
Screening of Small-Molecule Libraries Using SARS-CoV-2-Derived Sequences Identifies Novel Furin Inhibitors

 , ,
, ,  , , ,
, , ,  , , and
, , and
Abstract
1. Introduction
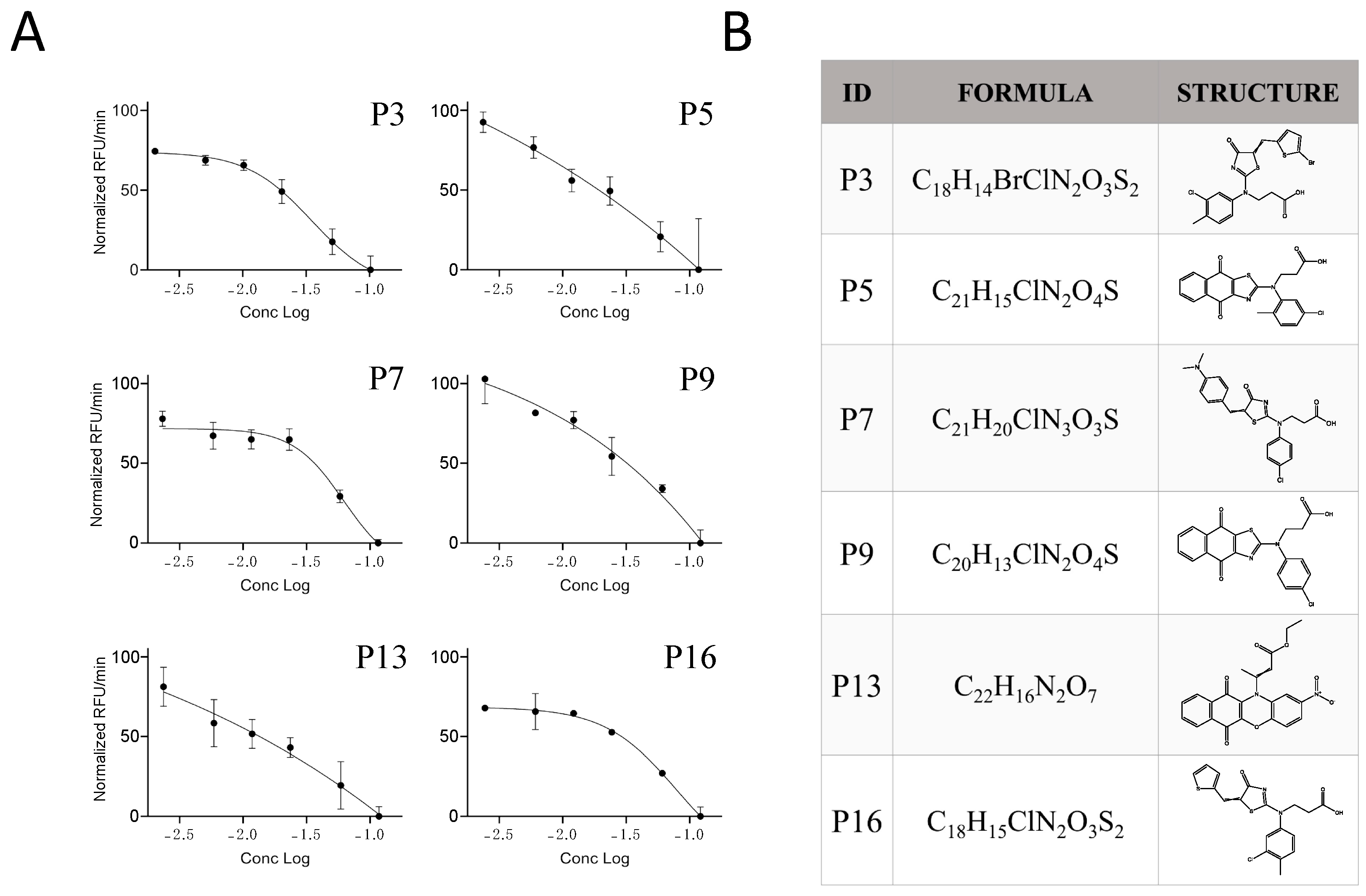
2. Results
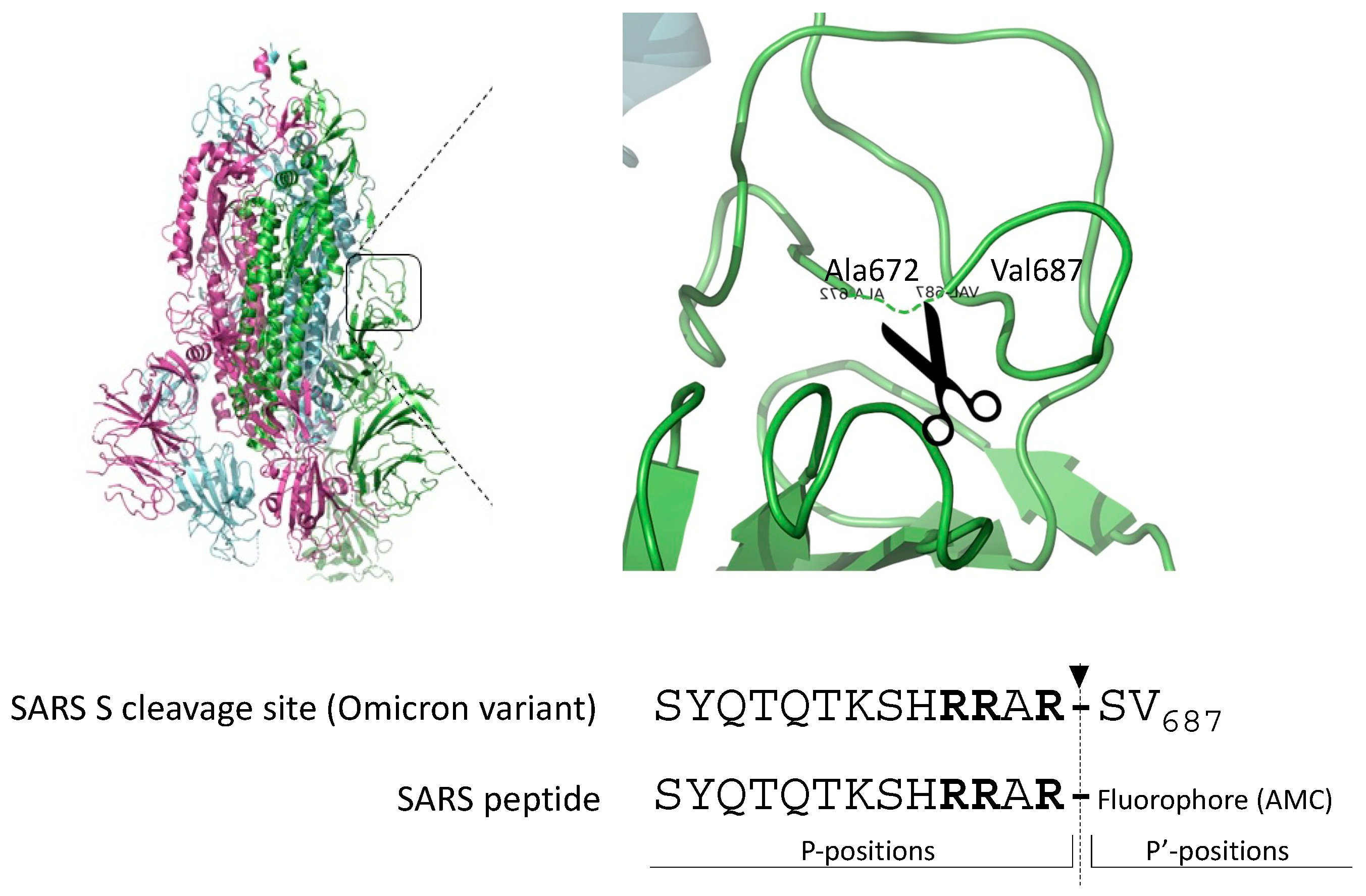
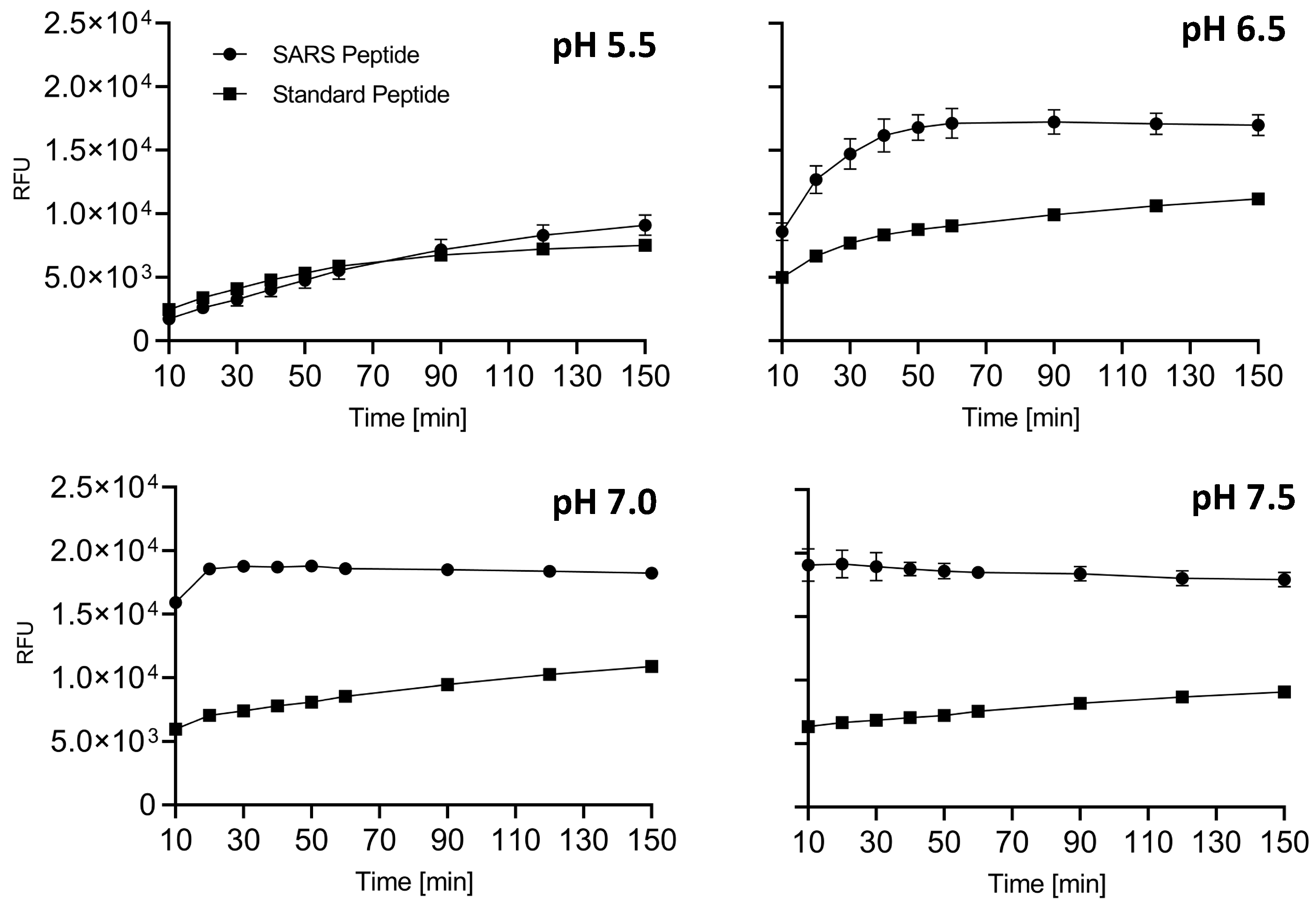
2.1. Omicron SARS-CoV-2-Derived Peptide Is a Superior Furin Substrate
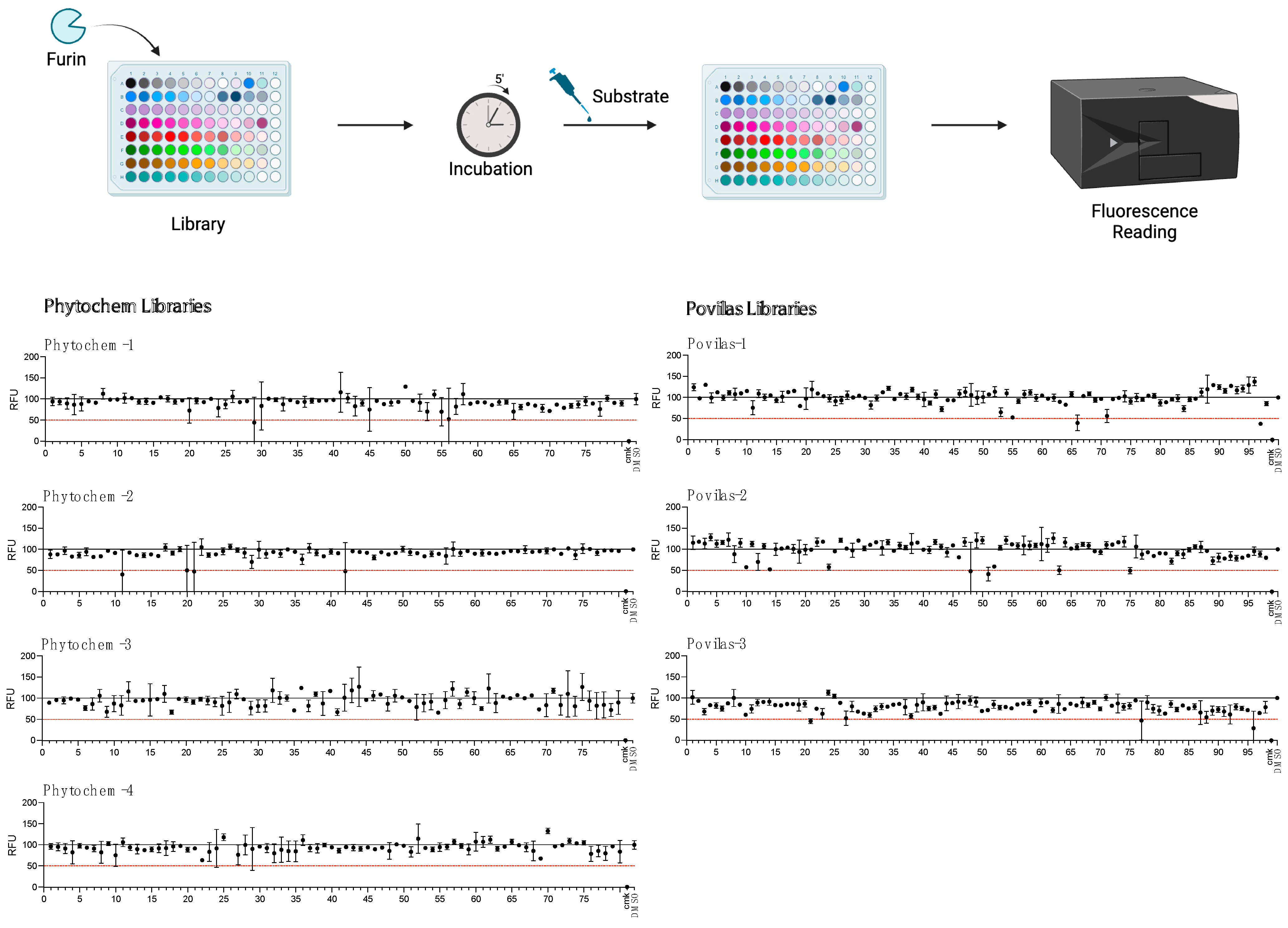
2.2. Screening of Small-Compound Libraries Using the SARS Peptide
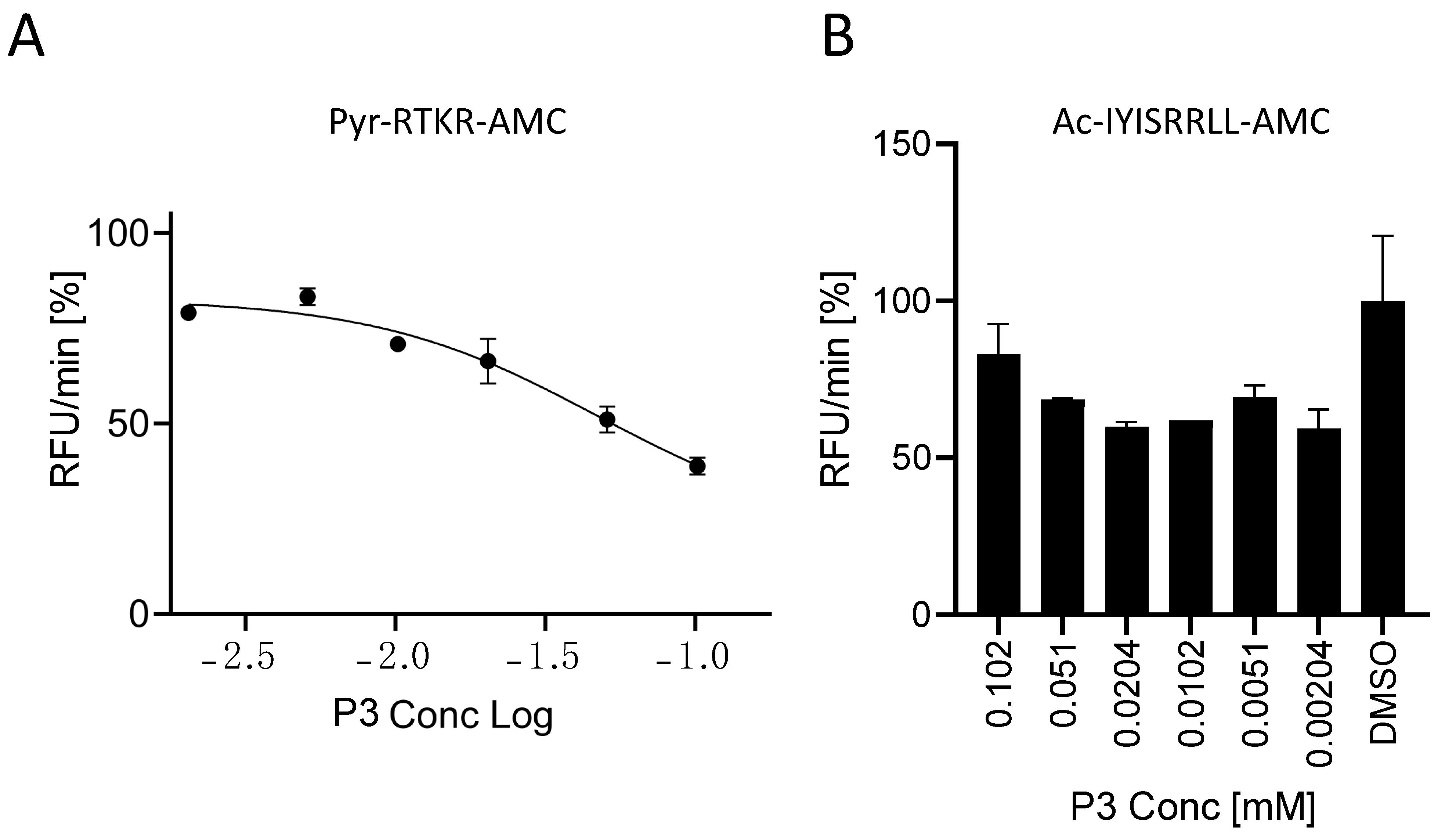
2.3. Characterization of the Furin Inhibitor P3
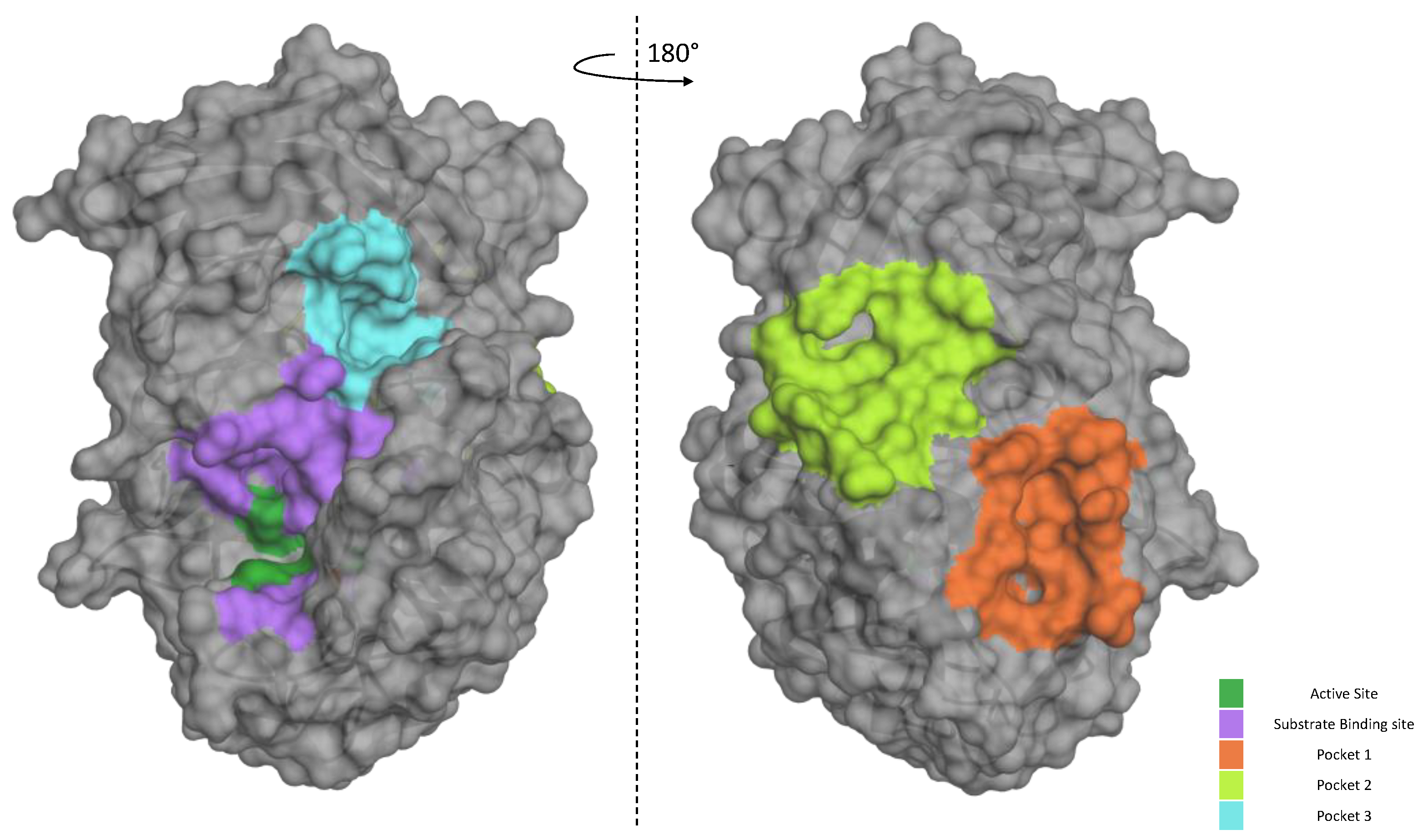
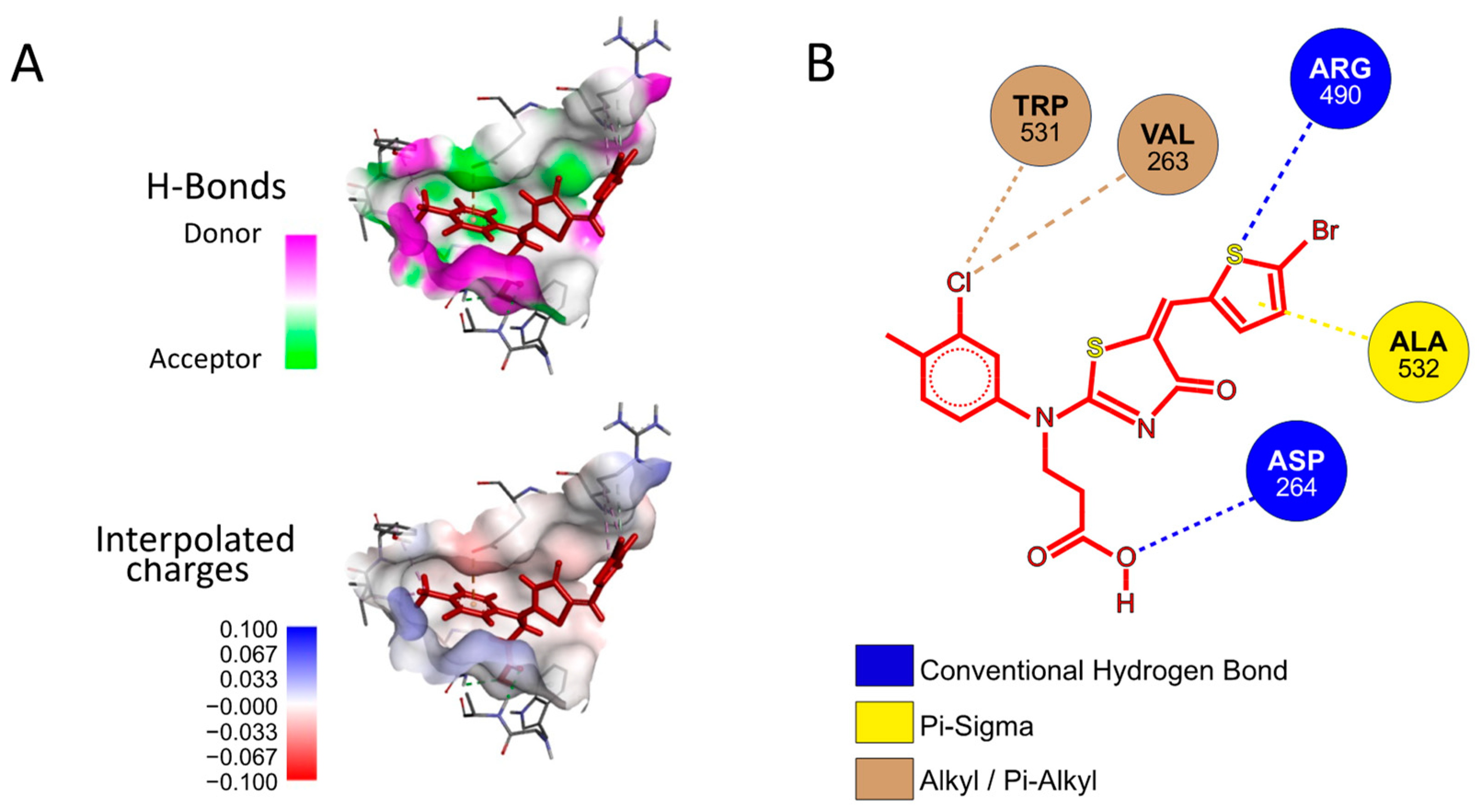
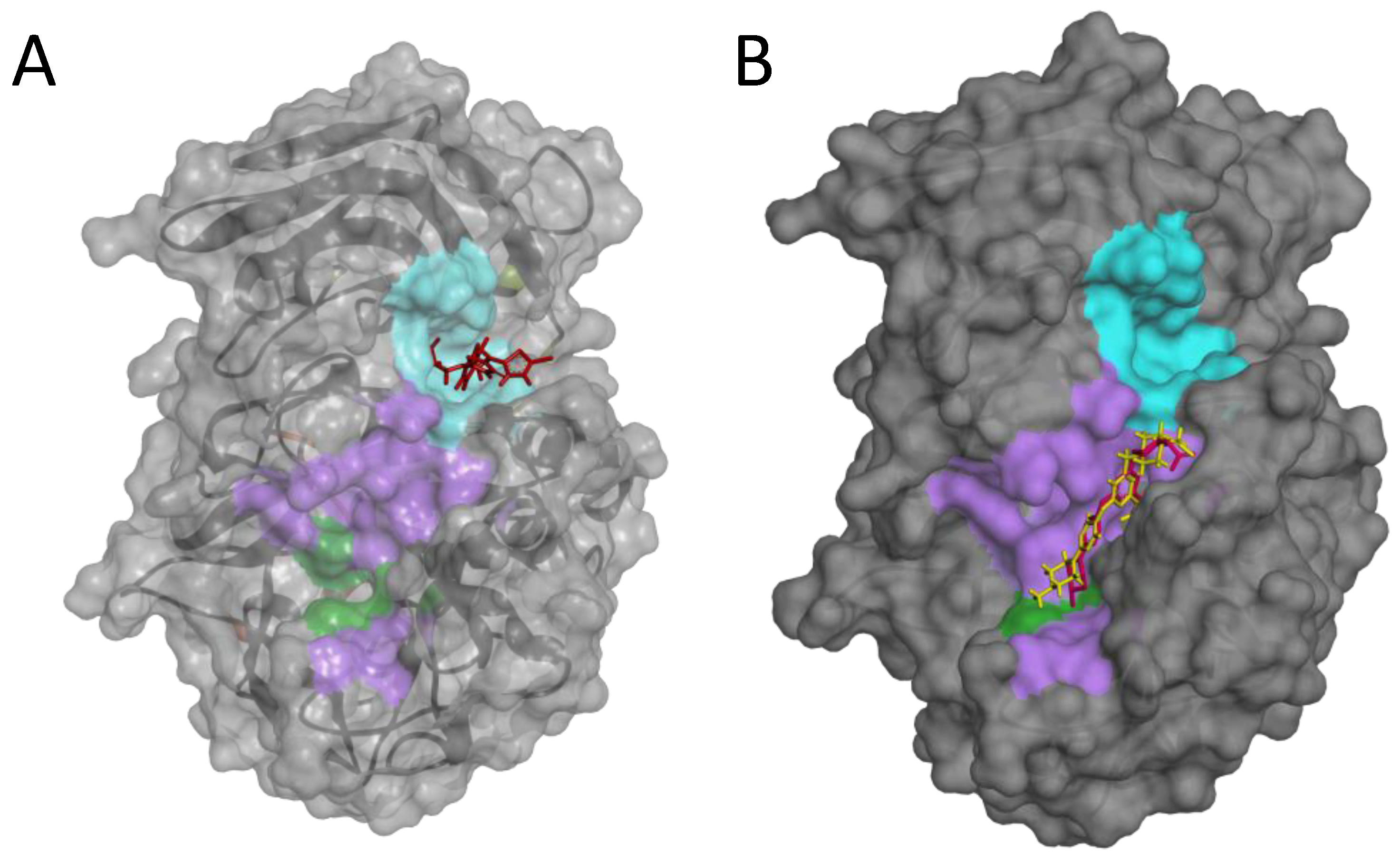
2.4. Possible Mechanism of Action of the P3 Furin Inhibitor
3. Discussion and Conclusions
4. Materials and Methods
4.1. Soluble Furin Production
4.2. In Vitro Assays
4.3. High-Throughput Screening
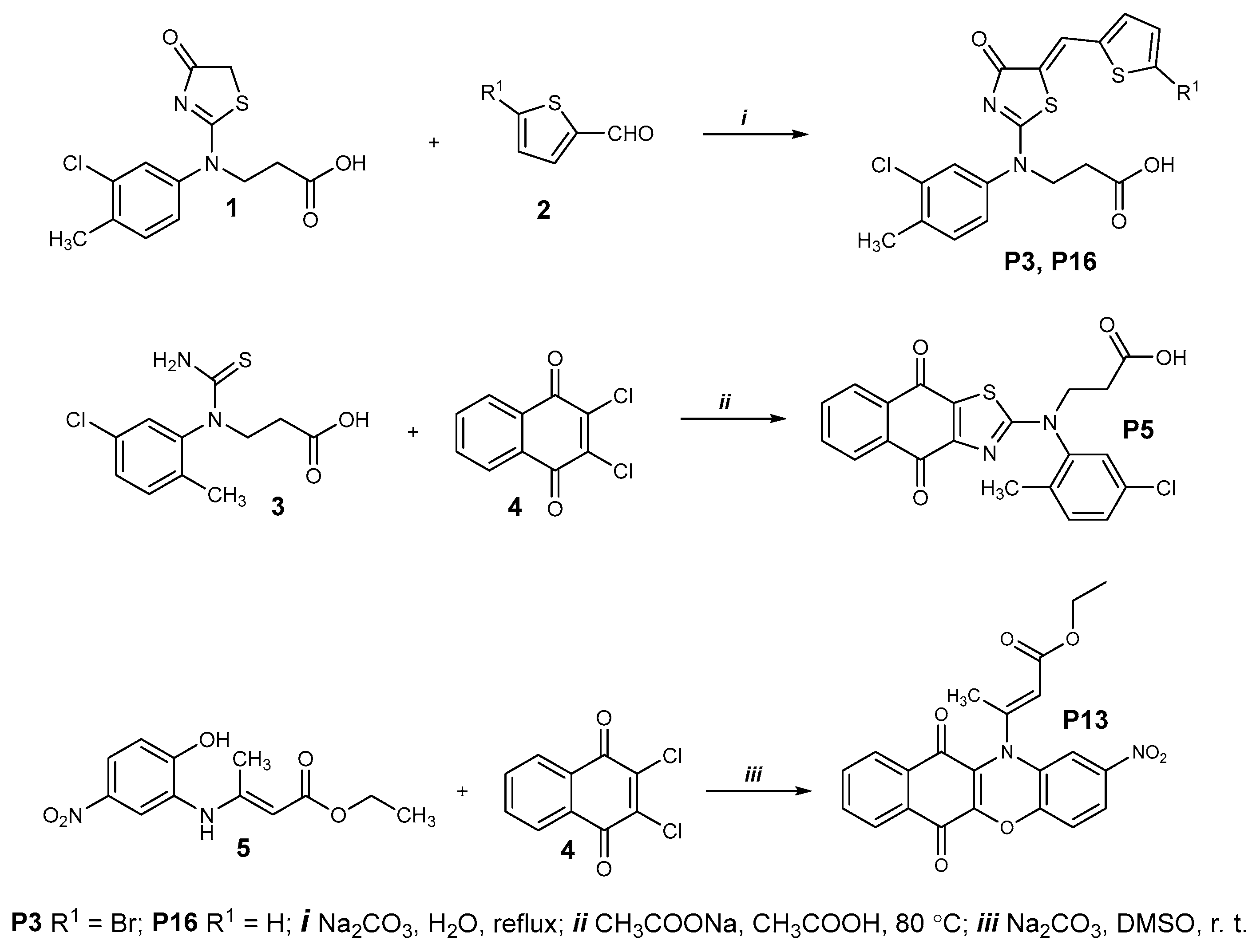
4.4. General Procedure for the Preparation of Compounds P3, P5, P7, P9, P13, and P16 and Their Characterization
4.4.1. Compounds P3 and P16
- 3-((5-((5-Bromothiophen-2-yl)methylene)-4-oxo-4,5-dihydrothiazol-2-yl)(3-chloro-4-methylphenyl)amino)propanoic acid (P3)
- 3-((3-Chloro-4-methylphenyl)(4-oxo-5-(thiophen-2-ylmethylene)-4,5-dihydrothiazol-2-yl)amino)propanoic acid (P16)
4.4.2. Compound P5
- 3-((5-Chloro-2-methylphenyl)(4,9-dioxo-4,9-dihydronaphtho[2,3-d]thiazol-2-yl)amino)propanoic acid (P5)
4.4.3. Compound P7 and P9
- 3-((4-Chlorophenyl)(5-(4-(dimethylamino)benzylidene)-4-oxo-4,5-dihydrothiazol-2-yl)amino)propanoic acid (P7)
- 3-((4-Chlorophenyl)(4,9-dioxo-4,9-dihydronaphtho[2,3-d]thiazol-2-yl)amino)propanoic acid (P9)
4.4.4. Compound P13
- Ethyl 3-(2-nitro-6,11-dioxo-6,11-dihydro-12H-benzo[b]phenoxazin-12-yl)but-2-enoate (P13)
4.5. In Silico Analyses
4.5.1. Furin Enzyme Structure
4.5.2. Molecular Docking
4.6. Sustainability
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eccleston-Turner, M.; Phelan, A.; Katz, R. Preparing for the Next Pandemic—The WHO’s Global Influenza Strategy. N. Engl. J. Med. 2019, 381, 2192–2194. [Google Scholar] [CrossRef]
- Poon, L.L.M.; Peiris, M. Emergence of a Novel Human Coronavirus Threatening Human Health. Nat. Med. 2020, 26, 317–319. [Google Scholar] [CrossRef]
- Wang, H.; Paulson, K.R.; Pease, S.A.; Watson, S.; Comfort, H.; Zheng, P.; Aravkin, A.Y.; Bisignano, C.; Barber, R.M.; Alam, T.; et al. Estimating Excess Mortality Due to the COVID-19 Pandemic: A Systematic Analysis of COVID-19-Related Mortality, 2020–2021. Lancet 2022, 399, 1513–1536. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Yang, Z.-R.; Jiang, Y.-W.; Li, F.-X.; Liu, D.; Lin, T.-F.; Zhao, Z.-Y.; Wei, C.; Jin, Q.-Y.; Li, X.-M.; Jia, Y.-X.; et al. Efficacy of SARS-CoV-2 Vaccines and the Dose–Response Relationship with Three Major Antibodies: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. Lancet Microbe 2023, 4, e236–e246. [Google Scholar] [CrossRef]
- Vasireddy, D.; Vanaparthy, R.; Mohan, G.; Malayala, S.V.; Atluri, P. Review of COVID-19 Variants and COVID-19 Vaccine Efficacy: What the Clinician Should Know? J. Clin. Med. Res. 2021, 13, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Aleem, A.; Akbar Samad, A.B.; Vaqar, S. Emerging Variants of SARS-CoV-2 and Novel Therapeutics Against Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The Spike Glycoprotein of the New Coronavirus 2019-nCoV Contains a Furin-like Cleavage Site Absent in CoV of the Same Clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The Biology and Therapeutic Targeting of the Proprotein Convertases. Nat. Rev. Drug Discov. 2012, 11, 367–383. [Google Scholar] [CrossRef]
- Cendron, L.; Rothenberger, S.; Cassari, L.; Dettin, M.; Pasquato, A. Chapter One—Proprotein Convertases Regulate Trafficking and Maturation of Key Proteins within the Secretory Pathway. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2023; Volume 133, pp. 1–54. ISBN 1876-1623. [Google Scholar]
- Seidah, N.G.; Pasquato, A.; Andréo, U. How Do Enveloped Viruses Exploit the Secretory Proprotein Convertases to Regulate Infectivity and Spread? Viruses 2021, 13, 1229. [Google Scholar] [CrossRef] [PubMed]
- Cassari, L.; Pavan, A.; Zoia, G.; Chinellato, M.; Zeni, E.; Grinzato, A.; Rothenberger, S.; Cendron, L.; Dettin, M.; Pasquato, A. SARS-CoV-2 S Mutations: A Lesson from the Viral World to Understand How Human Furin Works. Int. J. Mol. Sci. 2023, 24, 4791. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, B.; Jaimes, J.A.; Whittaker, G.R. Whittaker Intrinsic Furin-Mediated Cleavability of the Spike S1/S2 Site from SARS-CoV-2 Variant B.1.1.529 (Omicron). bioRxiv 2022. bioRxiv:2022.04.20.488969. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The Furin Cleavage Site in the SARS-CoV-2 Spike Protein Is Required for Transmission in Ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Hossain, G.; Tang, Y.; Akter, S.; Zheng, C. Roles of the Polybasic Furin Cleavage Site of Spike Protein in SARS-CoV-2 Replication, Pathogenesis, and Host Immune Responses and Vaccination. J. Med. Virol. 2022, 94, 1815–1820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mann, M.; Syed, Z.A.; Reynolds, H.M.; Tian, E.; Samara, N.L.; Zeldin, D.C.; Tabak, L.A.; Ten Hagen, K.G. Furin Cleavage of the SARS-CoV-2 Spike Is Modulated by O-Glycosylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2109905118. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.A.; Zhan, S.H. The Emergence of the Spike Furin Cleavage Site in SARS-CoV-2. Mol. Biol. Evol. 2022, 39, msab327. [Google Scholar] [CrossRef] [PubMed]
- Reuter, N.; Chen, X.; Kropff, B.; Peter, A.S.; Britt, W.J.; Mach, M.; Überla, K.; Thomas, M. SARS-CoV-2 Spike Protein Is Capable of Inducing Cell–Cell Fusions Independent from Its Receptor ACE2 and This Activity Can Be Impaired by Furin Inhibitors or a Subset of Monoclonal Antibodies. Viruses 2023, 15, 1500. [Google Scholar] [CrossRef] [PubMed]
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andréo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A.; et al. Distinctive Roles of Furin and TMPRSS2 in SARS-CoV-2 Infectivity. J. Virol. 2022, 96, e00128-22. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-W.; Chao, T.-L.; Li, C.-L.; Chiu, M.-F.; Kao, H.-C.; Wang, S.-H.; Pang, Y.-H.; Lin, C.-H.; Tsai, Y.-M.; Lee, W.-H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020, 33, 108254. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; de Silva, T.I.; Peacock, S.J.; Barclay, W.S.; de Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 Variant Biology: Immune Escape, Transmission and Fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar] [CrossRef]
- Spelios, M.G.; Capanelli, J.M.; Li, A.W. A Novel Antibody against the Furin Cleavage Site of SARS-CoV-2 Spike Protein: Effects on Proteolytic Cleavage and ACE2 Binding. Immunol. Lett. 2022, 242, 1–7. [Google Scholar] [CrossRef]
- Li, L.; Gao, M.; Li, J.; Xie, X.; Zhao, H.; Wang, Y.; Xu, X.; Zu, S.; Chen, C.; Wan, D.; et al. Identification of an Immunogenic Epitope and Protective Antibody against the Furin Cleavage Site of SARS-CoV-2. eBioMedicine 2023, 87, 104401. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Declercq, J.; Roucourt, B.; Ghassabeh, G.H.; Meulemans, S.; Kinne, J.; David, G.; Vermorken, A.J.M.; Van de Ven, W.J.M.; Lindberg, I.; et al. Generation and Characterization of Non-Competitive Furin-Inhibiting Nanobodies. Biochem. J. 2012, 448, 73–82. [Google Scholar] [CrossRef]
- Jean, F.; Stella, K.; Thomas, L.; Liu, G.; Xiang, Y.; Reason, A.J.; Thomas, G. A1-Antitrypsin Portland, a Bioengineered Serpin Highly Selective for Furin: Application as an Antipathogenic Agent. Proc. Natl. Acad. Sci. USA 1998, 95, 7293–7298. [Google Scholar] [CrossRef]
- Bai, X.; Schountz, T.; Buckle, A.M.; Talbert, J.L.; Sandhaus, R.A.; Chan, E.D. Alpha-1-Antitrypsin Antagonizes COVID-19: A Review of the Epidemiology, Molecular Mechanisms, and Clinical Evidence. Biochem. Soc. Trans. 2023, 51, 1361–1375. [Google Scholar] [CrossRef]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.-D. Processing of the Ebola Virus Glycoprotein by the Proprotein Convertase Furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; Rohde, C. TMPRSS2 and Furin Are Both Essential for Proteolytic Activation of SARS-CoV-2 in Human Airway Cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Douglas, L.E.J.; Reihill, J.A.; Ho, M.W.Y.; Axten, J.M.; Campobasso, N.; Schneck, J.L.; Rendina, A.R.; Wilcoxen, K.M.; Martin, S.L. A Highly Selective, Cell-Permeable Furin Inhibitor BOS-318 Rescues Key Features of Cystic Fibrosis Airway Disease. Cell Chem. Biol. 2022, 29, 947–957. [Google Scholar] [CrossRef]
- Thomas, G.; Couture, F.; Kwiatkowska, A. The Path to Therapeutic Furin Inhibitors: From Yeast Pheromones to SARS-CoV-2. Int. J. Mol. Sci. 2022, 23, 3435. [Google Scholar] [CrossRef]
- Pasquato, A.; Pullikotil, P.; Asselin, M.-C.; Vacatello, M.; Paolillo, L.; Ghezzo, F.; Basso, F.; Di Bello, C.; Dettin, M.; Seidah, N.G. The Proprotein Convertase SKI-1/S1P: In Vitro Analysis of Lassa Virus Glycoprotein-Derived Substrates and Ex Vivo Validation of Irreversible Peptide Inhibitors. J. Biol. Chem. 2006, 281, 23471–23481. [Google Scholar] [CrossRef]
- Pasquato, A.; Burri, D.J.; Traba, E.G.-I.; Hanna-El-Daher, L.; Seidah, N.G.; Kunz, S. Arenavirus Envelope Glycoproteins Mimic Autoprocessing Sites of the Cellular Proprotein Convertase Subtilisin Kexin Isozyme-1/Site-1 Protease. Virology 2011, 417, 18–26. [Google Scholar] [CrossRef]
- Remacle, A.G.; Shiryaev, S.A.; Oh, E.-S.; Cieplak, P.; Srinivasan, A.; Wei, G.; Liddington, R.C.; Ratnikov, B.I.; Parent, A.; Desjardins, R.; et al. Substrate Cleavage Analysis of Furin and Related Proprotein Convertases: A Comparative Study*. J. Biol. Chem. 2008, 283, 20897–20906. [Google Scholar] [CrossRef]
- Lamango, N.S.; Zhu, X.; Lindberg, I. Purification and Enzymatic Characterization of Recombinant Prohormone Convertase 2: Stabilization of Activity by 21 kDa 7B2. Arch. Biochem. Biophys. 1996, 330, 238–250. [Google Scholar] [CrossRef]
- Zhu, J.; Declercq, J.; Creemers, J.W.; Chen, C.; Cui, Y.; Van de Ven, W.J.; Vermorken, A.J. Limitations of Inhibitory Activities of Polyphenols on Furin-Mediated Substrate Processing. Curr. Med. Chem. 2012, 19, 3641–3650. [Google Scholar] [CrossRef]
- Kiba, Y.; Oyama, R.; Misawa, S.; Tanikawa, T.; Kitamura, M.; Suzuki, R. Screening for Inhibitory Effects of Crude Drugs on Furin-like Enzymatic Activities. J. Nat. Med. 2021, 75, 1080–1085. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. SLAS Discov. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Ren, L.; Wu, J.; Guo, L.; Han, Z.; Yang, J.; Xie, W.; Wang, Y.; Xu, F.; Su, X.; et al. Permethrin as a Potential Furin Inhibitor through a Novel Non-Competitive Allosteric Inhibition. Molecules 2023, 28, 1883. [Google Scholar] [CrossRef] [PubMed]
- Pandya, M.; Shah, S.; M, D.; Juneja, T.; Patel, A.; Gadnayak, A.; Dave, S.; Das, K.; Das, J. Unravelling Vitamin B12 as a Potential Inhibitor against SARS-CoV-2: A Computational Approach. Inform. Med. Unlocked 2022, 30, 100951. [Google Scholar] [CrossRef] [PubMed]
- Peiter, G.C.; De Souza, C.D.B.T.; De Oliveira, L.M.; Pagliarin, L.G.; Dos Anjos, V.N.F.; Da Silva, F.A.F.; De Melo, F.F.; Teixeira, K.N. COVID-19 Liver and Gastroenterology Findings: An in Silico Analysis of SARS-CoV-2 Interactions with Liver Molecules. WJH 2022, 14, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Rajak, P.; Ganguly, A. In Silico Study Unfolds Inhibitory Potential of Epicatechin Gallate against SARS-CoV-2 Entry and Replication within the Host Cell. Mechanobiol. Med. 2023, 1, 100015. [Google Scholar] [CrossRef]
- Ridgway, H.; Orbell, J.D.; Matsoukas, M.-T.; Kelaidonis, K.; Moore, G.J.; Tsiodras, S.; Gorgoulis, V.G.; Chasapis, C.T.; Apostolopoulos, V.; Matsoukas, J.M. W254 in Furin Functions as a Molecular Gate Promoting Anti-Viral Drug Binding: Elucidation of Putative Drug Tunneling and Docking by Non-Equilibrium Molecular Dynamics. Comput. Struct. Biotechnol. J. 2023, 21, 4589–4612. [Google Scholar] [CrossRef] [PubMed]
- Decroly, E.; Wouters, S.; Di Bello, C.; Lazure, C.; Ruysschaert, J.-M.; Seidah, N.G. Identification of the Paired Basic Convertases Implicated in HIV Gp160 Processing Based on in Vitro Assays and Expression in CD4+ Cell Lines. J. Biol. Chem. 1996, 271, 30442–30450. [Google Scholar] [CrossRef] [PubMed]
- Anusevičius, K.; Jonuškienė, I.; Mickevičius, V. Synthesis and Antimicrobial Activity of N-(4-Chlorophenyl)-β-Alanine Derivatives with an Azole Moiety. Monatshefte Chem. 2013, 144, 1883–1891. [Google Scholar] [CrossRef]
- Lowe, D.M.; Corbett, P.T.; Murray-Rust, P.; Glen, R.C. Chemical Name to Structure: OPSIN, an Open Source Solution. J. Chem. Inf. Model. 2011, 51, 739–753. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | Vmax RFU/min | Km [μM] |
|---|---|---|
| 5.5 | 2.27 | 17.54 |
| 6.5 | 11.69 | 19.90 |
| 7.0 | 21.04 | 14.70 |
| 7.5 | 24.2 | 8.94 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jorkesh, A.; Rothenberger, S.; Baldassar, L.; Grybaite, B.; Kavaliauskas, P.; Mickevicius, V.; Dettin, M.; Vascon, F.; Cendron, L.; Pasquato, A. Screening of Small-Molecule Libraries Using SARS-CoV-2-Derived Sequences Identifies Novel Furin Inhibitors. Int. J. Mol. Sci. 2024, 25, 5079. https://doi.org/10.3390/ijms25105079
Jorkesh A, Rothenberger S, Baldassar L, Grybaite B, Kavaliauskas P, Mickevicius V, Dettin M, Vascon F, Cendron L, Pasquato A. Screening of Small-Molecule Libraries Using SARS-CoV-2-Derived Sequences Identifies Novel Furin Inhibitors. International Journal of Molecular Sciences. 2024; 25(10):5079. https://doi.org/10.3390/ijms25105079
Chicago/Turabian StyleJorkesh, Alireza, Sylvia Rothenberger, Laura Baldassar, Birute Grybaite, Povilas Kavaliauskas, Vytautas Mickevicius, Monica Dettin, Filippo Vascon, Laura Cendron, and Antonella Pasquato. 2024. "Screening of Small-Molecule Libraries Using SARS-CoV-2-Derived Sequences Identifies Novel Furin Inhibitors" International Journal of Molecular Sciences 25, no. 10: 5079. https://doi.org/10.3390/ijms25105079
APA StyleJorkesh, A., Rothenberger, S., Baldassar, L., Grybaite, B., Kavaliauskas, P., Mickevicius, V., Dettin, M., Vascon, F., Cendron, L., & Pasquato, A. (2024). Screening of Small-Molecule Libraries Using SARS-CoV-2-Derived Sequences Identifies Novel Furin Inhibitors. International Journal of Molecular Sciences, 25(10), 5079. https://doi.org/10.3390/ijms25105079