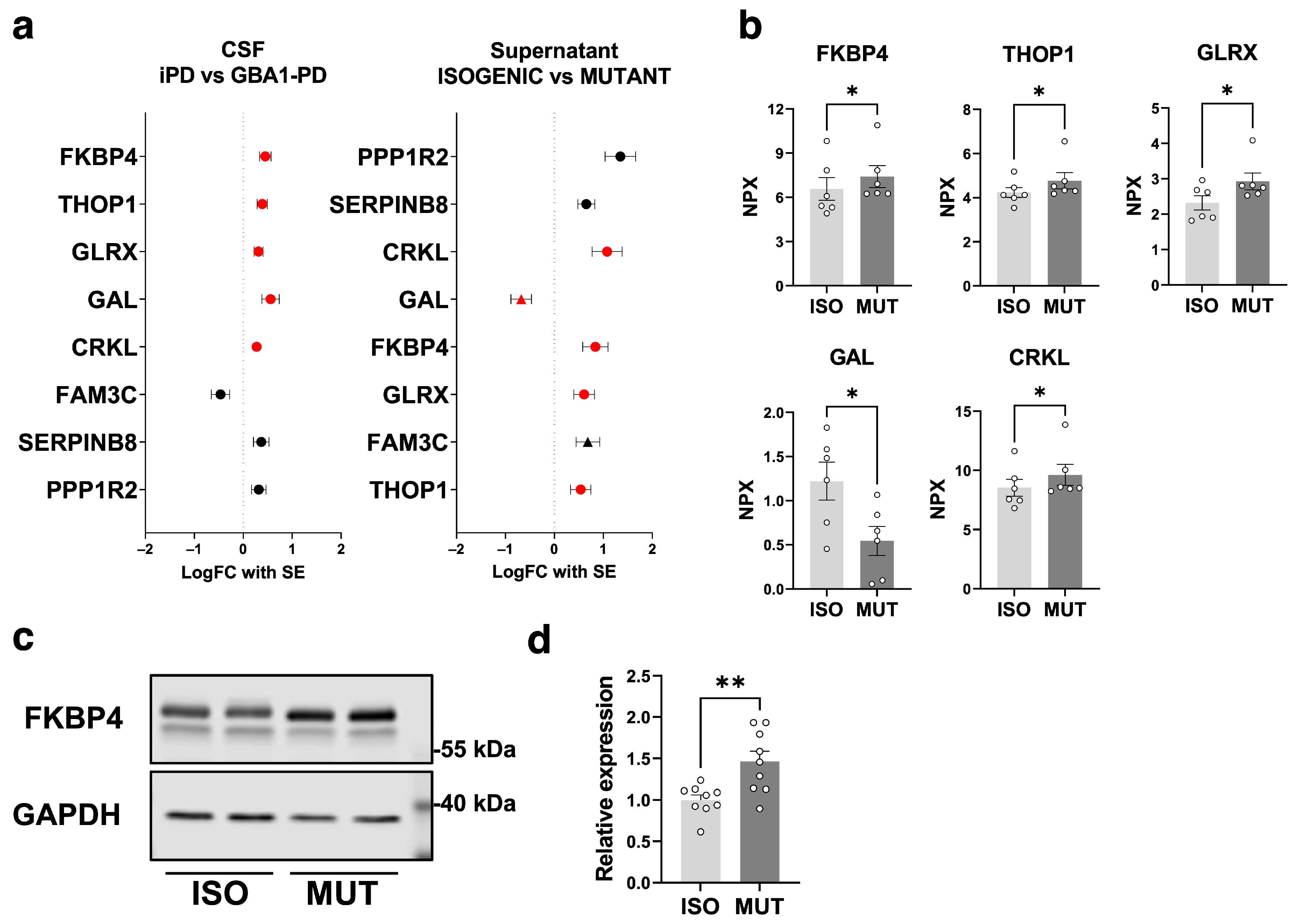
2.3. Secretome Changes in GBA1-PD iPSC-Derived mDA Neuron Culture Supernatant
2.4. Secretome Analysis on GBA1-PD iPSC-Derived mDA Neurons Highlights the GBA1-PD CSF Secretome Alterations Attributed to the mDA Neuronal Population
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Patients with Parkinson’s Disease
4.3. CSF Collection
4.4. Cell Lines
4.5. Midbrain Dopaminergic Neuron Differentiation
4.6. Conditioned Cell Culture Media Collection
4.7. Protein Quantification
4.8. Western Blot
4.9. Immunocytochemistry
4.10. Genotyping of iPSCs
4.11. GCase Activity Assay
4.12. Proximity Extension Assay (PEA)
4.13. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and Its Relevance to Parkinson Disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.J.; Bandres-Ciga, S.; Saez-Atienzar, S.; Singleton, A.B. Genetic Risk Factors in Parkinson’s Disease. Cell Tissue Res. 2018, 373, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Kaur, G.; Fratila, O.; Buhas, C.; Judea-Pusta, C.T.; Negrut, N.; Bustea, C.; Bungau, S. Cross-Talks among GBA Mutations, Glucocerebrosidase, and α-Synuclein in GBA-Associated Parkinson’s Disease and Their Targeted Therapeutic Approaches: A Comprehensive Review. Transl. Neurodegener. 2021, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Mullin, S.; Schapira, A.H.V. Insights into the Structural Biology of Gaucher Disease. Exp. Neurol. 2017, 298, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher Disease: Mutation and Polymorphism Spectrum in the Glucocerebrosidase Gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Gelbart, T.; Scott, C.R. Hematologically Important Mutations: Gaucher Disease. Blood Cells. Mol. Dis. 2005, 35, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Goldstein, O.; Gana-Weisz, M.; Cohen-Avinoam, D.; Shiner, T.; Thaler, A.; Cedarbaum, J.M.; John, S.; Lalioti, M.; Gurevich, T.; Bar-Shira, A.; et al. Revisiting the Non-Gaucher-GBA-E326K Carrier State: Is It Sufficient to Increase Parkinson’s Disease Risk? Mol. Genet. Metab. 2019, 128, 470–475. [Google Scholar] [CrossRef]
- Berge-Seidl, V.; Pihlstrøm, L.; Maple-Grødem, J.; Forsgren, L.; Linder, J.; Larsen, J.P.; Tysnes, O.-B.; Toft, M. The GBA Variant E326K Is Associated with Parkinson’s Disease and Explains a Genome-Wide Association Signal. Neurosci. Lett. 2017, 658, 48–52. [Google Scholar] [CrossRef]
- Duran, R.; Mencacci, N.E.; Angeli, A.V.; Shoai, M.; Deas, E.; Houlden, H.; Mehta, A.; Hughes, D.; Cox, T.M.; Deegan, P.; et al. The Glucocerobrosidase E326K Variant Predisposes to Parkinson’s Disease, but Does Not Cause Gaucher’s Disease. Mov. Disord. 2013, 28, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase Activity in Parkinson’s Disease with and without GBA Mutations. Brain 2015, 138, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, B.; Balwani, M.; Bronstein, J.M.; Kolodny, E.; Sathe, S.; Gwosdow, A.R.; Taylor, J.S.; Cole, J.A.; Zimran, A.; Weinreb, N.J. The Incidence of Parkinsonism in Patients with Type 1 Gaucher Disease: Data from the ICGG Gaucher Registry. Blood Cells. Mol. Dis. 2011, 46, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zurbruegg, M.; Chan, M.Y.; Svenningsson, P. GBA RNAi but Not Catalytic Inhibition of Glucocerebrosidase with Conduritol-β-Epoxide Increases Levels of Total α-Synuclein in SH-SY5Y Cells. Neurosci. Lett. 2019, 706, 217–222. [Google Scholar] [CrossRef] [PubMed]
- te Vruchte, D.; Sturchio, A.; Priestman, D.A.; Tsitsi, P.; Hertz, E.; Andréasson, M.; Markaki, I.; Wallom, K.L.; Platt, F.; Svenningsson, P. Glycosphingolipid Changes in Plasma in Parkinson’s Disease Independent of Glucosylceramide Levels. Mov. Disord. 2022, 37, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, T.O.; Damkier, H.H.; Pedersen, M. A Brief Overview of the Cerebrospinal Fluid System and Its Implications for Brain and Spinal Cord Diseases. Front. Hum. Neurosci. 2022, 15, 737217. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal Fluid and Plasma Biomarkers in Alzheimer Disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef]
- Magdalinou, N.; Lees, A.J.; Zetterberg, H. Cerebrospinal Fluid Biomarkers in Parkinsonian Conditions: An Update and Future Directions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1065–1075. [Google Scholar] [CrossRef]
- Parnetti, L.; Gaetani, L.; Eusebi, P.; Paciotti, S.; Hansson, O.; El-Agnaf, O.; Mollenhauer, B.; Blennow, K.; Calabresi, P. CSF and Blood Biomarkers for Parkinson’s Disease. Lancet Neurol. 2019, 18, 573–586. [Google Scholar] [CrossRef]
- Paslawski, W.; Khosousi, S.; Hertz, E.; Markaki, I.; Boxer, A.; Svenningsson, P. Large-Scale Proximity Extension Assay Reveals CSF Midkine and DOPA Decarboxylase as Supportive Diagnostic Biomarkers for Parkinson’s Disease. Transl. Neurodegener. 2023, 12, 42. [Google Scholar] [CrossRef]
- Avazzadeh, S.; Baena, J.M.; Keighron, C.; Feller-Sanchez, Y.; Quinlan, L.R. Modelling Parkinson’s Disease: Ipscs towards Better Understanding of Human Pathology. Brain Sci. 2021, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Arenas, E.; Denham, M.; Villaescusa, J.C. How to Make a Midbrain Dopaminergic Neuron. Dev. Camb. 2015, 142, 1918–1936. [Google Scholar] [CrossRef] [PubMed]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. IPSC-Derived Neurons from GBA1-Associated Parkinson’s Disease Patients Show Autophagic Defects and Impaired Calcium Homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [PubMed]
- Woodard, C.M.; Campos, B.A.; Kuo, S.-H.; Nirenberg, M.J.; Nestor, M.W.; Zimmer, M.; Mosharov, E.V.; Sulzer, D.; Zhou, H.; Paull, D.; et al. iPSC-Derived Dopamine Neurons Reveal Differences between Monozygotic Twins Discordant for Parkinson’s Disease. Cell Rep. 2014, 9, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.H.; Lee, H.; et al. GBA1 Deficiency Negatively Affects Physiological α-Synuclein Tetramers and Related Multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef]
- Bogetofte, H.; Ryan, B.J.; Jensen, P.; Schmidt, S.I.; Vergoossen, D.L.E.; Barnkob, M.B.; Kiani, L.N.; Chughtai, U.; Heon-Roberts, R.; Caiazza, M.C.; et al. Post-Translational Proteomics Platform Identifies Neurite Outgrowth Impairments in Parkinson’s Disease GBA-N370S Dopamine Neurons. Cell Rep. 2023, 42, 112180. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced Pluripotent Stem Cell Technology: A Decade of Progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Whelan, C.D.; Mattsson, N.; Nagle, M.W.; Vijayaraghavan, S.; Hyde, C.; Janelidze, S.; Stomrud, E.; Lee, J.; Fitz, L.; Samad, T.A.; et al. Multiplex Proteomics Identifies Novel CSF and Plasma Biomarkers of Early Alzheimer’s Disease. Acta Neuropathol. Commun. 2019, 7, 169. [Google Scholar] [CrossRef]
- Jabbari, E.; Woodside, J.; Guo, T.; Magdalinou, N.K.; Chelban, V.; Athauda, D.; Lees, A.J.; Foltynie, T.; Houlden, H.; Church, A.; et al. Proximity Extension Assay Testing Reveals Novel Diagnostic Biomarkers of Atypical Parkinsonian Syndromes. J. Neurol. Neurosurg. Psychiatry 2019, 90, 768–773. [Google Scholar] [CrossRef]
- Yang, S.Y.; Beavan, M.; Chau, K.Y.; Taanman, J.W.; Schapira, A.H.V. A Human Neural Crest Stem Cell-Derived Dopaminergic Neuronal Model Recapitulates Biochemical Abnormalities in GBA1 Mutation Carriers. Stem Cell Rep. 2017, 8, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Mubariz, F.; Saadin, A.; Lingenfelter, N.; Sarkar, C.; Banerjee, A.; Lipinski, M.M.; Awad, O. Deregulation of mTORC1-TFEB Axis in Human iPSC Model of GBA1-Associated Parkinson’s Disease. Front. Neurosci. 2023, 17, 1152503. [Google Scholar] [CrossRef] [PubMed]
- Tüshaus, J.; Müller, S.A.; Kataka, E.S.; Zaucha, J.; Sebastian Monasor, L.; Su, M.; Güner, G.; Jocher, G.; Tahirovic, S.; Frishman, D.; et al. An Optimized Quantitative Proteomics Method Establishes the Cell Type-resolved Mouse Brain Secretome. EMBO J. 2020, 39, e105693. [Google Scholar] [CrossRef] [PubMed]
- Peattie, D.A.; Harding, M.W.; Fleming, M.A.; DeCenzo, M.T.; Lippke, J.A.; Livingston, D.J.; Benasutti, M. Expression and Characterization of Human FKBP52, an Immunophilin That Associates with the 90-kDa Heat Shock Protein and Is a Component of Steroid Receptor Complexes. Proc. Natl. Acad. Sci. USA 1992, 89, 10974–10978. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Peptidylproline Cis-trans-isomerases: Immunophilins. Eur. J. Biochem. 1993, 216, 689–707. [Google Scholar] [CrossRef] [PubMed]
- Pirkl, F.; Buchner, J. Functional Analysis of the Hsp90-Associated Human Peptidyl Prolyl Cis/Trans Isomerases FKBP51, FKBP52 and Cyp40. J. Mol. Biol. 2001, 308, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Guy, N.C.; Garcia, Y.A.; Sivils, J.C.; Galigniana, M.D.; Cox, M.B. Functions of the Hsp90-Binding FKBP Immunophilins. Subcell. Biochem. 2015, 78, 1–34. [Google Scholar] [CrossRef]
- Labrador-Garrido, A.; Cejudo-Guillén, M.; Daturpalli, S.; Leal, M.M.; Klippstein, R.; De Genst, E.J.; Villadiego, J.; Toledo-Aral, J.J.; Dobson, C.M.; Jackson, S.E.; et al. Chaperome Screening Leads to Identification of Grp94/Gp96 and FKBP4/52 as Modulators of the a-Synuclein-Elicited Immune Response. FASEB J. 2016, 30, 564–577. [Google Scholar] [CrossRef]
- Gerard, M.; Deleersnijder, A.; Daniëls, V.; Schreurs, S.; Munck, S.; Reumers, V.; Pottel, H.; Engelborghs, Y.; Van Den Haute, C.; Taymans, J.M.; et al. Inhibition of FK506 Binding Proteins Reduces α-Synuclein Aggregation and Parkinson’s Disease-like Pathology. J. Neurosci. 2010, 30, 2454–2463. [Google Scholar] [CrossRef]
- Fusco, D.; Vargiolu, M.; Vidone, M.; Mariani, E.; Pennisi, L.F.; Bonora, E.; Capellari, S.; Dirnberger, D.; Baumeister, R.; Martinelli, P.; et al. The RET51/FKBP52 Complex and Its Involvement in Parkinson Disease. Hum. Mol. Genet. 2010, 19, 2804–2816. [Google Scholar] [CrossRef]
- Van Dijk, K.D.; Berendse, H.W.; Drukarch, B.; Fratantoni, S.A.; Pham, T.V.; Piersma, S.R.; Huisman, E.; Brevé, J.J.; Groenewegen, H.J.; Jimenez, C.R.; et al. The Proteome of the Locus Ceruleus in Parkinson’s Disease: Relevance to Pathogenesis. Brain Pathol. 2012, 22, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Choi, G.E.; Chae, C.W.; Lim, J.R.; Jung, Y.H.; Yoon, J.H.; Park, J.Y.; Han, H.J. Melatonin-Mediated FKBP4 Downregulation Protects against Stress-Induced Neuronal Mitochondria Dysfunctions by Blocking Nuclear Translocation of GR. Cell Death Dis. 2023, 14, 146. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.G.; Dando, P.M.; Barrett, A.J. Thimet Oligopeptidase Specificity: Evidence of Preferential Cleavage near the C-Terminus and Product Inhibition from Kinetic Analysis of Peptide Hydrolysis. Biochem. J. 1995, 308, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Huber, G.; Malherbe, P. Cloning and Functional Expression of a Metalloendopeptidase from Human Brain with the Ability to Cleave a β-APP Substrate Peptide. Biochem. Biophys. Res. Commun. 1995, 213, 66–73. [Google Scholar] [CrossRef]
- Pollio, G.; Hoozemans, J.J.M.; Andersen, C.A.; Roncarati, R.; Rosi, M.C.; van Haastert, E.S.; Seredenina, T.; Diamanti, D.; Gotta, S.; Fiorentini, A.; et al. Increased Expression of the Oligopeptidase THOP1 Is a Neuroprotective Response to Aβ Toxicity. Neurobiol. Dis. 2008, 31, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Hok-A-Hin, Y.S.; Bolsewig, K.; Ruiters, D.N.; Lleó, A.; Alcolea, D.; Lemstra, A.W.; van der Flier, W.M.; del Campo, M.; Teunissen, C.E. Thimet Oligopeptidase Is a Potential CSF Biomarker for Alzheimer’s Disease. Alzheimers Dement. 2022, 18, e065528. [Google Scholar] [CrossRef]
- Sathe, G.; Na, C.H.; Renuse, S.; Madugundu, A.K.; Albert, M.; Moghekar, A.; Pandey, A. Quantitative Proteomic Profiling of Cerebrospinal Fluid to Identify Candidate Biomarkers for Alzheimer’s Disease. Proteomics-Clin. Appl. 2019, 13, 1800105. [Google Scholar] [CrossRef]
- Ogata, F.T.; Branco, V.; Vale, F.F.; Coppo, L. Glutaredoxin: Discovery, Redox Defense and Much More. Redox Biol. 2021, 43, 101975. [Google Scholar] [CrossRef]
- Gorelenkova Miller, O.; Mieyal, J.J. Critical Roles of Glutaredoxin in Brain Cells—Implications for Parkinson’s Disease. Antioxid. Redox Signal. 2019, 30, 1352–1368. [Google Scholar] [CrossRef]
- Sabens, E.A.; Distler, A.M.; Mieyal, J.J. Levodopa Deactivates Enzymes That Regulate Thiol-Disulfide Homeostasis and Promotes Neuronal Cell Death: Implications for Therapy of Parkinson’s Disease. Biochemistry 2010, 49, 2715–2724. [Google Scholar] [CrossRef]
- Allen, E.M.G.; Mieyal, J.J. Protein-Thiol Oxidation and Cell Death: Regulatory Role of Glutaredoxins. Antioxid. Redox Signal. 2012, 17, 1748–1763. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Yao, C.; Siedlak, S.L.; Wang, W.; Zhu, X.; Caldwell, G.A.; Wilson-Delfosse, A.L.; Mieyal, J.J.; Chen, S.G. Glutaredoxin Deficiency Exacerbates Neurodegeneration in C. Elegans Models of Parkinson’s Disease. Hum. Mol. Genet. 2015, 24, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.J.R.; Patikas, N.; Foskolou, S.; Field, S.F.; Park, J.E.; Byrne, M.L.; Bassett, A.R.; Metzakopian, E. Single-Cell Transcriptomics of Parkinson’s Disease Human In Vitro Models Reveals Dopamine Neuron-Specific Stress Responses. Cell Rep. 2020, 33, 108263. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. SH2/SH3 Signaling Proteins. Curr. Opin. Genet. Dev. 1994, 4, 25–30. [Google Scholar] [CrossRef]
- Feller, S.M. CrK Family Adaptors-Signalling Complex Formation and Biological Roles. Oncogene 2001, 20, 6348–6371. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kwei, K.A.; Girard, L.; Salari, K.; Kao, J.; Pacyna-Gengelbach, M.; Wang, P.; Hernandez-Boussard, T.; Gazdar, A.F.; Petersen, I.; et al. Genomic and Functional Analysis Identifies CRKL as an Oncogene Amplified in Lung Cancer. Oncogene 2010, 29, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Natsume, H.; Shinmura, K.; Tao, H.; Igarashi, H.; Suzuki, M.; Nagura, K.; Goto, M.; Yamada, H.; Maeda, M.; Konno, H.; et al. The CRKL Gene Encoding an Adaptor Protein Is Amplified, Overexpressed, and a Possible Therapeutic Target in Gastric Cancer. J. Transl. Med. 2012, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Siletti, K.; Hodge, R.; Albiach, A.M.; Hu, L.; Lee, K.W.; Lönnerberg, P.; Bakken, T.; Ding, S.-L.; Clark, M.; Casper, T.; et al. Transcriptomic Diversity of Cell Types across the Adult Human Brain. bioRxiv 2022. bioRxiv:2022.10.12.511898. [Google Scholar] [CrossRef]
- Depeursinge, A.; Racoceanu, D.; Iavindrasana, J.; Cohen, G.; Platon, A.; Poletti, P.A.; Müller, H. Fusing Visual and Clinical Information for Lung Tissue Classification in High-Resolution Computed Tomography. Artif. Intell. Med. 2010, 50, 13–21. [Google Scholar] [CrossRef]
- Park, T.J.; Curran, T. Crk and Crk-like Play Essential Overlapping Roles Downstream of Disabled-1 in the Reelin Pathway. J. Neurosci. 2008, 28, 13551–13562. [Google Scholar] [CrossRef]
- Vaswani, A.R.; Weykopf, B.; Hagemann, C.; Fried, H.U.; Brüstle, O.; Blaess, S. Correct Setup of the Substantia Nigra Requires Reelin-Mediated Fast, Laterally-Directed Migration of Dopaminergic Neurons. eLife 2019, 8, e41623. [Google Scholar] [CrossRef] [PubMed]
- Tatemoto, K.; Rökaeus, Å.; Jörnvall, H.; McDonald, T.J.; Mutt, V. Galanin—A Novel Biologically Active Peptide from Porcine Intestine. FEBS Lett. 1983, 164, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Gundlach, A.L.; Kofler, B. The Galanin Peptide Family: Receptor Pharmacology, Pleiotropic Biological Actions, and Implications in Health and Disease. Pharmacol. Ther. 2007, 115, 177–207. [Google Scholar] [CrossRef] [PubMed]
- Webling, K.E.B.; Runesson, J.; Bartfai, T.; Langel, Ü. Galanin Receptors and Ligands. Front. Endocrinol. 2012, 3, 146. [Google Scholar] [CrossRef] [PubMed]
- Hökfelt, T.; Wiesenfeld-Hallin, Z.; Villar, M.; Melander, T. Increase of Galanin-like Immunoreactivity in Rat Dorsal Root Ganglion Cells after Peripheral Axotomy. Neurosci. Lett. 1987, 83, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Villar, M.J.; Cortés, R.; Theodorsson, E.; Wiesenfeld-Hallin, Z.; Schalling, M.; Fahrenkrug, J.; Emson, P.C.; Hökfelt, T. Neuropeptide Expression in Rat Dorsal Root Ganglion Cells and Spinal Cord after Peripheral Nerve Injury with Special Reference to Galanin. Neuroscience 1989, 33, 587–604. [Google Scholar] [CrossRef]
- Counts, S.E.; Perez, S.E.; Ginsberg, S.D.; Mufson, E.J. Neuroprotective Role for Galanin in Alzheimer’s Disease. EXS 2010, 102, 143–162. [Google Scholar] [CrossRef]
- Ding, X.; MacTavish, D.; Kar, S.; Jhamandas, J.H. Galanin Attenuates β-Amyloid (Aβ) Toxicity in Rat Cholinergic Basal Forebrain Neurons. Neurobiol. Dis. 2006, 21, 413–420. [Google Scholar] [CrossRef]
- Pirondi, S.; Giuliani, A.; Del Vecchio, G.; Giardino, L.; Hökfelt, T.; Calzà, L. The Galanin Receptor 2/3 Agonist Gal2-11 Protects the SN56 Cells against β-Amyloid25-35 Toxicity. J. Neurosci. Res. 2010, 88, 1064–1073. [Google Scholar] [CrossRef]
- Wrenn, C.C.; Crawley, J.N. Pharmacological Evidence Supporting a Role for Galanin in Cognition and Affect. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 283–299. [Google Scholar] [CrossRef]
- Steiner, R.A.; Hohmann, J.G.; Holmes, A.; Wrenn, C.C.; Cadd, G.; Juréus, A.; Clifton, D.K.; Luo, M.; Gutshall, M.; Ma, S.Y.; et al. Galanin Transgenic Mice Display Cognitive and Neurochemical Deficits Characteristic of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2001, 98, 4184–4189. [Google Scholar] [CrossRef] [PubMed]
- Nordström, Ö.; Melander, T.; Hökfelt, T.; Bartfai, T.; Goldstein, M. Evidence for an Inhibitory Effect of the Peptide Galanin on Dopamine Release from the Rat Median Eminence. Neurosci. Lett. 1987, 73, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Bonsall, R.W.; Demetrikopoulos, M.K.; Emery, M.S.; West, C.H.K. Galanin: A Significant Role in Depression? In Annals of the New York Academy of Sciences; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1998; Volume 863, pp. 364–382. [Google Scholar]
- Maître, E.L.; Barde, S.S.; Palkovits, M.; Diaz-Heijtz, R.; Hökfelt, T.G.M. Distinct Features of Neurotransmitter Systems in the Human Brain with Focus on the Galanin System in Locus Coeruleus and Dorsal Raphe. Proc. Natl. Acad. Sci. USA 2013, 110, E536–E545. [Google Scholar] [CrossRef] [PubMed]
- Berrettini, W.H.; Kaye, W.H.; Sunderland, T.; May, C.; Gwirtsman, H.E.; Mellow, A.; Albright, A. Galanin Immunoreactivity in Human Csf: Studies in Eating Disorders and Alzheimer’s Disease. Neuropsychobiology 1988, 19, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Sunderland, T.; Berrettini, W.H.; Molchan, S.E.; Lawlor, B.A.; Martinez, R.A.; Vitiello, B.; Tariot, P.N.; Cohen, R.M. Reduced Cerebrospinal Fluid Dynorphin A1-8 in Alzheimer’s Disease. Biol. Psychiatry 1991, 30, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, S.; Zhang, L.; Mollenhauer, B.; Jacob, J.; Longerich, S.; Del-Aguila, J.; Marcus, J.; Raghavan, N.; Stone, D.; Fagboyegun, O.; et al. A Proteogenomic View of Parkinson’s Disease Causality and Heterogeneity. npj Park. Dis. 2023, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Lerche, S.; Zimmermann, M.; Roeben, B.; Wurster, I.; Fries, F.L.; Deuschle, C.; Waniek, K.; Lachmann, I.; Jakobi, M.; Joos, T.O.; et al. Inflammatory CSF Profiles and Longitudinal Development of Cognitive Decline in Sporadic and GBA-Associated PD. NPJ Park. Dis. 2023, 9, 38. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS Clinical Diagnostic Criteria for Parkinson’s Disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Markaki, I.; Ntetsika, T.; Sorjonen, K.; Svenningsson, P. Euglycemia Indicates Favorable Motor Outcome in Parkinson’s Disease. Mov. Disord. 2021, 36, 1430–1434. [Google Scholar] [CrossRef]
- Goetz, C.G.; Tilley, B.C.; Shaftman, S.R.; Stebbins, G.T.; Fahn, S.; Martinez-Martin, P.; Poewe, W.; Sampaio, C.; Stern, M.B.; Dodel, R.; et al. Movement Disorder Society-Sponsored Revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale Presentation and Clinimetric Testing Results. Mov. Disord. 2008, 23, 2129–2170. [Google Scholar] [CrossRef]
- Nasreddine, Z.S.; Phillips, N.A.; Bédirian, V.; Charbonneau, S.; Whitehead, V.; Collin, I.; Cummings, J.L.; Chertkow, H. The Montreal Cognitive Assessment, MoCA: A Brief Screening Tool for Mild Cognitive Impairment. J. Am. Geriatr. Soc. 2005, 53, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, C.L.; Stowe, R.; Patel, S.; Rick, C.; Gray, R.; Clarke, C.E. Systematic Review of Levodopa Dose Equivalency Reporting in Parkinson’s Disease. Mov. Disord. 2010, 25, 2649–2653. [Google Scholar] [CrossRef] [PubMed]
- Paslawski, W.; Zareba-Paslawska, J.; Zhang, X.; Hölzl, K.; Wadensten, H.; Shariatgorji, M.; Janelidze, S.; Hansson, O.; Forsgren, L.; Andrén, P.E.; et al. α-Synuclein−lipoprotein Interactions and Elevated ApoE Level in Cerebrospinal Fluid from Parkinson’s Disease Patients. Proc. Natl. Acad. Sci. USA 2019, 116, 15226–15235. [Google Scholar] [CrossRef] [PubMed]
- Paslawski, W.; Bergström, S.; Zhang, X.; Remnestål, J.; He, Y.; Boxer, A.; Månberg, A.; Nilsson, P.; Svenningsson, P. Cerebrospinal Fluid Proteins Altered in Corticobasal Degeneration. Mov. Disord. 2021, 36, 1278–1280. [Google Scholar] [CrossRef]
- Kim, T.W.; Piao, J.; Koo, S.Y.; Kriks, S.; Chung, S.Y.; Betel, D.; Socci, N.D.; Choi, S.J.; Zabierowski, S.; Dubose, B.N.; et al. Biphasic Activation of WNT Signaling Facilitates the Derivation of Midbrain Dopamine Neurons from hESCs for Translational Use. Cell Stem Cell 2021, 28, 343–355.e5. [Google Scholar] [CrossRef]
- Nishimura, K.; Yang, S.; Lee, K.W.; Ásgrímsdóttir, E.S.; Nikouei, K.; Paslawski, W.; Gnodde, S.; Lyu, G.; Hu, L.; Saltó, C.; et al. Single-Cell Transcriptomics Reveals Correct Developmental Dynamics and High-Quality Midbrain Cell Types by Improved hESC Differentiation. Stem Cell Rep. 2023, 18, 337–353. [Google Scholar] [CrossRef]
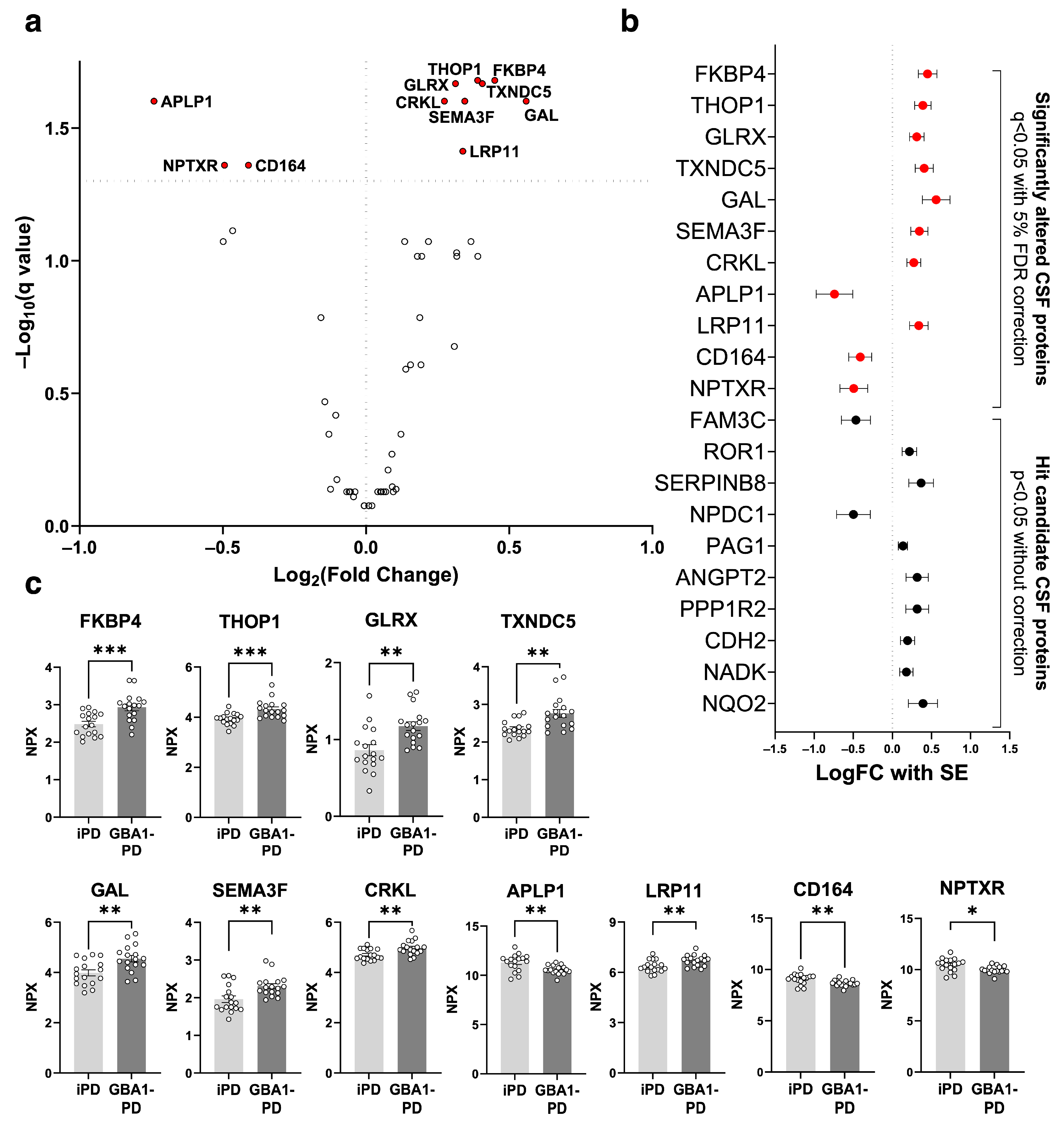


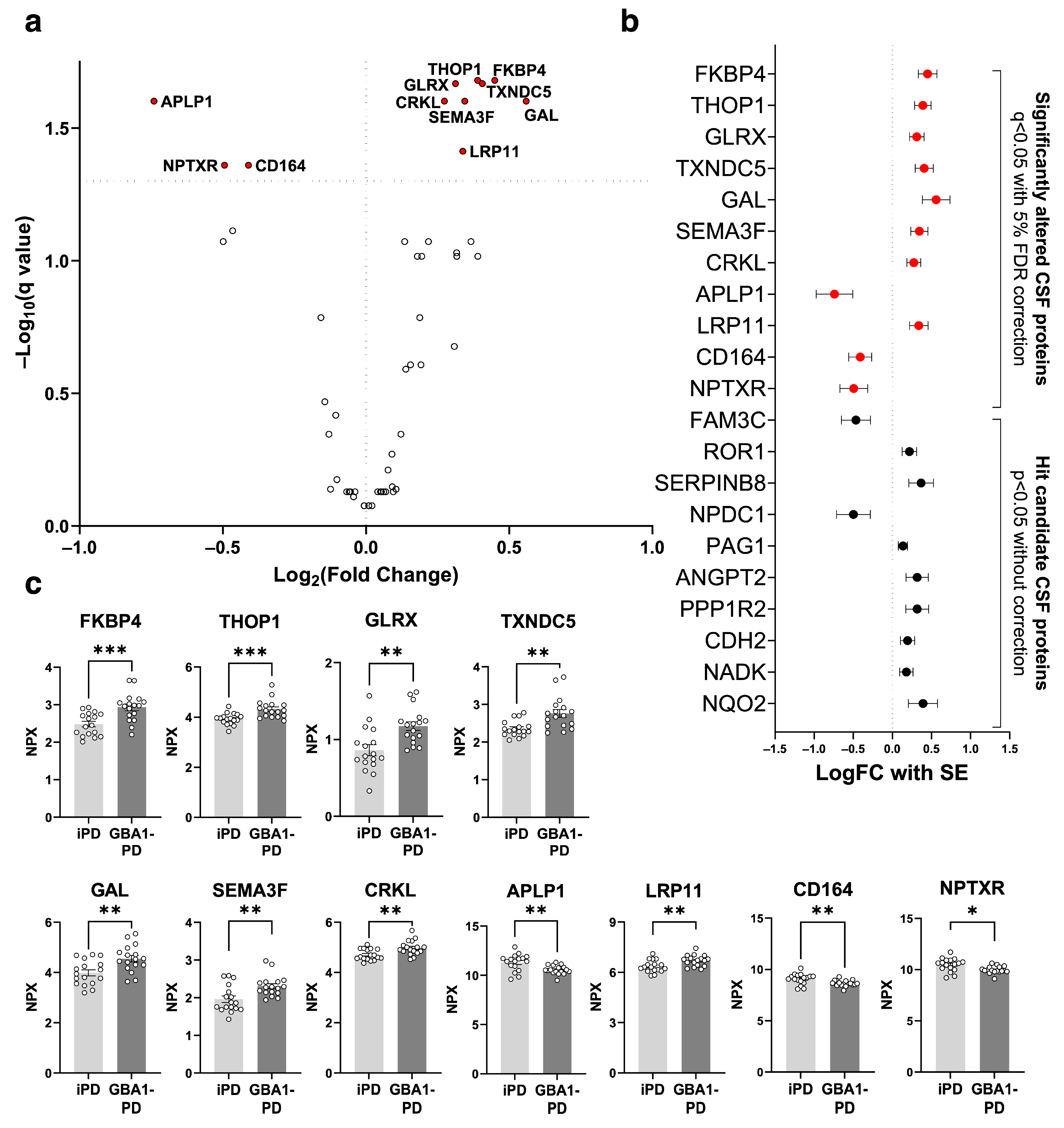{kind=link}
{kind=link}
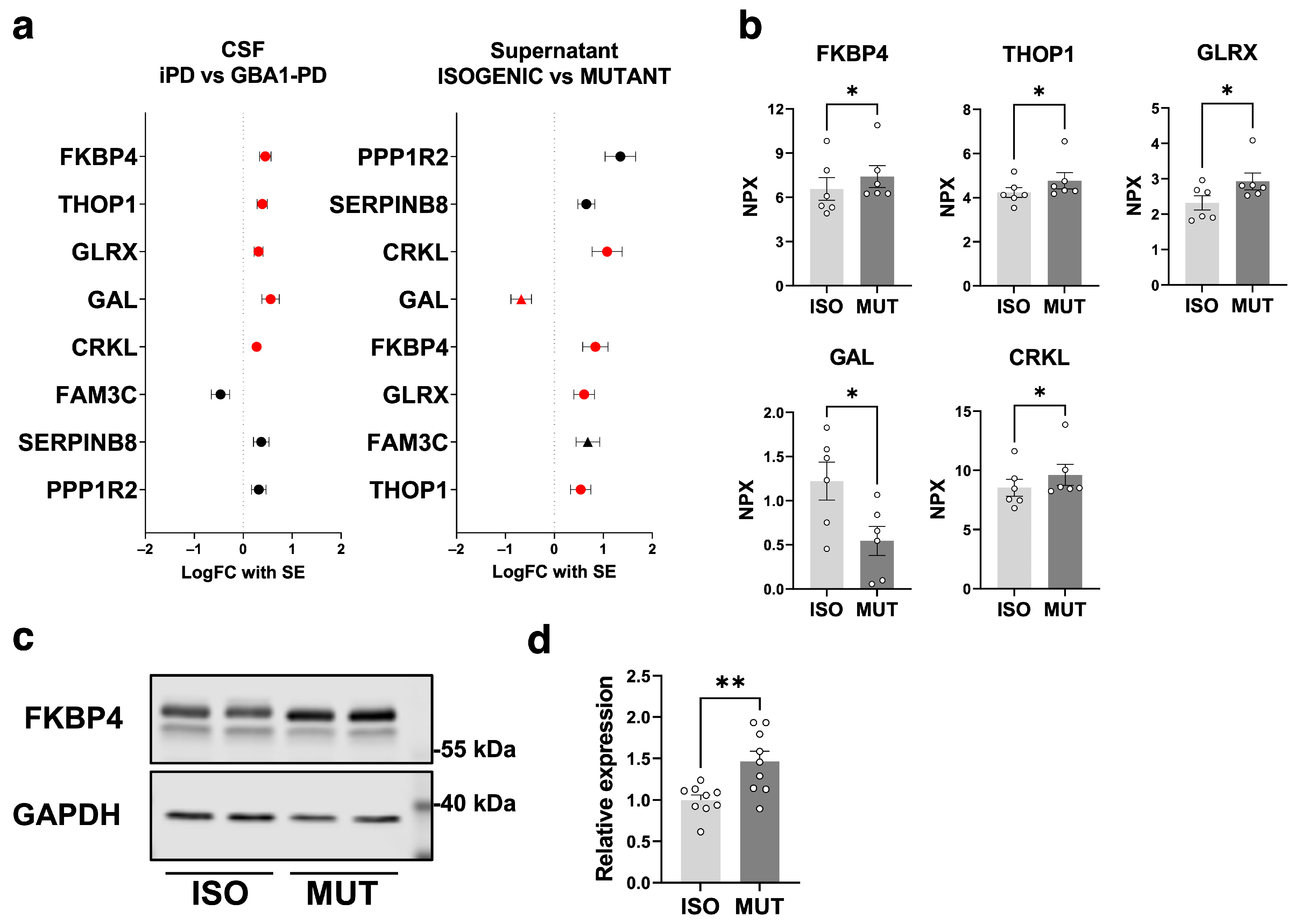{kind=link}
| GBA1-PD | iPD | p-Value 1 | |
|---|---|---|---|
| n | 17 | 17 | >0.99 |
| Female:Male | 5:12 | 5:12 | >0.99 |
| Age mean (SD) | 63.29 (9.726) n = 17 | 63.29 (8.908) N = 17 | 0.7658 |
| Duration of disease in years | 4.0 (0–10) n = 17 | 2.29 (0.04–18.29) N = 17 | 0.6768 |
| HY 2 | 2 (1–3) n = 17 | 2 (1–4) N = 17 | 0.9562 |
| UPDRS-III 3 | 26 (5–62) n = 15 | 27.5 (10–54) N = 16 | 0.4287 |
| MoCA 4 | 26 (9–29) n = 16 | 26.0 (16–29) N = 16 | 0.5800 |
| LEDD 5 | 710 (0–1805) n = 17 | 425.6 (0–1080) N = 17 | 0.1724 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kojima, R.; Paslawski, W.; Lyu, G.; Arenas, E.; Zhang, X.; Svenningsson, P. Secretome Analyses Identify FKBP4 as a GBA1-Associated Protein in CSF and iPS Cells from Parkinson’s Disease Patients with GBA1 Mutations. Int. J. Mol. Sci. 2024, 25, 683. https://doi.org/10.3390/ijms25010683
Kojima R, Paslawski W, Lyu G, Arenas E, Zhang X, Svenningsson P. Secretome Analyses Identify FKBP4 as a GBA1-Associated Protein in CSF and iPS Cells from Parkinson’s Disease Patients with GBA1 Mutations. International Journal of Molecular Sciences. 2024; 25(1):683. https://doi.org/10.3390/ijms25010683
Chicago/Turabian StyleKojima, Rika, Wojciech Paslawski, Guochang Lyu, Ernest Arenas, Xiaoqun Zhang, and Per Svenningsson. 2024. "Secretome Analyses Identify FKBP4 as a GBA1-Associated Protein in CSF and iPS Cells from Parkinson’s Disease Patients with GBA1 Mutations" International Journal of Molecular Sciences 25, no. 1: 683. https://doi.org/10.3390/ijms25010683
APA StyleKojima, R., Paslawski, W., Lyu, G., Arenas, E., Zhang, X., & Svenningsson, P. (2024). Secretome Analyses Identify FKBP4 as a GBA1-Associated Protein in CSF and iPS Cells from Parkinson’s Disease Patients with GBA1 Mutations. International Journal of Molecular Sciences, 25(1), 683. https://doi.org/10.3390/ijms25010683